
Synthesis and Biological Activity of N-acyl Anabasine and Cytisine Derivatives with Adamantane, Pyridine and 1,2-Azole Fragments
 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Evaluation of the Biological Activity
2.2.1. Antimicrobial Activity
2.2.2. Analgesic Activity In Vivo
2.2.3. Antiviral Activity
Cytotoxicity and Chicken-Embryo Lethality of Test Compounds
Virucidal Activity
3. Materials and Methods
3.1. General Chemistry Section
3.2. In Vitro Biological Assays
3.2.1. Antimicrobial Activity
3.2.2. Analgesic Activity In Vivo
3.2.3. Antiviral Activity
The Study of Drug Toxicity
Study of the Virus-Inhibiting Activity of the Samples against Influenza Viruses (H1N1, H3N2 Strains) on a Chicken Embryo Model
Evaluation of the Ability to Suppress the Infectivity of the Influenza Virus (on a Model of 2 Strains)
3.3. Quantum Chemical Calculations
3.4. Experimental Section
3.4.1. General Procedure for the Synthesis of anabasine Derivatives 1a–c,f
3.4.2. General Procedure for the Synthesis of Compounds 1d,e
3.4.3. General Procedure for the Synthesis of Methyliodides 2a–e
3.4.4. General Procedure for the Synthesis of Cytisine Derivatives 3a–c,f
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Prakash, B.; Kujur, A.; Yadav, A. Drug synthesis from natural products: A historical overview and future perspective. In Synthesis of Medicinal Agents from Plant; Tewari, A., Tiwari, S., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; pp. 25–46. [Google Scholar]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Mah, J.; Amirkia, V. Alkaloids Used as Medicines: Structural Phytochemistry Meets Biodiversity—An Update and Forward Look. Molecules 2021, 26, 1836. [Google Scholar] [CrossRef] [PubMed]
- Abookleesh, F.L.; Al-Anzi, B.S.; Ullah, A. Potential Antiviral Action of Alkaloids. Molecules 2022, 27, 903. [Google Scholar] [CrossRef]
- Fielding, B.C.; da Silva Maia Bezerra Filho, C.; Ismail, N.S.M.; Sousa, D.P. Alkaloids: Therapeutic Potential against Human Coronaviruses. Molecules 2020, 25, 5496. [Google Scholar] [CrossRef] [PubMed]
- Kletskov, A.V.; Bumagin, N.A.; Zubkov, F.I.; Grudinin, D.G.; Potkin, V.I. Isothiazoles in the Design and Synthesis of Biologically Active Substances and Ligands for Metal Complexes. Synthesis 2020, 52, 159–188. [Google Scholar] [CrossRef]
- Clerici, F.; Gelmi, M.L.; Pellegrino, S.; Pocar, D. Chemistry of Biologically Active Isothiazoles. In Bioactive Heterocycles III; Khan, M.T.H., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 9, pp. 179–264. [Google Scholar]
- Zhu, J.; Mo, J.; Lin, H.; Chen, Y.; Sun, H. The recent progress of isoxazole in medicinal chemistry. Bioorg. Med. Chem. 2018, 26, 3065–3075. [Google Scholar] [CrossRef]
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. Review on the Medicinal Importance of Pyridine Derivatives. J. Drug Des. Med. Chem. 2015, 1, 1–11. [Google Scholar] [CrossRef]
- Sysak, A.; Obmińska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef]
- Klimochkin, Y.N.; Shiryaev, V.A.; Leonova, M.V. Antiviral properties of cage compounds. New prospects. Russ. Chem. Bull. 2015, 64, 1473–1496. [Google Scholar] [CrossRef]
- Mukusheva, G.K.; Zhasymbekova, A.R.; Seidakhmetova, R.B.; Nurkenov, O.A.; Akishina, E.A.; Petkevich, S.K.; Dikusar, E.A.; Potkin, V.I. Quinine Esters with 1,2-Azole, Pyridine and Adamantane Fragments. Molecules 2022, 27, 3476. [Google Scholar] [CrossRef]
- Ng, Y.P.; Or, T.C.T.; Ip, N.Y. Plant alkaloids as drug leads for Alzheimer’s disease. Neurochem. Int. 2015, 89, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F. Cytisine and cytisine derivatives. More than smoking cessation aids. Pharmacol. Res. 2021, 170, 105700. [Google Scholar] [CrossRef] [PubMed]
- Boido, C.C.; Tasso, B.; Boido, V.; Sparatore, F. Cytisine derivatives as ligands for neuronal nicotine receptors and with various pharmacological activities. Farmaco 2003, 58, 265–277. [Google Scholar] [CrossRef]
- Sobarzo, S.E.; Castedo, L.; De la Fuente, J.R. Synthesis of Anabaseine and Anabasine Derivatives: Structural Modifications of Possible Nicotinic Agonists. Synth. Commun. 2007, 37, 1331–1338. [Google Scholar] [CrossRef]
- Kulakov, I.V.; Nurkenov, O.A.; Zhambekov, Z.M.; Akhmetova, S.B.; Seidakhmetova, R.B. Synthesis and antibacterial and antifungal activities of thiourea derivatives of the alkaloid anabasine. Pharm. Chem. J. 2011, 45, 15–18. [Google Scholar] [CrossRef]
- Tsypysheva, I.P.; Lai, H.C.; Kiu, Y.T.; Koval’skaya, A.V.; Tsypyshev, D.O.; Huang, S.H.; Lin, C.W. Synthesis and antiviral evaluation of cytisine derivatives against dengue virus types 1 and 2. Bioorg. Med. Chem. Lett. 2021, 54, 128437. [Google Scholar] [CrossRef]
- Przybył, A.K.; Maj, E.; Wietrzyk, J.; Kubicki, M. Spectroscopic, structural and anticancer activity studies of (−)-cytisine halogenated N-benzyl derivatives. J. Mol. Struct. 2019, 1176, 871. [Google Scholar] [CrossRef]
- Molyneux, R.J.; Panter, K.E. Alkaloids Toxic to Livestock. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2009; Volume 63, pp. 143–216. [Google Scholar]
- Kamarul Zaman, M.A.; Azzeme, A.M. Plant toxins: Alkaloids and their toxicities. GSC Biol. Pharm. Sci. 2019, 6, 21–29. [Google Scholar] [CrossRef]
- Krasnov, K.A.; Kartsev, V.G. Chemical modification of plant alkaloids. T-reactions of anabasine derivatives. Chem. Nat. Compd. 2011, 46, 915–919. [Google Scholar] [CrossRef]
- Tang, S.; Li, Y.; Bao, Y.; Dai, Z.; Niu, T.; Wang, K.; He, H.; Song, D. Novel cytisine derivatives exert anti-liver fibrosis effect via PI3K/Akt/Smad pathway. Bioorg. Chem. 2019, 90, 103032. [Google Scholar] [CrossRef]
- Rouden, J.; Lasne, M.-C.; Blanchet, J.; Baudoux, J. (−)-Cytisine and Derivatives: Synthesis, Reactivity, and Applications. Chem. Rev. 2013, 114, 712–778. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yang, J.; Dai, Z.; Yu, H.-F.; Ding, C.-F.; Khan, A.; Zhao, Y.-L.; Liu, Y.-P.; Luo, X.-D. Antitumor Pyridine Alkaloids Hybrid with Diverse Units from Alangium chinense. Tetrahedron Lett. 2020, 61, 151502. [Google Scholar] [CrossRef]
- Sowmiah, S.; Esperança, J.M.S.S.; Rebelo, L.P.N.; Afonso, C.A.M. Pyridinium salts: From synthesis to reactivity and applications. Org. Chem. Front. 2018, 5, 453. [Google Scholar] [CrossRef]
- Aoki-Utsubo, C.; Chen, M.; Hotta, H. Virucidal and Neutralizing Activity Tests for Antiviral Substances and Antibodies. Bio Protoc. 2018, 8, e2855. [Google Scholar] [CrossRef]
- Reed, L.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493. [Google Scholar]
- State Pharmacopoeia of the Republic of Kazakhstan; Publishing House “Zhibek Zholy”: Almaty, Kazakhstan, 2015; Volume I, 720p.
- Mironov, A.N. (Ed.) Guidelines for Conducting Preclinical Studies of Drugs—Part 1; GRIF-K: Moscow, Russia, 2012; p. 206. [Google Scholar]
- Council, N.R. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; 220p. [Google Scholar]
- Réus, G.Z.; Stringari, R.B.; de Souza, B.; Petronilho, F.; Dal-Pizzol, F.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; Quevedo, J. Harmine and Imipramine Promote Antioxidant Activities in Prefrontal Cortex and Hippocampus. Oxid. Med. Cell. Longev. 2010, 3, 325–331. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General Atomic and Molecular Electronic-Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Huzinaga, S.; Andzelm, J.M.; Klobukowski, M. Gaussian Basis Sets for Molecular Calculations; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Sadykov, A. Syntheses with anabasine. II. Derivatives of nicotinic acid. Russ. J. Gen. Chem. 1945, 15, 255–258. [Google Scholar]
- Sadykov, A.S.; Tumur, B. Syntheses on the basis of anabasine and N-methyl-α-aminoanabasine. Dokl. Akad. Nauk UzSSR 1960, 8, 38–41. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Compound | Etot1, a.u. | Etot2, a.u. | ΔH, kJ/mol |
|---|---|---|---|
| 1a | −2088.40644 | −2088.39805 | 22.05 |
| 1b | −1081.84594 | −1081.84657 | 1.65 |
| 1c | −1120.93407 | −1120.94068 | 17.35 |
| 1f | −995.98394 | −995.98390 | 0.13 |
| 3a | −2201.09723 | −2201.09734 | 0.27 |
| 3b | −1194.54020 | −1194.54023 | 0.09 |
| 3c | −1233.62838 | −1233.62839 | 0.02 |
| 3f | −1108.68401 | −1108.67736 | 17.45 |
| Compound (c = 1 mg/1 mL) | The Growth Inhibition Zone Diameter (mm) | ||||
|---|---|---|---|---|---|
| Staphylococcus aureus ATCC 6538 | Bacillus subtilis ATCC 6633 | Escherichia coli ATCC 25922 | Pseudomonas aeruginosa ATCC 27853 | Candida albicans ATCC 10231 | |
| anabasine | 17.1 ± 0.1 | 16.1 ± 0.1 | 15.2 ± 0.1 | – | 14.1 ± 0.3 |
| cytisine | 15.0 ± 0.2 | – | 14.1 ± 0.1 | – | 12.2 ± 0.2 |
| 1a | 21.0 ± 0.1 | 14.2 ± 0.1 | – | – | – |
| 1b | 24.0 ± 0.2 | – | 15.1 ± 0.1 | – | – |
| 1c | 20.0 ± 0.2 | 15.1 ± 0.1 | – | – | 14.2 ± 0.1 |
| 1d | 18.2 ± 0.1 | 14.2 ± 0.2 | – | – | – |
| 1e | 16.2 ± 0.1 | – | – | – | – |
| 1f | 23.0 ± 0.2 | – | 16.1 ± 0.1 | – | – |
| 2a | – | – | – | – | – |
| 2b | 20.1 ± 0.1 | 14.0 ± 0.2 | – | – | – |
| 2c | 23.0 ± 0.1 | – | – | – | – |
| 2d | 21.0 ± 0.1 | 17.0 ± 0.1 | – | – | 14.1 ± 0.1 |
| 2e | 19.1 ± 0.2 | – | 14.0 ± 0.2 | – | – |
| 3a | 11.2 ± 0.2 | – | – | – | – |
| 3b | 17.2 ± 0.1 | – | – | – | – |
| 3c | 15.1 ± 0.1 | – | 16.0 ± 0.1 | – | – |
| 3f | 12.2 ± 0.1 | – | – | – | – |
| Benzylpenicillin sodium salt | 16.2 ± 0.1 | 14.2 ± 0.1 | 15.1 ± 0.1 | – | – |
| Gentamicin | 24.0 ± 0.1 | 21.0 ± 0.2 | 26.0 ± 0.1 | 27.0 ± 0.1 | – |
| Nystatin | – | – | – | – | 21.0 ± 0.2 |
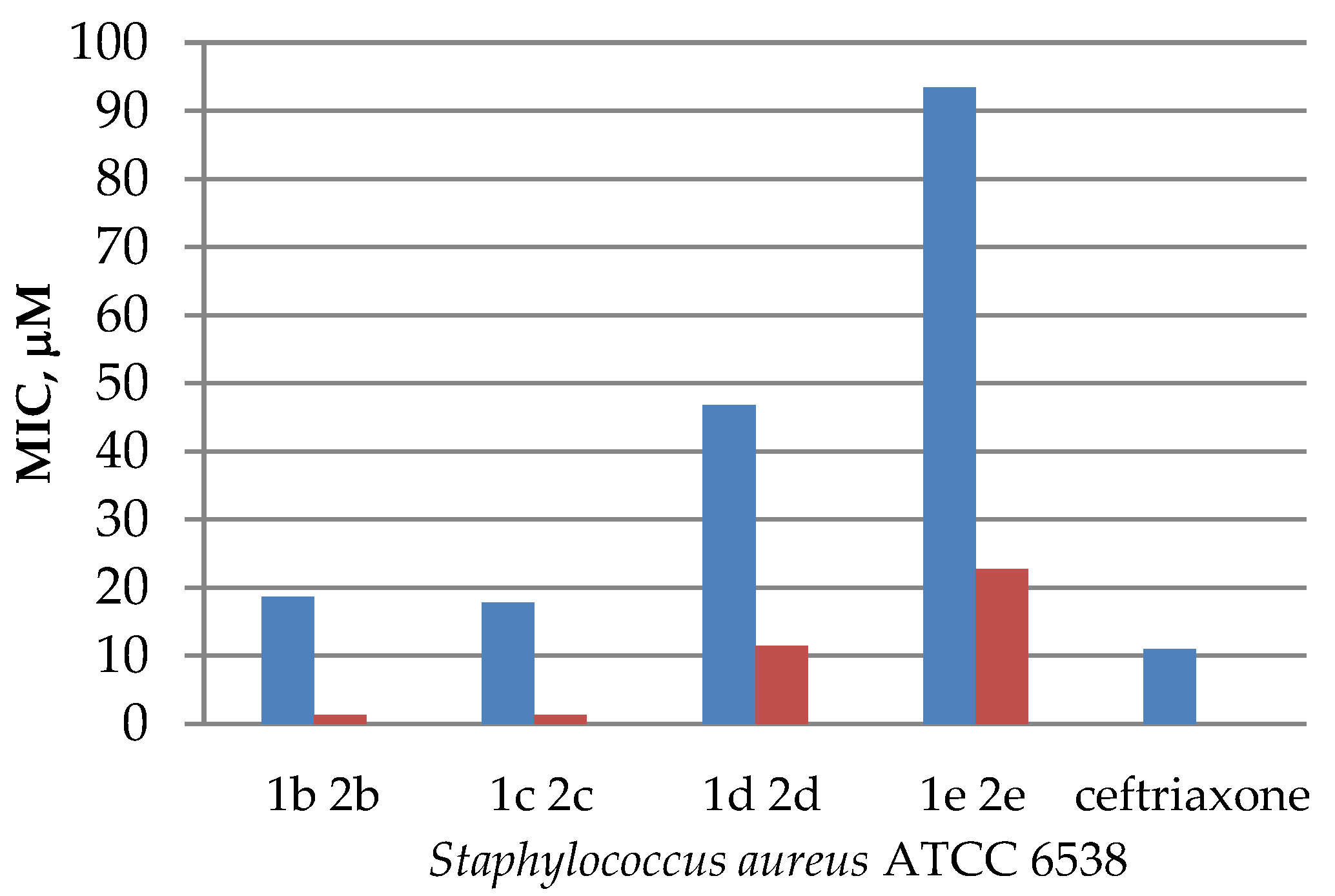
| Compound | MIC (μM) | ||||
|---|---|---|---|---|---|
| Staphylococcus aureus ATCC 6538 | Bacillus subtilis ATCC 6633 | Escherichia coli ATCC 25922 | Pseudomonas aeruginosa ATCC 27853 | Candida albicans ATCC 10231 | |
| anabasine | 77.1 | 77.1 | 154.1 | – | 50 |
| cytisine | 131.4 | – | 262.8 | – | – |
| 1a | 18.1 | 146.1 | – | – | – |
| 1b | 18.6 | – | 75.0 | – | – |
| 1c | 17.8 | 72.0 | – | – | 144.0 |
| 1d | 46.8 | 187.0 | – | – | – |
| 1e | 93.5 | – | – | – | – |
| 1f | 19.1 | – | 38.5 | – | – |
| 2a | – | – | – | – | – |
| 2b | 1.3 | 10.5 | – | – | – |
| 2c | 1.3 | – | – | – | – |
| 2d | 11.4 | 45.4 | – | – | 90.7 |
| 2e | 22.7 | – | 90.7 | – | – |
| 3a | – | – | – | – | – |
| 3b | 3.5 | – | – | – | – |
| 3c | 6.7 | – | 3.3 | – | – |
| 3f | – | – | – | – | – |
| Ceftriaxone | 11 | 22 | 11 | 22 | – |
| Nystatin | – | – | – | – | 14 |
| Compound | Dose, mg/kg | Writhing Number | Decrease in the Vinegar Writhing Number (%) |
|---|---|---|---|
| control | - | 106.2 ± 11.2 | 100 |
| sodium diclofenac | 8 | 49.5 ± 10.7 | 53.3 |
| 1a | 25 | 61.2 ± 8.5 | 42.4 |
| 1b | 25 | 60.6 ± 8.7 | 42.9 |
| 1c | 25 | 75.7 ± 8.2 | 28.7 |
| 1d | 25 | 54.1 ± 11.7 | 49.0 |
| 1e | 25 | 65.4 ± 11.5 | 38.4 |
| 1f | 25 | 76.8 ± 10.2 | 27.7 |
| 2a | 25 | 59.4 ± 6.1 | 44.1 |
| 2b | 25 | 56.7 ± 9.5 | 46.6 |
| 2c | 25 | 58.2 ± 9.3 | 45.2 |
| 2d | 25 | 58.5 ± 10.6 | 44.9 |
| 2e | 25 | 59.3 ± 10.4 | 44.2 |
| 3a | 25 | 82.5 ± 10.4 | 22.3 |
| 3b | 25 | 77.4 ± 10.1 | 27.1 |
| 3c | 25 | 69.6 ± 9.2 | 34.5 |
| 3f | 25 | 71.3 ± 8.4 | 32.8 |
| Compound | Chemical Therapeutic Index of the Drug | |
|---|---|---|
| A/Almaty/8/98 (H3N2) | A/Vladivostok/2/09 (H1N1) | |
| 1b | 6.0 | 5.7 |
| 1c | 8.0 | 8.1 |
| 1f | 60.0 | 60.5 |
| 2c | 4.0 | 3.8 |
| 3b | 6.0 | 6.1 |
| 3c | 8.0 | 7.8 |
| 3f | 50.0 | 52.3 |
| Tamiflu | 10.3 | 11.0 |
| Remantadine | 29.9 | 30.0 |
| Compound 0.2%, 0.4 mg/Chick Embryo (8 mg/kg) | Neutralization Index: (Titer Control—Titer Experiment), lg | |
|---|---|---|
| H3N2 | H1N1 | |
| 1b | 0.75 | 0.8 |
| 1c | 0.75 | 0.8 |
| 1f | 1.25 | 1.3 |
| 2c | 0.25 | 0.3 |
| 3b | 0.75 | 0.8 |
| 3c | 0.75 | 0.8 |
| 3f | 1.0 | 1.1 |
| Tamiflu | 0.7 | 0.75 |
| Remantadine | 0.6 | 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukusheva, G.K.; Zhasymbekova, A.R.; Zhumagalieva, Z.Z.; Seidakhmetova, R.B.; Nurkenov, O.A.; Akishina, E.A.; Petkevich, S.K.; Dikusar, E.A.; Potkin, V.I. Synthesis and Biological Activity of N-acyl Anabasine and Cytisine Derivatives with Adamantane, Pyridine and 1,2-Azole Fragments. Molecules 2022, 27, 7387. https://doi.org/10.3390/molecules27217387
Mukusheva GK, Zhasymbekova AR, Zhumagalieva ZZ, Seidakhmetova RB, Nurkenov OA, Akishina EA, Petkevich SK, Dikusar EA, Potkin VI. Synthesis and Biological Activity of N-acyl Anabasine and Cytisine Derivatives with Adamantane, Pyridine and 1,2-Azole Fragments. Molecules. 2022; 27(21):7387. https://doi.org/10.3390/molecules27217387
Chicago/Turabian StyleMukusheva, Gulim K., Aigerym R. Zhasymbekova, Zharkyn Zh. Zhumagalieva, Roza B. Seidakhmetova, Oralgazy A. Nurkenov, Ekaterina A. Akishina, Sergey K. Petkevich, Evgenij A. Dikusar, and Vladimir I. Potkin. 2022. "Synthesis and Biological Activity of N-acyl Anabasine and Cytisine Derivatives with Adamantane, Pyridine and 1,2-Azole Fragments" Molecules 27, no. 21: 7387. https://doi.org/10.3390/molecules27217387
APA StyleMukusheva, G. K., Zhasymbekova, A. R., Zhumagalieva, Z. Z., Seidakhmetova, R. B., Nurkenov, O. A., Akishina, E. A., Petkevich, S. K., Dikusar, E. A., & Potkin, V. I. (2022). Synthesis and Biological Activity of N-acyl Anabasine and Cytisine Derivatives with Adamantane, Pyridine and 1,2-Azole Fragments. Molecules, 27(21), 7387. https://doi.org/10.3390/molecules27217387