
Anti-α-Glucosidase and Antiglycation Activities of α-Mangostin and New Xanthenone Derivatives: Enzymatic Kinetics and Mechanistic Insights through In Vitro Studies


 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
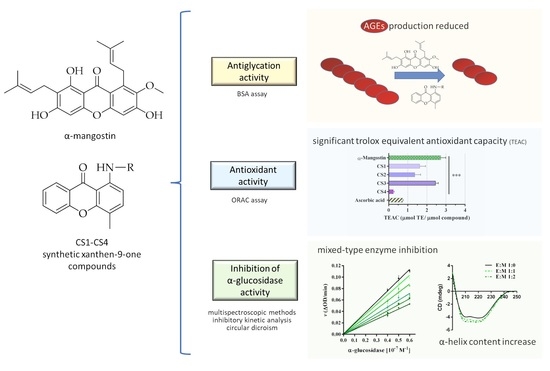
2. Results and Discussion
2.1. α-Mangostin and Synthetic Compounds CS1–CS4: Chemistry
2.2. α-Mangostin and Synthetic Compounds CS1–CS4: Antiglycation Activity
2.3. α-Mangostin and Synthetic Compounds CS1–CS4: Antioxidant Activity
2.4. Yeast α-Glucosidase Inhibitory Activity
2.4.1. α-Mangostin and Synthetic Compounds CS1–CS4
2.4.2. α-Mangostin Inhibition with Different Concentrations of pNPG
2.4.3. α-Glucosidase Fluorescence Quenching by α-Mangostin
2.4.4. Circular Dichroism Measurements
3. Materials and Methods
3.1. Reagents
3.2. Chemistry
3.2.1. General Information
3.2.2. General Synthesis of Compounds CS1–CS4
3.3. Detection of Advanced Glycation End Products
3.4. Measurement of the Oxygen Radical Absorbance Capacity
3.5. Measurement of α-Glucosidase Activity
3.5.1. Fluorescence Quenching Analysis
3.5.2. Circular Dichroism Measurements
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Paul, S.; Ali, A.; Katare, R. Molecular complexities underlying the vascular complications of diabetes mellitus–A comprehensive review. J. Diabetes Its Complicat. 2020, 34, 107613. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef]
- Ceriello, A.; Catrinoiu, D.; Chandramouli, C.; Cosentino, F.; Dombrowsky, A.C.; Itzhak, B.; Lalic, N.M.; Prattichizzo, F.; Schnell, O.; Seferović, P.M.; et al. Heart failure in type 2 diabetes: Current perspectives on screening, diagnosis and management. Cardiovasc. Diabetol. 2021, 20, 1–19. [Google Scholar] [CrossRef]
- Fournet, M.; Bonté, F.; Desmoulière, A. Glycation damage: A possible hub for major pathophysiological disorders and aging. Aging Dis. 2018, 9, 880–900. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.Y.; Lu, C.H.; Wu, C.H.; Li, K.J.; Kuo, Y.M.; Hsieh, S.C.; Yu, C.L. The development of Maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with age-related diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef]
- Padhi, S.; Nayak, A.K.; Behera, A. Type II diabetes mellitus: A review on recent drug based therapeutics. Biomed. Pharmacother. 2020, 131, 110708. [Google Scholar] [CrossRef]
- Kusunoki, M.; Wakazono, N.; Wakazono, N.; Tsutsumi, K.; Tsutsumi, K.; Oshida, Y.; Oshida, Y.; Miyata, T.; Miyata, T. Combination therapy of α-glucosidase inhibitor, thiazolidinedione and sodium glucose co-transporter-2 inhibitor in Japanese type 2 diabetes patients. J. Endocrinol. Metab. 2020, 10, 6. [Google Scholar] [CrossRef]
- Blahova, J.; Martiniakova, M.; Babikova, M.; Kovacova, V.; Mondockova, V.; Omelka, R. Pharmaceutical drugs and natural therapeutic products for the treatment of type 2 diabetes mellitus. Pharmaceuticals 2021, 14, 806. [Google Scholar] [CrossRef] [PubMed]
- Liandhajani; Immaculata Iwo, M.; Sukrasno; Soemardji, A.A.; Hanafi, M. Sunscreen activity of α-mangostin from the pericarps of Garcinia mangostana Linn. J. Appl. Pharm. Sci. 2013, 3, 70–73. [Google Scholar] [CrossRef]
- Kumar, V.; Bhatt, P.C.; Kaithwas, G.; Rashid, M.; Al-abbasi, F.A.; Khan, J.A.J.; Anwar, F.; Verma, A. α-Mangostin mediated pharmacological modulation of hepatic carbohydrate metabolism in diabetes induced Wistar rat. Beni-Suef Univ. J. Basic Appl. Sci. 2016, 5, 255–276. [Google Scholar] [CrossRef]
- Usman, F.; Shah, H.S.; Zaib, S.; Manee, S.; Mudassir, J.; Khan, A.; Batiha, G.E.; Abualnaja, K.M.; Alhashmialameer, D.; Khan, I. Fabrication and biological assessment of antidiabetic α-Mangostin loaded nanosponges: In vitro, in vivo, and in silico studies. Molecules 2021, 26, 6633. [Google Scholar] [CrossRef]
- Ryu, H.W.; Cho, J.K.; Curtis-Long, M.J.; Yuk, H.J.; Kim, Y.S.; Jung, S.; Kim, Y.S.; Lee, B.W.; Park, K.H. α-Glucosidase inhibition and antihyperglycemic activity of prenylated xanthones from Garcinia mangostana. Phytochemistry 2011, 72, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Vongsak, B.; Kongkiatpaiboon, S.; Jaisamut, S.; Machana, S.; Pattarapanich, C. In vitro α-glucosidase inhibition and free-radical scavenging activity of propolis from Thai stingless bees in mangosteen orchard. Rev. Bras. Farmacogn. 2015, 25, 445–450. [Google Scholar] [CrossRef]
- Gutierrez-Orozco, F.; Failla, M.L. Biological activities and bioavailability of mangosteen xanthones: A critical review of the current evidence. Nutrients 2013, 5, 3163–3183. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Cho, J. Mangosteen pericarp and its bioactive xanthones: Potential therapeutic value in Alzheimer’s disease, Parkinson’s disease, and depression with pharmacokinetic and safety profiles. Int. J. Mol. Sci. 2020, 21, 6211. [Google Scholar] [CrossRef]
- Chayaburakul, K.; Sukplang, P.; Pinpart, T.; Kengkoom, K. Acute and subchronic oral toxicity of xanthones extracted from the pericarp of Garcinia mangostana Linn. in Rat. Asian Arch. Pathol. 2015, 11, 57–70. [Google Scholar]
- Santos, C.M.M.; Freitas, M.; Fernandes, E. A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 2018, 157, 1460–1479. [Google Scholar] [CrossRef]
- Ye, G.J.; Lan, T.; Huang, Z.X.; Cheng, X.N.; Cai, C.Y.; Ding, S.M.; Xie, M.L.; Wang, B. Design and synthesis of novel xanthone-triazole derivatives as potential antidiabetic agents: α-Glucosidase inhibition and glucose uptake promotion. Eur. J. Med. Chem. 2019, 177, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Mauss, H. Über basisch substituierte xanthon- und thioxanthon-abkömmlinge; miracil, ein neues chemotherapeuticum. Chem. Ber. 1948, 81, 19–31. [Google Scholar] [CrossRef]
- Wei, Y.; Han, C.S.; Zhou, J.; Liu, Y.; Chen, L.; He, R.Q. D-ribose in glycation and protein aggregation. Biochimica Biophysica Acta–Gen. Subj. 2012, 1820, 488–494. [Google Scholar] [CrossRef]
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Kim, Y.S. The role of advanced glycation end products in diabetic vascular complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar] [CrossRef]
- Abdallah, H.M.; El-Bassossy, H.M.; Mohamed, G.A.; El-Halawany, A.M.; Alshali, K.Z.; Banjar, Z.M. Mangostana xanthones III and IV: Advanced glycation end-product inhibitors from the pericarp of Garcinia mangostana. J. Nat. Med. 2017, 71, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.M.; El-Bassossy, H.; Mohamed, G.A.; El-Halawany, A.M.; Alshali, K.Z.; Banjar, Z.M. Phenolics from Garcinia mangostana inhibit advanced glycation endproducts formation: Effect on Amadori products, cross-linked structures and protein thiols. Molecules 2016, 21, 251. [Google Scholar] [CrossRef]
- Cao, G. Oxygen-radical absorbance capacity assay for antioxidants. Free Radic. Biol. Med. 1993, 14, 303. [Google Scholar] [CrossRef]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J. Agric. Food Chem. 2005, 53, 4290–4302. [Google Scholar] [CrossRef]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant and prooxidant behavior of flavonoids: Structure-activity relationships. Free Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar] [CrossRef]
- Minami, H.; Kuwayama, A.; Yoshizawa, T.; Fukuyama, Y. Novel prenylated xanthones with antioxidant property from the wood of Garcinia subelliptica. Chem. Pharm. Bull. 1996, 2103–2106. [Google Scholar] [CrossRef]
- Thong, N.M.; Quang, D.T.; Bui, N.H.T.; Dao, D.Q.; Nam, P.C. Antioxidant properties of xanthones extracted from the pericarp of Garcinia mangostana (Mangosteen): A theoretical study. Chem. Phys. Lett. 2015, 625, 30–35. [Google Scholar] [CrossRef]
- Panda, S.S.; Chand, M.; Sakhuja, R.; Jain, S.C. Xanthones as potential antioxidants. Chem. Phys. Lett. 2013, 20, 4481–4507. [Google Scholar] [CrossRef]
- Anwar, S.; Khan, S.; Almatroudi, A.; Khan, A.A.; Alsahli, M.A.; Almatroodi, S.A.; Rahmani, A.H. A review on mechanism of inhibition of advanced glycation end products formation by plant derived polyphenolic compounds. Mol. Biol. Rep. 2021, 48, 787–805. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.I.; Apostolidis, E.; Shetty, K. In vitro studies of eggplant (Solanum melongena) phenolics as inhibitors of key enzymes relevant for type 2 diabetes and hypertension. Bioresour. Technol. 2008, 99, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.P.; Song, Y.H.; Uddin, Z.; Wang, Y.; Park, K.H. Inhibition of protein tyrosine phosphatase 1B (PTP1B) and α-glucosidase by xanthones from Cratoxylum cochinchinense, and their kinetic characterization. Bioorganic Med. Chem. 2018, 26, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.K.; Truong, X.A.; Bui, T.Q.; Bui, D.N.; Nguyen, H.X.; Tran, P.T.; Nguyen, L.H.D. α-Glucosidase inhibitory xanthones from the roots of Garcinia fusca. Chem. Biodivers. 2017, 14, e1700232. [Google Scholar] [CrossRef]
- Grant, G.A. The many faces of partial inhibition: Revealing imposters with graphical analysis. Arch. Biochem. Biophys. 2018, 653, 10–23. [Google Scholar] [CrossRef]
- Han, L.; Fang, C.; Zhu, R.; Peng, Q.; Li, D.; Wang, M. Inhibitory effect of phloretin on α-glucosidase: Kinetics, interaction mechanism and molecular docking. Int. J. Biol. Macromol. 2017, 95, 520–527. [Google Scholar] [CrossRef]
- Dai, T.; Li, T.; He, X.; Li, X.; Liu, C.; Chen, J.; McClements, D.J. Analysis of inhibitory interaction between epigallocatechin gallate and α-glucosidase: A spectroscopy and molecular simulation study. Spectrochimica Acta–Part A Mol. Biomol. Spectrosc. 2020, 230, 118023. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. A probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef]
- Kohlmann, T.; Goez, M. Combined static and dynamic intramicellar fluorescence quenching: Effects on stationary and time-resolved Stern-Volmer experiments. Phys. Chem. Chem. Phys. 2019, 21, 10075–10085. [Google Scholar] [CrossRef]
- Zhao, L.; Wen, L.; Lu, Q.; Liu, R. Interaction mechanism between α-glucosidase and A-type trimer procyanidin revealed by integrated spectroscopic analysis techniques. Int. J. Biol. Macromol. 2020, 143, 173–180. [Google Scholar] [CrossRef]
- Lakowicz, J. Principles of Fluorescence Spectroscopy; Lakowicz, J.R., Ed.; Springer: Boston, MA, USA, 2013; ISBN 9781475787702, 1475787707. [Google Scholar]
- Zeng, L.; Zhang, G.; Lin, S.; Gong, D. Inhibitory mechanism of apigenin on α-glucosidase and synergy analysis of flavonoids. J. Agric. Food Chem. 2016, 64, 6939–6949. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Ding, H.; Hu, X.; Zhang, G.; Gong, D. Galangin inhibits α-glucosidase activity and formation of non-enzymatic glycation products. Food Chem. 2019, 271, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Hu, X.; Xu, X.; Zhang, G.; Gong, D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Int. J. Biol. Macromol. 2018, 107, 1844–1855. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Hu, P.; Cao, X.; Chen, Y.; Xu, Y.; He, T.; Wei, Y.; He, R. Comparison of bovine serum albumin glycation by ribose and fructose in vitro and in vivo. Biochimica Biophysica Acta–Mol. Basis Dis. 2022, 1868, 166283. [Google Scholar] [CrossRef]
- Chen, Z.; Bertin, R.; Marin, R.; Medjiofack Djeujo, F.; Froldi, G. Effects of Croton lechleri sap (Sangre de Drago) on AGEs formation, LDL oxidation and oxidative stress related to vascular diseases. Nat. Prod. Res. 2021. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Medjiofack Djeujo, F.; Cusinato, F.; Ragazzi, E.; Froldi, G. α-Glucosidase and advanced glycation end products inhibition with Vernonia amygdalina root and leaf extracts: New data supporting the antidiabetic properties. J. Pharm. Pharmacol. 2021, 73, 1240–1249. [Google Scholar] [CrossRef]
- Iqbal, S.; Rehman, N.U.; Kortz, U.; Iqbal, J. Development of a fast and efficient CE enzyme assay for the characterization and inhibition studies of α-glucosidase inhibitors. J. Sep. Sci. 2013, 36, 3623–3628. [Google Scholar] [CrossRef]
- Ghisaidoobe, A.B.T.; Chung, S.J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Förster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Green, R.J.; Frazier, R.A. Interaction of flavonoids with bovine serum albumin: A fluorescence quenching study. J. Agric. Food Chem. 2005, 53, 158–163. [Google Scholar] [CrossRef]
- Yang, J.; Wang, X.; Zhang, C.; Ma, L.; Wei, T.; Zhao, Y.; Peng, X. Comparative study of inhibition mechanisms of structurally different flavonoid compounds on α-glucosidase and synergistic effect with acarbose. Food Chem. 2021, 347, 129056. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.E.; Bruzzone, L. Synchronous fluorescence spectrometry: Conformational investigation or inner filter effect? J. Lumin. 2013, 137, 138–142. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2007, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α-Mangostin (µM) | Km (mM) | Vmax (ΔOD/min) | Vmax (µM/min) |
|---|---|---|---|
| -- | 0.454 ± 0.043 | 0.118 ± 0.003 | 11.80 ± 0.30 |
| 1 | 0.490 ± 0.058 | 0.111 ± 0.004 | 11.10 ± 0.40 |
| 5 | 0.504 ± 0.079 | 0.097 ± 0.005 | 9.70 ± 0.50 |
| 10 | 0.492 ± 0.079 | 0.083 ± 0.004 | 8.30 ± 0.40 |
| 25 | 0.524 ± 0.075 | 0.068 ± 0.003 | 6.80 ± 0.30 |
| 50 | 0.651 ± 0.126 | 0.057 ± 0.004 | 5.70 ± 0.40 |
| α-Mangostin (µM) | Km (mM) | Kcat (10−3 sec−1) | Kcat/Km (sec−1 mM−1) | Ki (µM) | Ki’ (µM) |
|---|---|---|---|---|---|
| 0 | 0.454 ± 0.043 | 3.90 ± 1.10 | 0.087 ± 0.03 | 30.33 | 45.60 |
| 1 | 0.490 ± 0.058 | 3.70 ± 1.43 | 0.076 ± 0.02 | ||
| 5 | 0.504 ± 0.079 | 3.24 ± 1.62 | 0.064 ± 0.01 | ||
| 10 | 0.492 ± 0.079 | 2.76 ± 1.38 | 0.056 ± 0.02 | ||
| 25 | 0.524 ± 0.075 | 2.26 ± 1.05 | 0.043 ± 0.01 | ||
| 50 | 0.585 ± 0.126 | 1.85 ± 1.76 | 0.032 ± 0.01 |
| T (K) | Ksv (105 M−1) | Kq (1013 M−1 s−1) | R |
|---|---|---|---|
| 298 | 1.92 ± 0.01 | 1.92 ± 0.01 | 0.9733 |
| 304 | 2.30 ± 0.01 | 2.30 ± 0.01 | 0.9731 |
| 310 | 2.62 ± 0.01 | 262 ± 0.01 | 0.9718 |
| Molar Ratios [E]:[M] | α-Helix (%) | β-Sheet (%) | β-Turn (%) | Random Coil (%) |
|---|---|---|---|---|
| 1:0 | 43.22 | 14.05 | 17.56 | 25.17 |
| 1:1 | 51.76 | 5.77 | 15.26 | 27.20 |
| 1:2 | 52.95 | 5.99 | 4.70 | 36.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djeujo, F.M.; Francesconi, V.; Gonella, M.; Ragazzi, E.; Tonelli, M.; Froldi, G. Anti-α-Glucosidase and Antiglycation Activities of α-Mangostin and New Xanthenone Derivatives: Enzymatic Kinetics and Mechanistic Insights through In Vitro Studies. Molecules 2022, 27, 547. https://doi.org/10.3390/molecules27020547
Djeujo FM, Francesconi V, Gonella M, Ragazzi E, Tonelli M, Froldi G. Anti-α-Glucosidase and Antiglycation Activities of α-Mangostin and New Xanthenone Derivatives: Enzymatic Kinetics and Mechanistic Insights through In Vitro Studies. Molecules. 2022; 27(2):547. https://doi.org/10.3390/molecules27020547
Chicago/Turabian StyleDjeujo, Francine Medjiofack, Valeria Francesconi, Maddalena Gonella, Eugenio Ragazzi, Michele Tonelli, and Guglielmina Froldi. 2022. "Anti-α-Glucosidase and Antiglycation Activities of α-Mangostin and New Xanthenone Derivatives: Enzymatic Kinetics and Mechanistic Insights through In Vitro Studies" Molecules 27, no. 2: 547. https://doi.org/10.3390/molecules27020547
APA StyleDjeujo, F. M., Francesconi, V., Gonella, M., Ragazzi, E., Tonelli, M., & Froldi, G. (2022). Anti-α-Glucosidase and Antiglycation Activities of α-Mangostin and New Xanthenone Derivatives: Enzymatic Kinetics and Mechanistic Insights through In Vitro Studies. Molecules, 27(2), 547. https://doi.org/10.3390/molecules27020547