
Simultaneous Quantification of Opioids in Blood and Urine by Gas Chromatography-Mass Spectrometer with Modified Dispersive Solid-Phase Extraction Technique

 ,
,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Methodology
2.1. Chemicals and Reagents
2.2. Preparation of Standard Solutions
2.3. Preparation of Standards
2.4. Preparation of Quality Controls Checks
2.5. Sample Collection and Storage
2.6. Extraction Procedure
2.7. Instrument Programming
2.8. Target Analytes with Respective SIM Ions
2.9. Method Validation
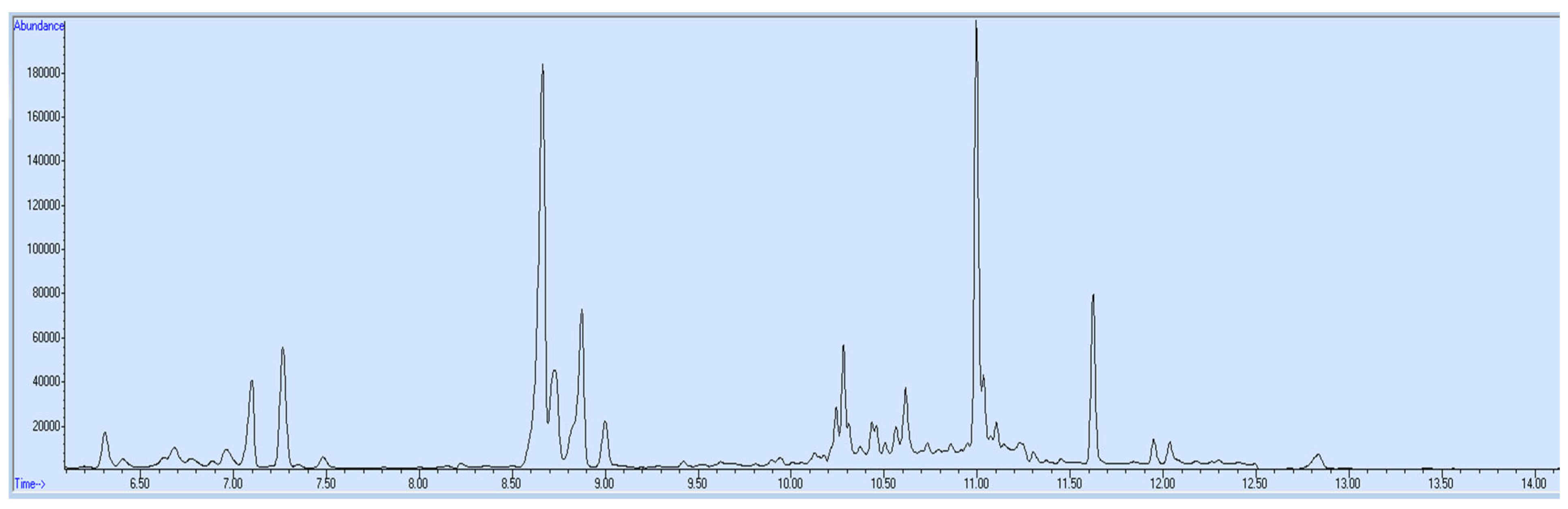
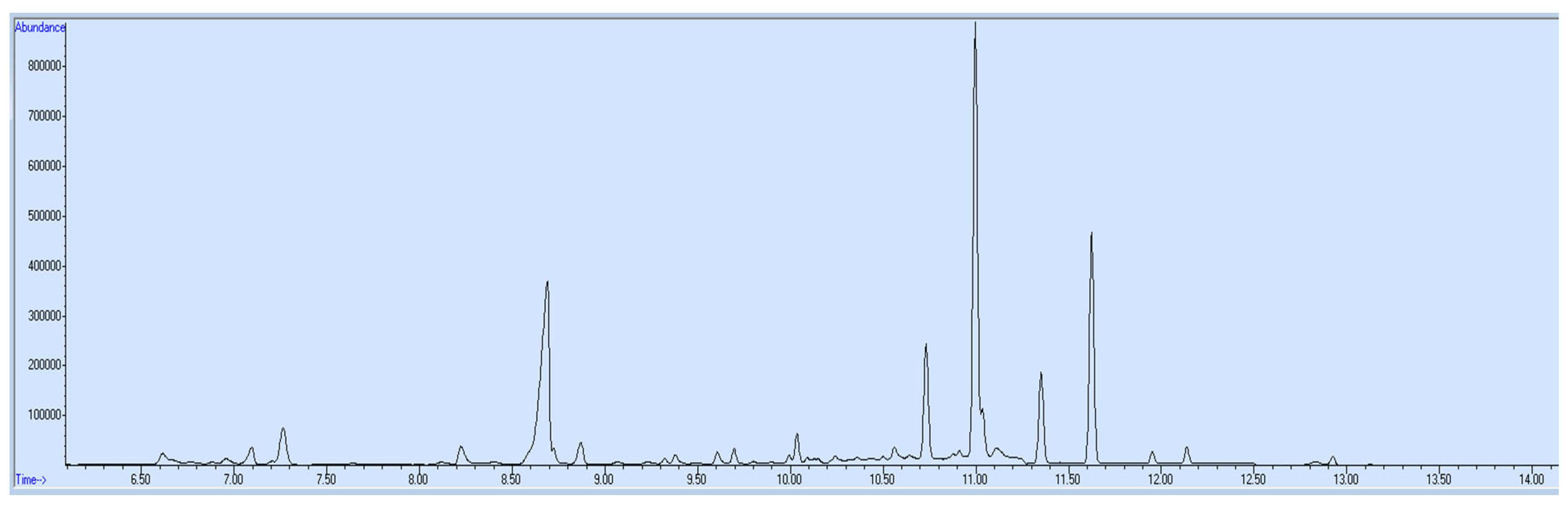
3. Results and Discussion
4. Application
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Høiseth, G.; Bernard, J.P.; Karinen, R.; Johnsen, L.; Helander, A.; Christophersen, A.S.; Mørland, J. A pharmacokinetic study of ethyl glucuronide in blood and urine: Applications to forensic toxicology. Forensic Sci. Int. 2007, 172, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Busardo, F.P.; Jones, A.W. GHB pharmacology and toxicology: Acute intoxication, concentrations in blood and urine in forensic cases and treatment of the withdrawal syndrome. Curr. Neuropharmacol. 2015, 13, 47–70. [Google Scholar] [CrossRef]
- Tenore, P.L. Advanced urine toxicology testing. J. Addict. Dis. 2010, 29, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Finoulst, I.; Pinkse, M.; Van Dongen, W.; Verhaert, P. Sample preparation techniques for the untargeted LC-MS-based discovery of peptides in complex biological matrices. J. Biomed. Biotechnol. 2011, 2011, 245291. [Google Scholar] [CrossRef] [PubMed]
- Santana, M.F.; Kroon, F.J.; van Herwerden, L.; Vamvounis, G.; Motti, C.A. An assessment workflow to recover microplastics from complex biological matrices. Mar. Pollut. Bull. 2022, 179, 113676. [Google Scholar] [CrossRef]
- Choi, S.; Kim, S.; Shin, J.Y.; Kim, M.; Kim, J.H. Development and verification for analysis of pesticides in eggs and egg products using QuEChERS and LC–MS/MS. Food Chem. 2015, 173, 1236–1242. [Google Scholar] [CrossRef]
- Koesukwiwat, U.; Lehotay, S.J.; Mastovska, K.; Dorweiler, K.J.; Leepipatpiboon, N. Extension of the QuEChERS method for pesticide residues in cereals to flaxseeds, peanuts, and doughs. J. Agric. Food Chem. 2010, 58, 5950–5958. [Google Scholar] [CrossRef]
- Manav, Ö.G.; Dinç-Zor, Ş.; Alpdoğan, G. Optimization of a modified QuEChERS method by means of experimental design for multiresidue determination of pesticides in milk and dairy products by GC–MS. Microchem. J. 2019, 144, 124–129. [Google Scholar] [CrossRef]
- Lawal, A.; Wong, R.C.; Tan, G.H.; Abdulra’uf, L.B.; Alsharif, A.M. Recent modifications and validation of QuEChERS-dSPE coupled to LC–MS and GC–MS instruments for determination of pesticide/agrochemical residues in fruits and vegetables. J. Chromatogr. Sci. 2018, 56, 656–669. [Google Scholar] [CrossRef]
- Kusano, M.; Sakamoto, Y.; Natori, Y.; Miyagawa, H.; Tsuchihashi, H.; Ishii, A.; Zaitsu, K. Development of “quick-DB forensic”: A total workflow from QuEChERS-dSPE method to GC–MS/MS quantification of forensically relevant drugs and pesticides in whole blood. Forensic Sci. Int. 2019, 300, 125–135. [Google Scholar] [CrossRef]
- Csonka, C.; Páli, T.; Bencsik, P.; Görbe, A.; Ferdinandy, P.; Csont, T. Measurement of NO in biological samples. Br. J. Pharmacol. 2015, 172, 1620–1632. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.C.; Harding, W.R.; Archibald, C.G. A methods manual for the collection, preparation and analysis of diatom samples. Version 2007, 1, 60. [Google Scholar]
- Pavlović, D.M.; Babić, S.; Horvat, A.J.; Kaštelan-Macan, M. Sample preparation in analysis of pharmaceuticals. TrAC Trends Anal. Chem. 2007, 26, 1062–1075. [Google Scholar] [CrossRef]
- Álvarez-Sánchez, B.; Priego-Capote, F.; de Castro, M.L. Metabolomics analysis II. Preparation of biological samples prior to detection. TrAC Trends Anal. Chem. 2010, 29, 120–127. [Google Scholar] [CrossRef]
- Feist, P.; Hummon, A.B. Proteomic challenges: Sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef]
- Liakh, I.; Pakiet, A.; Sledzinski, T.; Mika, A. Modern methods of sample preparation for the analysis of oxylipins in biological samples. Molecules 2019, 24, 1639. [Google Scholar] [CrossRef]
- Poole, C.F. New trends in solid-phase extraction. TrAC Trends Anal. Chem. 2003, 22, 362–373. [Google Scholar] [CrossRef]
- Buszewski, B.; Szultka, M. Past, present, and future of solid phase extraction: A review. Crit. Rev. Anal. Chem. 2012, 42, 198–213. [Google Scholar] [CrossRef]
- Thurman, E.M.; Mills, M.S. Solid-Phase Extraction: Principles and Practice; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Simpson, N.J. Solid-Phase Extraction: Principles, Techniques, and Applications; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Żwir-Ferenc, A.; Biziuk, M. Solid Phase Extraction Technique—Trends, Opportunities and Applications. Pol. J. Environ. Stud. 2006, 15, 677–690. [Google Scholar]
- Mazzola, P.G.; Lopes, A.M.; Hasmann, F.A.; Jozala, A.F.; Penna, T.C.; Magalhaes, P.O.; Rangel-Yagui, C.O.; Pessoa, A., Jr. Liquid–liquid extraction of biomolecules: An overview and update of the main techniques. J. Chem. Technol. Biotechnol. Int. Res. Process Environ. Clean Technol. 2008, 83, 143–157. [Google Scholar] [CrossRef]
- Müller, E.; Berger, R.; Blass, E.; Sluyts, D.; Pfennig, A. Liquid–liquid extraction. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley Online Library: Hoboken, NJ, USA, 2000. [Google Scholar]
- Silvestre, C.I.; Santos, J.L.; Lima, J.L.; Zagatto, E.A. Liquid–liquid extraction in flow analysis: A critical review. Anal. Chim. Acta 2009, 652, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kralj, J.G.; Sahoo, H.R.; Jensen, K.F. Integrated continuous microfluidic liquid–liquid extraction. Lab Chip 2007, 7, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Remane, D.; Meyer, M.R.; Peters, F.T.; Wissenbach, D.K.; Maurer, H.H. Fast and simple procedure for liquid–liquid extraction of 136 analytes from different drug classes for development of a liquid chromatographic-tandem mass spectrometric quantification method in human blood plasma. Anal. Bioanal. Chem. 2010, 397, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Morales, L.; Vazquez, C.; Bermejo, A.M.; Tabernero, M.J. HPLC–DAD determination of opioids, cocaine and their metabolites in plasma. Forensic Sci. Int. 2006, 161, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, T.; El-Shihi, T.H.; Emara, M.M.; Giorgi, M. New HPLC method to detect individual opioids (heroin and tramadol) and their metabolites in the blood of rats on combination treatment. J. Chromatogr. Sci. 2012, 50, 658–665. [Google Scholar] [CrossRef]
- Pragst, F.; Herzler, M.; Erxleben, B.T. Systematic toxicological analysis by high-performance liquid chromatography with diode array detection (HPLC-DAD). Clin. Chem. Lab. Med. (CCLM) 2004, 42, 1325–1340. [Google Scholar] [CrossRef]
- French, D. The challenges of LC–MS/MS analysis of opiates and opioids in urine. Bioanalysis 2013, 5, 2803–2820. [Google Scholar] [CrossRef]
- Musshoff, F.; Trafkowski, J.; Kuepper, U.; Madea, B. An automated and fully validated LC-MS/MS procedure for the simultaneous determination of 11 opioids used in palliative care, with 5 of their metabolites. J. Mass Spectrom. 2006, 41, 633–640. [Google Scholar] [CrossRef]
- Valdez, C.A. Gas chromatography-mass spectrometry analysis of synthetic opioids belonging to the fentanyl class: A review. Crit. Rev. Anal. Chem. 2021, 1–31. [Google Scholar] [CrossRef]
- Amorim Alves, E.; Sofia Agonia, A.; Manuela Cravo, S.; Manuel Afonso, C.; Duarte Pereira Netto, A.; de Lourdes Bastos, M.; Carvalho, F.; Jorge Dinis-Oliveira, R. GC-MS method for the analysis of thirteen opioids, cocaine and cocaethylene in whole blood based on a modified quechers extraction. Curr. Pharm. Anal. 2017, 13, 215–223. [Google Scholar] [CrossRef]
- Meatherall, R. GC-MS confirmation of codeine, morphine, 6-acetylmorphine, hydrocodone, hydromorphone, oxycodone, and oxymorphone in urine. J. Anal. Toxicol. 1999, 23, 177–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sisco, E.; Burns, A.; Moorthy, A.S. Development and evaluation of a synthetic opioid targeted gas chromatography mass spectrometry (GC-MS) method. J. Forensic Sci. 2021, 66, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Schummer, C.; Delhomme, O.; Appenzeller, B.M.; Wennig, R.; Millet, M. Comparison of MTBSTFA and BSTFA in derivatization reactions of polar compounds prior to GC/MS analysis. Talanta 2009, 77, 1473–1482. [Google Scholar] [CrossRef]
- Valverde, S.; Ares, A.M.; Bernal, J.L.; Nozal, M.J.; Bernal, J. Fast determination of neonicotinoid insecticides in beeswax by ultra-high performance liquid chromatography-tandem mass spectrometry using an enhanced matrix removal-lipid sorbent for clean-up. Microchem. J. 2018, 142, 70–77. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Opioid | Half Life (h) | Analgesic Duration (h) | Protein Binding (%) |
|---|---|---|---|
| Morphine | 1.3–6.7 | 4–5 | 35 |
| Codeine | 1.2–3.9 | 3–4 | 7–25 |
| Tramadol | 4.3–6.7 | 2.6–2.9 | 15–20 |
| Nalbuphine | 1.9–7.7 | 3–6 | 25–40 |
| Dextromethorphan | 3–4 | 2–3 | 60–70 |
| Opioid Name | Quantifier Ion (m/z) | Qualifier Ion 1 (m/z) | Qualifier Ion 2 (m/z) |
|---|---|---|---|
| Tramadol | 335 | 320 | 245 |
| Dextromethorphan | 271 | 270 | 214 |
| Codeine | 371 | 178 | 234 |
| Morphine | 429 | 236 | 287 |
| 6-Acetylmorphine | 399 | 287 | 340 |
| Nalbuphine | 574 | 518 | 428 |
| Nalorphine (IS) | 455 | 414 | - |
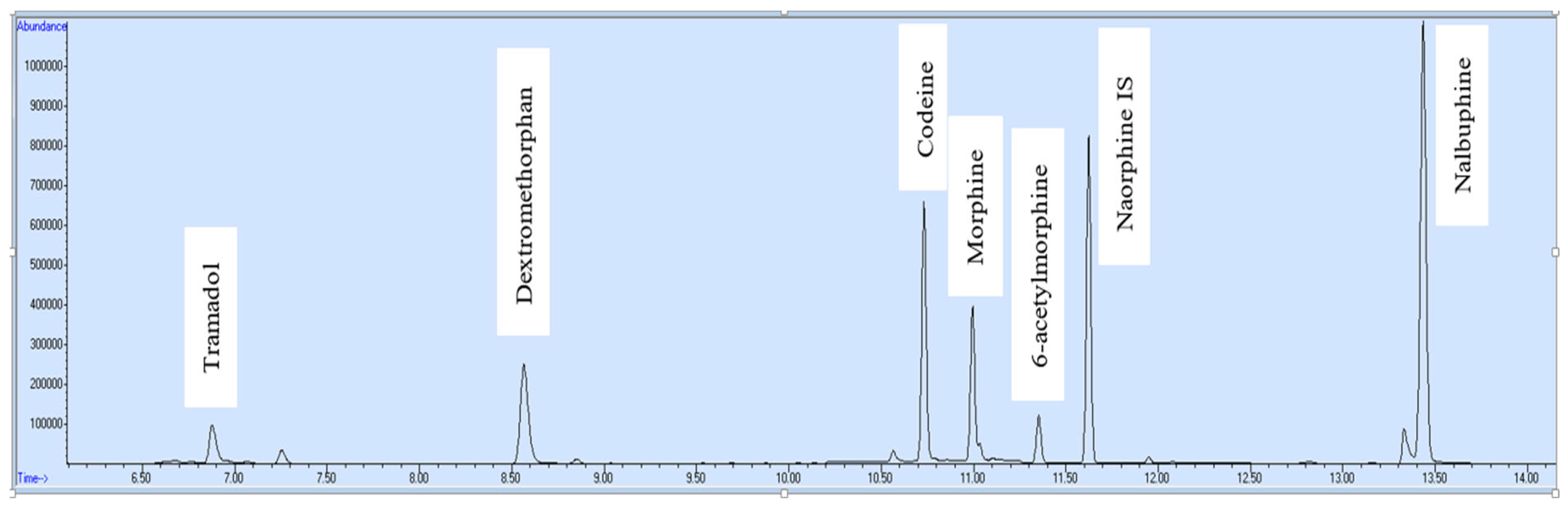
| Opioid Name | Retention Time (minutes) |
|---|---|
| Tramadol | 6.88 |
| Dextromethorphan | 8.56 |
| Codeine | 10.73 |
| Morphine | 10.99 |
| 6-Acetylmorphine | 11.35 |
| Nalbuphine | 13.43 |
| Nalorphine (IS) | 11.62 |
| Target Opioid | Actual Concentrations (µg/L) of Three Standards | Accuracy (% bias) Range | Precision (% CV) Range |
|---|---|---|---|
| Tramadol | 10 | 0.02–0.253 | 0.418–2.40 |
| 800 | 0.081–0.133 | 0.10–0.016 | |
| 1500 | 0.142–0.237 | 0.009–0.015 | |
| Dextromethorphan | 10 | 0.041–0.133 | 0.41–1.24 |
| 800 | 0.081–0.133 | 0.01–0.015 | |
| 1500 | 0.203–0.268 | 0.013–0.017 | |
| Codeine | 10 | 0.028–0.342 | 0.287–3.26 |
| 800 | 0.081–0.133 | 0.01–0.016 | |
| 1500 | 0.142–0.257 | 0.008–0.015 | |
| Morphine | 10 | 0.020–0.214 | 0.287–326 |
| 800 | 0.081–0.133 | 0.01–0.016 | |
| 1500 | 0.142–0.257 | 0.008–0.015 | |
| 6-acetylmorphine | 5 | 0.041–0.253 | 0.379–2.65 |
| 100 | 0.081–0.133 | 0.01–0.016 | |
| 150 | 0.142–0.237 | 0.009–0.015 | |
| Nalbuphine | 10 | 0.077–0.315 | 0.006–5.67 |
| 800 | 0.59–0.247 | 0.057–0.253 | |
| 1500 | 0.124–0.132 | 0.080–0.20 |
| Days | Rotator Speed (rpm) | Rotation Time (min) | Centrifuge Speed (rpm) | Centrifugation Time (min) | Derivatisation Temperature (°C) |
|---|---|---|---|---|---|
| Day1 | 15 | 20 | 2000 | 15 | 65 |
| Day2 | 20 | 16 | 2500 | 12 | 70 |
| Day3 | 25 | 12 | 3000 | 9 | 75 |
| Day4 | 30 | 8 | 3500 | 6 | 80 |
| Day5 | 35 | 4 | 4000 | 3 | 85 |
| Parameter | Actual Results |
|---|---|
| No. of target opioid | 6 |
| Extraction time per sample | Almost 35 min |
| GC-MSrun time | 14 min |
| Limit of detection (lod) | 5ng/mL for 6-acetylmorphine |
| Limit of quantitation (loq) | 5–150 ng/mL for 6-acetylmorphine, and 10–1500 ng/mL for all other opioids |
| Linearity | 5–150 ng/mL, r2 ≥ 0.985 for 6-acetylmorphine, and 10–1500 ng/mL for all other opioids, r2 ≥ 0.985 |
| Precision (% cv) | It was less than 5 for all analytes |
| Accuracy (% bias) | It was less than 1 for all analytes |
| Recovery | Greater than 80% |
| Ruggedness | The method proved to be rugged |
| Interference | Absence of any interference in quantification |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasien, S.; Ali, E.; Javed, M.; Iqbal, M.M.; Iqbal, S.; Alrbyawi, H.; Aljazzar, S.O.; Elkaeed, E.B.; Dera, A.A.; Pashameah, R.A.; et al. Simultaneous Quantification of Opioids in Blood and Urine by Gas Chromatography-Mass Spectrometer with Modified Dispersive Solid-Phase Extraction Technique. Molecules 2022, 27, 6761. https://doi.org/10.3390/molecules27196761
Yasien S, Ali E, Javed M, Iqbal MM, Iqbal S, Alrbyawi H, Aljazzar SO, Elkaeed EB, Dera AA, Pashameah RA, et al. Simultaneous Quantification of Opioids in Blood and Urine by Gas Chromatography-Mass Spectrometer with Modified Dispersive Solid-Phase Extraction Technique. Molecules. 2022; 27(19):6761. https://doi.org/10.3390/molecules27196761
Chicago/Turabian StyleYasien, Sara, Ejaz Ali, Mohsin Javed, Muhammad Muntazir Iqbal, Shahid Iqbal, Hamad Alrbyawi, Samar O. Aljazzar, Eslam B. Elkaeed, Ayed A. Dera, Rami Adel Pashameah, and et al. 2022. "Simultaneous Quantification of Opioids in Blood and Urine by Gas Chromatography-Mass Spectrometer with Modified Dispersive Solid-Phase Extraction Technique" Molecules 27, no. 19: 6761. https://doi.org/10.3390/molecules27196761
APA StyleYasien, S., Ali, E., Javed, M., Iqbal, M. M., Iqbal, S., Alrbyawi, H., Aljazzar, S. O., Elkaeed, E. B., Dera, A. A., Pashameah, R. A., Alzahrani, E., & Farouk, A.-E. (2022). Simultaneous Quantification of Opioids in Blood and Urine by Gas Chromatography-Mass Spectrometer with Modified Dispersive Solid-Phase Extraction Technique. Molecules, 27(19), 6761. https://doi.org/10.3390/molecules27196761