
Structural Variations in the Central Heterocyclic Scaffold of Tripartite 2,6-Difluorobenzamides: Influence on Their Antibacterial Activity against MDR Staphylococcus aureus

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
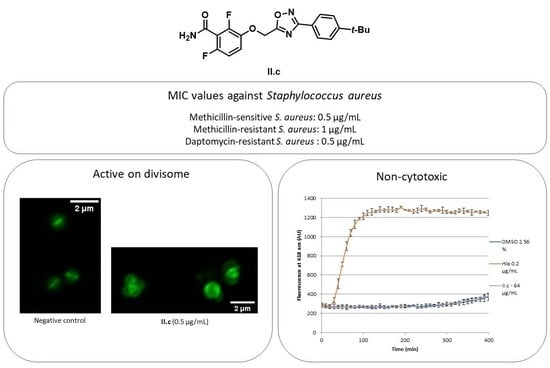
2.2. Biological Evaluation
2.3. Optimization of Series II
2.4. Mechanism of Action
2.5. Molecular Docking
2.6. Cytotoxicity Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. Series I
3.1.2. Series II
3.1.3. Series III
3.1.4. Series IV
3.1.5. Series V
3.2. Biological Evaluation
3.2.1. Minimum Inhibitory Concentration (MIC) Evaluation
3.2.2. Divisome Inhibition Assay
3.2.3. Cytotoxicity Assay
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morehead, M.S.; Scarbrough, C. Emergence of Global Antibiotic Resistance. Prim. Care 2018, 45, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Frees, D.; Ingmer, H. Antibiotic Resistance and the MRSA Problem. Microbiol. Spectr. 2019, 7, GPP3-0057-2018. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Chijiiwa, Y.; Suzuki, K.; Takahashi, K.; Nanamiya, H.; Sato, T.; Hosoya, Y.; Ochi, K.; Kawamura, F. The lethal effect of a benzamide derivative, 3-methoxybenzamide, can be suppressed by mutations within a cell division gene, ftsZ, in Bacillus subtilis. J. Bacteriol. 1999, 181, 1348–1351. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; Lyu, Y.L.; Pawlak, J.; Saravolatz, S.; Saravolatz, L.D.; Weinstein, M.P.; LaVoie, E.J.; et al. TXA709, an FtsZ-Targeting Benzamide Prodrug with Improved Pharmacokinetics and Enhanced In Vivo Efficacy against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 4845–4855. [Google Scholar] [CrossRef]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Chauhan, P.K.; Collins, I.; Davies, D.T.; Gavade, M.; Kumar, D.; Lancett, P.; Macdonald, R.; et al. Design, synthesis and structure-activity relationships of substituted oxazole-benzamide antibacterial inhibitors of FtsZ. Bioorg. Med. Chem. Lett. 2014, 24, 353–359. [Google Scholar] [CrossRef]
- Bi, F.; Song, D.; Zhang, N.; Liu, Z.; Gu, X.; Hu, C.; Cai, X.; Venter, H.; Ma, S. Design, synthesis and structure-based optimization of novel isoxazole-containing benzamide derivatives as FtsZ modulators. Eur. J. Med. Chem. 2018, 159, 90–103. [Google Scholar] [CrossRef]
- Song, D.; Bi, F.; Zhang, N.; Qin, Y.; Liu, X.; Teng, Y.; Ma, S. Design, synthesis of novel 4,5-dihydroisoxazole-containing benzamide derivatives as highly potent FtsZ inhibitors capable of killing a variety of MDR Staphylococcus aureus. Bioorg. Med. Chem. 2020, 28, 115729. [Google Scholar] [CrossRef]
- Bi, F.; Song, D.; Qin, Y.; Liu, X.; Teng, Y.; Zhang, N.; Zhang, P.; Zhang, N.; Ma, S. Discovery of 1,3,4-oxadiazol-2-one-containing benzamide derivatives targeting FtsZ as highly potent agents of killing a variety of MDR bacteria strains. Bioorg. Med. Chem. 2019, 27, 3179–3193. [Google Scholar] [CrossRef]
- Brown, D.R.; Collins, I.; Czaplewski, L.G.; Hayden, D.J. Preparation of Substituted Benzamides and Pyridylamides as Antibacterial Agents. Patent WO2007107758, 27 September 2007. [Google Scholar]
- Barral, K.; Moorhouse, A.D.; Moses, J.E. Efficient conversion of aromatic amines into azides: A one-pot synthesis of triazole linkages. Org. Lett. 2007, 9, 1809–1811. [Google Scholar] [CrossRef]
- Gerfaud, T.; Wei, H.-L.; Neuville, L.; Zhu, J. Unexpected C-C bond cleavage: Synthesis of 1,2,4-oxadiazol-5-ones from amidoximes with pentafluorophenyl or trifluoromethyl anion acting as leaving group. Org. Lett. 2011, 13, 6172–6175. [Google Scholar] [CrossRef]
- Dai, H.; Chen, J.; Li, G.; Ge, S.; Shi, Y.; Fang, Y.; Ling, Y. Design, synthesis, and bioactivities of novel oxadiazole-substituted pyrazole oximes. Bioorg. Med. Chem. Lett. 2017, 27, 950–953. [Google Scholar] [CrossRef]
- Haydon, D.J.; Bennett, J.M.; Brown, D.; Collins, I.; Galbraith, G.; Lancett, P.; Macdonald, R.; Stokes, N.R.; Chauhan, P.K.; Sutariya, J.K.; et al. Creating an antibacterial with in vivo efficacy: Synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J. Med. Chem. 2010, 53, 3927–3936. [Google Scholar] [CrossRef]
- Kashid, B.B.; Salunkhe, P.H.; Dongare, B.B.; More, K.R.; Khedkar, V.M.; Ghanwat, A.A. Synthesis of novel of 2, 5-disubstituted 1, 3, 4-oxadiazole derivatives and their in vitro anti-inflammatory, anti-oxidant evaluation, and molecular docking study. Bioorg. Med. Chem. Lett. 2020, 30, 127136. [Google Scholar] [CrossRef]
- Diep, B.A.; Stone, G.G.; Basuino, L.; Graber, C.J.; Miller, A.; des Etages, S.-A.; Jones, A.; Palazzolo-Ballance, A.M.; Perdreau-Remington, F.; Sensabaugh, G.F.; et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: Convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2008, 197, 1523–1530. [Google Scholar] [CrossRef]
- Barbier, T.; Barbry, A.; Magand, J.; Badiou, C.; Davy, F.; Baudouin, A.; Queneau, Y.; Dumitrescu, O.; Lina, G.; Soulère, L. Synthesis and Biological Evaluation of Benzo[b]thiophene Acylhydrazones as Antimicrobial Agents against Multidrug-Resistant Staphylococcus aureus. Biomolecules 2022, 12, 131. [Google Scholar] [CrossRef]
- Vollmer, W. The prokaryotic cytoskeleton: A putative target for inhibitors and antibiotics? Appl. Microbiol. Biotechnol. 2006, 73, 37–47. [Google Scholar] [CrossRef]
- Barrows, J.M.; Goley, E.D. FtsZ dynamics in bacterial division: What, how, and why? Curr. Opin. Cell Biol. 2021, 68, 163–172. [Google Scholar] [CrossRef]
- Haranahalli, K.; Tong, S.; Ojima, I. Recent advances in the discovery and development of antibacterial agents targeting the cell-division protein FtsZ. Bioorg. Med. Chem. 2016, 24, 6354–6369. [Google Scholar] [CrossRef]
- Hurley, K.A.; Santos, T.M.A.; Nepomuceno, G.M.; Huynh, V.; Shaw, J.T.; Weibel, D.B. Targeting the Bacterial Division Protein FtsZ. J. Med. Chem. 2016, 59, 6975–6998. [Google Scholar]
- Panda, D.; Bhattacharya, D.; Gao, Q.H.; Oza, P.M.; Lin, H.-Y.J.; Hawkins, B.; Hibbs, D.E.; Groundwater, P.W. Identification of agents targeting FtsZ assembly. Future Med. Chem. 2016, 8, 1111–1132. [Google Scholar] [CrossRef]
- Tripathy, S.; Sahu, S.K. FtsZ inhibitors as a new genera of antibacterial agents. Bioorg. Chem. 2019, 91, 103169. [Google Scholar] [CrossRef]
- Pradhan, P.; Margolin, W.; Beuria, T.K. Targeting the Achilles Heel of FtsZ: The Interdomain Cleft. Front. Microbiol. 2021, 12, 732796. [Google Scholar] [CrossRef]
- Carro, L. Recent Progress in the Development of Small-Molecule FtsZ Inhibitors as Chemical Tools for the Development of Novel Antibiotics. Antibiotics 2019, 8, 217. [Google Scholar] [CrossRef]
- Dumitrescu, O.; Choudhury, P.; Boisset, S.; Badiou, C.; Bes, M.; Benito, Y.; Wolz, C.; Vandenesch, F.; Etienne, J.; Cheung, A.L.; et al. Beta-lactams interfering with PBP1 induce Panton-Valentine leukocidin expression by triggering sarA and rot global regulators of Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 3261–3271. [Google Scholar] [CrossRef]
- Ferrer-González, E.; Fujita, J.; Yoshizawa, T.; Nelson, J.M.; Pilch, A.J.; Hillman, E.; Ozawa, M.; Kuroda, N.; Al-Tameemi, H.M.; Boyd, J.M.; et al. Structure-Guided Design of a Fluorescent Probe for the Visualization of FtsZ in Clinically Important Gram-Positive and Gram-Negative Bacterial Pathogens. Sci. Rep. 2019, 9, 20092. [Google Scholar] [CrossRef]
- Kusuma, K.D.; Griffith, R.; Harry, E.J.; Bottomley, A.L.; Ung, A.T. In silico Analysis of FtsZ Crystal Structures towards a New Target for Antibiotics. Aust. J. Chem. 2019, 72, 184–193. [Google Scholar] [CrossRef]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; De Giuli Morghen, C.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhu, N.; Han, Y.; Jiang, J.; Si, S. Identification of anti-tuberculosis agents that target the cell-division protein FtsZ. J. Antibiot. 2014, 67, 671–676. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, L.-L.; Wu, Y.-F.; Wang, L.; Li, C.-C.; Li, L.; Di, B.; You, Q.-D.; Jiang, Z.-Y. Structure-activity and structure-property relationships of novel Nrf2 activators with a 1,2,4-oxadiazole core and their therapeutic effects on acetaminophen (APAP)-induced acute liver injury. Eur. J. Med. Chem. 2018, 157, 1376–1394. [Google Scholar] [CrossRef]
- Cai, J.; Wei, H.; Hong, K.H.; Wu, X.; Cao, M.; Zong, X.; Li, L.; Sun, C.; Chen, J.; Ji, M. Discovery and preliminary evaluation of 2-aminobenzamide and hydroxamate derivatives containing 1,2,4-oxadiazole moiety as potent histone deacetylase inhibitors. Eur. J. Med. Chem. 2015, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.A.; Kers, A.; Malmberg, J.; Oscarsson, K.; Edwards, L.; Isaac, M.; Slassi, A.; Stormann, T.M.; Wensbo, D.; Xin, T.; et al. Preparation of Heterocyclic Compounds Having an Activity at Metabotropic Glutamate Receptors (mGluRs). Patent WO2004014370, 19 February 2004. [Google Scholar]
- Ye, J.; Yang, X.; Xu, M.; Chan, P.K.-S.; Ma, C. Novel N-Substituted oseltamivir derivatives as potent influenza neuraminidase inhibitors: Design, synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2019, 182, 111635. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Tripathi, A.; Tripathi, P.N.; Singh, S.S.; Singh, S.P.; Shrivastava, S.K. Novel Molecular Hybrids of N-Benzylpiperidine and 1,3,4-Oxadiazole as Multitargeted Therapeutics to Treat Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 4361–4384. [Google Scholar] [CrossRef]
- Natero, R.; Koltun, D.O.; Zablocki, J.A. Microwave-Assisted One-Step Synthesis of Substituted 2-Chloromethyl-1,3,4-oxadiazoles. Synth. Commun. 2004, 34, 2523–2529. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Compound | R | cLogP | MIC (µg/mL) | ||
|---|---|---|---|---|---|---|
| ATCC 29213 a | SF8300 b | ST20171643 c | ||||
 | I.a | H | 2.02 | >256 | >256 | >256 |
| I.b | CF3 | 2.92 | >256 | >256 | >256 | |
| I.c | t-Bu | 3.73 | 256 | 128 | 256 | |
 | II.a | H | 2.70 | 32 | 64 | 32 |
| II.b | CF3 | 3.60 | 16 | 32 | 8 | |
| II.c | t-Bu | 4.41 | 0.5 | 1 | 0.5 | |
 | III.a | H | 2.37 | 128 | 128 | >256 |
| III.b | CF3 | 3.27 | >256 | >256 | 128 | |
| III.c | t-Bu | 4.08 | 8 | 8 | 8 | |
 | IV.a | H | 2.01 | >256 | >256 | >256 |
| IV.b | CF3 | 2.91 | >256 | >256 | >256 | |
| IV.c | t-Bu | 3.72 | 256 | 256 | 256 | |
| PC190723 | 0.5 | 0.5 | 0.5 | |||
| Structure | Compound | R | cLogP | MIC (µg/mL) | ||
|---|---|---|---|---|---|---|
| ATCC 29213 a | SF8300 b | ST20171643 c | ||||
 | V.a | H | 3.06 | 16 | 16 | 16 |
| V.b | CF3 | 3.96 | 1 | 1 | 1 | |
| V.c | t-Bu | 4.77 | 1 | 1 | 1 | |
| PC190723 | 0.5 | 0.5 | 0.5 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbier, T.; Badiou, C.; Davy, F.; Queneau, Y.; Dumitrescu, O.; Lina, G.; Soulère, L. Structural Variations in the Central Heterocyclic Scaffold of Tripartite 2,6-Difluorobenzamides: Influence on Their Antibacterial Activity against MDR Staphylococcus aureus. Molecules 2022, 27, 6619. https://doi.org/10.3390/molecules27196619
Barbier T, Badiou C, Davy F, Queneau Y, Dumitrescu O, Lina G, Soulère L. Structural Variations in the Central Heterocyclic Scaffold of Tripartite 2,6-Difluorobenzamides: Influence on Their Antibacterial Activity against MDR Staphylococcus aureus. Molecules. 2022; 27(19):6619. https://doi.org/10.3390/molecules27196619
Chicago/Turabian StyleBarbier, Thibaut, Cédric Badiou, Floriane Davy, Yves Queneau, Oana Dumitrescu, Gérard Lina, and Laurent Soulère. 2022. "Structural Variations in the Central Heterocyclic Scaffold of Tripartite 2,6-Difluorobenzamides: Influence on Their Antibacterial Activity against MDR Staphylococcus aureus" Molecules 27, no. 19: 6619. https://doi.org/10.3390/molecules27196619
APA StyleBarbier, T., Badiou, C., Davy, F., Queneau, Y., Dumitrescu, O., Lina, G., & Soulère, L. (2022). Structural Variations in the Central Heterocyclic Scaffold of Tripartite 2,6-Difluorobenzamides: Influence on Their Antibacterial Activity against MDR Staphylococcus aureus. Molecules, 27(19), 6619. https://doi.org/10.3390/molecules27196619