
A Mini Review on Molecules Inducing Caspase-Independent Cell Death: A New Route to Cancer Therapy


Abstract
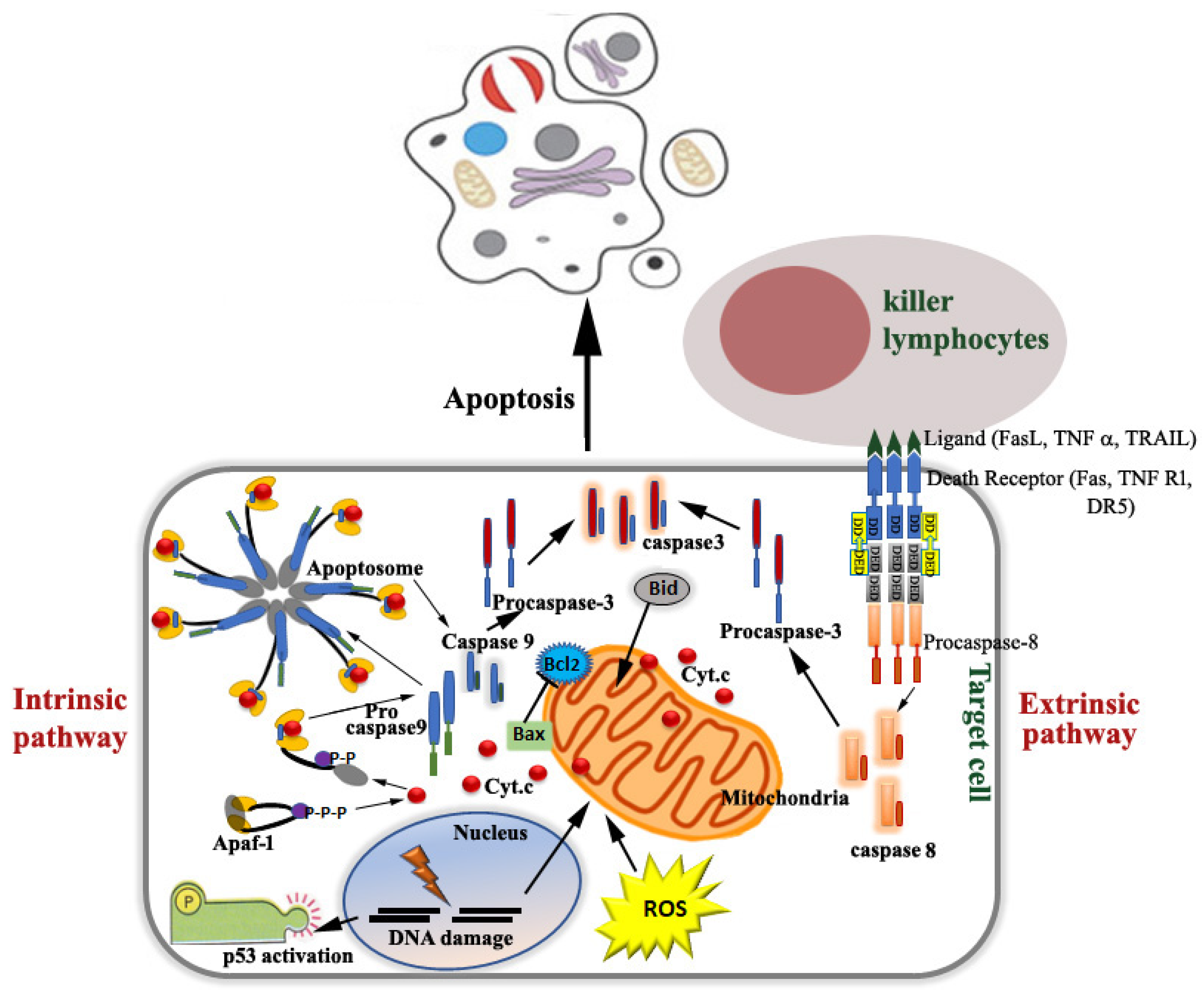
1. Introduction
2. Molecular Pathways of Caspase-Independent Programmed Death of Cell
3. Molecular Agents Inducing Caspase-Independent Programmed Cell Death
3.1. Synthetic Molecules
3.2. Inorganic Molecules
3.3. Natural Compounds
3.4. Repurposing Drugs
4. Summary
Funding
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Ann. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645. [Google Scholar] [CrossRef]
- Plaza, S.C.; Su, T.T. Non-Apoptotic Role of Apoptotic Caspases in the Drosophila Nervous System. Front. Cell Dev. Biol. 2022, 10, 839358. [Google Scholar] [CrossRef]
- Kaya, H.E.K.; Ditzel, M.; Meier, P.; Bergmann, A. An inhibitory mono-ubiquitylation of the Drosophila initiator caspase Dronc functions in both apoptotic and non-apoptotic pathways. PLoS Genet. 2017, 13, e1006438. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a026716. [Google Scholar] [CrossRef]
- Sarkar, S.; Bhadra, K. Therapeutic role of harmalol targeting nucleic acids: Biophysical perspective and in vitro cytotoxicity. Mini-Rev. Med. Chem. 2018, 18, 1624–1639. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Sarkar, S.; Shmatova, O.I.; Nenajdenko, V.G.; Pandya, P.; Bhadra, K. Synthetic carboline compounds targeting protein: Biophysical and biological perspective. J. Biomol. Struc. Dyn. 2021, 39, 3703–3720. [Google Scholar] [CrossRef]
- Sarkar, S.; Trebedi, P.; Bhadra, K. Structure-activity insights of harmine targeting DNA, ROS inducing cytotoxicity with PARP mediated apoptosis against cervical cancer, anti-biofilm formation and in vivo therapeutic study. J. Biomol. Struc. Dyn. 2022, 40, 5880–5902. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. The Mitochondrial Pathway of Apoptosis: Part I: MOMP and Beyond. Cold Spring Harb. Perspect. Biol. 2022, 14, a041038. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. The Mitochondrial Pathway of Apoptosis Part II: The BCL-2 Protein Family. Cold Spring Harb. Perspect. Biol. 2022, 14, a041046. [Google Scholar] [CrossRef] [PubMed]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J. Mol. Biol. 2022, 434, 167378. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Lim, B. Targeting apoptosis in cancer. Curr. Oncol. Rep. 2022, 24, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi RK, S.; Kanneganti, T.-D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features. World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Hu, X.-M.; Li, Z.-X.; Lin, R.-H.; Shan, J.-Q.; Yu, Q.-W.; Wang, R.-X.; Liao, L.-S.; Yan, W.-T.; Wang, Z.; Shang, L.; et al. Guidelines for regulated cell death assays: A systematic summary, a categorical comparison, a prospective. Front. Cell Dev. Biol. 2021, 9, 634690. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-S.; Ku, J.M.; Choi, H.-S.; Choi, Y.K.; Woo, J.-K.; Kim, M.; Kim, I.; Na, C.H.; Hur, H.; Jang, B.-H.; et al. Quercetin induces caspase-dependent extrinsic apoptosis through inhibition of signal transducer and activator of transcription 3 signaling in HER2-overexpressing BT-474 breast cancer cells. Oncol. Rep. 2016, 36, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Yanumula, A.; Cusick, J.K. Biochemistry, Extrinsic Pathway of Apoptosis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Bhutia, S.K.; Praharaj, P.P.; Bhol, C.S.; Panigrahi, D.P.; Mahapatra, K.K.; Patra, S.; Saha, S.; Das, D.N.; Mukhopadhyay, S.; Sinha, N.; et al. Monitoring and measuring mammalian autophagy. Methods Mol. Biol 2019, 1854, 209–222. [Google Scholar] [CrossRef]
- Guo, G.F.; Wang, Y.X.; Zhang, Y.J.; Chen, X.X.; Lu, J.B.; Wang, H.H.; Jiang, C.; Qiu, H.-Q.; Xia, L.-P. Predictive and prognostic implications of 4E-BP1, Beclin-1 and LC3 for cetuximab treatment combined with chemotherapy in advanced colorectal cancer with wild-type KRAS: Analysis from real-world data. World J. Gastroenterol. 2019, 25, 1840–1853. [Google Scholar] [CrossRef]
- Haun, F.; Neumann, S.; Peintner, L.; Wieland, K.; Habicht, J.; Schwan, C.; Østevold, K.; Koczorowska, M.M.; Biniossek, M.; Kist, M.; et al. Identification of a novel anoikis signaling pathway using the fungal virulence factor gliotoxin. Nat. Commun. 2018, 9, 3524. [Google Scholar] [CrossRef]
- Adeshakin, F.O.; Adeshakin, A.O.; Afolabi, L.O.; Yan, D.; Zhang, G.; Wan, X. Mechanisms for modulating anoikis resistance in cancer and the relevance of metabolic reprogramming. Front. Oncol. 2021, 11, 626577. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Zu, Y.; Mu, Y.; Li, Q.; Zhang, S.T.; Yan, H.J. Icariin alleviates osteoarthritis by inhibiting NLRP3-mediated pyroptosis. J. Orthop. Surg. Res. 2019, 14, 307. [Google Scholar] [CrossRef]
- Kitanaka, C.; Kuchino, Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999, 6, 508–515. [Google Scholar] [CrossRef]
- Wimmer, K.; Sachet, M.; Oehler, R. Circulating biomarkers of cell death. Clin. Chim. Acta 2020, 500, 87–97. [Google Scholar] [CrossRef]
- Lou, J.; Zhou, Y.; Feng, Z.; Ma, M.; Yao, Y.; Wang, Y.; Deng, Y.; Wu, Y. Caspase-Independent Regulated Necrosis Pathways as Potential Targets in Cancer Management. Front. Oncol. 2021, 10, 616952. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Liu, R.; Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019, 109, 2043–2053. [Google Scholar] [CrossRef]
- Jie Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88–101. [Google Scholar]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Lee, H.-J.; Jung, Y.M.; Jung, Y.-J. Regulated Necrotic Cell Death in Alternative Tumor Therapeutic Strategies. Cells 2020, 9, 2709. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.M.; Smith, S.W.; Jones, M.E.; Osborne, B.A. Do all programmed cell deaths occur via apoptosis? Proc. Natl. Acad. Sci. USA 1993, 90, 980–984. [Google Scholar] [CrossRef]
- Jurgensmeier, J.M.; Krajewski, S.; Armstrong, R.C.; Wilson, G.M.; Oltersdorf, T.; Fritz, L.C.; Reed, J.C.; Ottilie, S. Bax- and Bak-induced cell death in the fission yeast Schizosaccharomyces pombe. Mol Biol. Cell 1997, 8, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Mishra, A.K. The tale of caspase homologues and their evolutionary outlook: Deciphering programmed cell death in cyanobacteria. J. Exp. Bot. 2020, 71, 4639–4657. [Google Scholar] [CrossRef]
- Dixit, V.; Tiwari, R.; Katyal, R.; Pandey, A. Metacaspases: Potential Drug Target Against Protozoan Parasites. Front. Pharmacol. 2019, 10, 790. [Google Scholar] [CrossRef]
- Okuno, S.; Shimizu, S.; Ito, T.; Nomura, M.; Hamada, E.; Tsujimoto, Y.; Matsuda, H. Bcl-2 prevents caspase-independent cell death. J. Biol. Chem. 1998, 273, 34272–34277. [Google Scholar] [CrossRef]
- Mateo, V.; Brown, E.J.; Biron, G.; Rubio, M.; Fischer, A.; Deist FLe Sarfati, M. Mechanisms of CD47-induced caspase-independent cell death in normal and leukemic cells: Link between phosphatidylserine exposure and cytoskeleton organization. Blood 2002, 100, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Londei, H.M. CD47 ligation induces a rapid caspase-independent apoptosis-like cell death in human monocytes and dendritic cells. Scand. J. Immunol. 2003, 59, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Maianski, N.A.; Roos, D.; Kuijpers, T.W. Tumor necrosis factor alpha induces a caspase-independent death pathway in human neutrophils. Blood 2003, 101, 1987–1995. [Google Scholar] [CrossRef]
- Cawles, A.; Janssen, B.; Waeytens, A.; Cuvelier, C.; Brouckaert, P. Caspase inhibition causes hyperacute tumor necrosis factor-induced heat shock via oxidative stress and phospholipase A2. Nat. Immunol. 2003, 4, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Shrivastana, A.; Tiwari, M.; Sinha, R.A.; Kumar, A.; Balapure, A.K.; Bajpai, V.K.; Sharma, R.; Mitra, K.; Tandon, A.; Godbole, M.M. Molecular iodine induces caspase-independent apoptosis in human breast carcinoma cells involving the mitochondria-mediated pathway. J. Biol. Chem. 2006, 281, 19762–19771. [Google Scholar]
- Constantinou, C.; Papas, K.A.; Constantinou, A.I. Caspase-Independent Pathways of Programmed Cell Death: The Unraveling of New Targets of Cancer Therapy? Curr. Cancer Drug Targets 2009, 9, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.C.M.; Ruiz, A.R.; Londoño, M.B.; Molina, M.A.F.; Padilla, C.R. IMMUNEPOTENT CRP induces cell cycle arrest and caspase-independent regulated cell death in HeLa cells through reactive oxygen species production. BMC Cancer 2018, 18, 13. [Google Scholar] [CrossRef]
- Kolenko, V.M.; Uzzo, R.G.; Bukowski, R.; Finke, J.H. Caspase-dependent and independent death pathways in cancer therapy. Apoptosis 2000, 5, 17–20. [Google Scholar] [CrossRef]
- Smyth, M.J.; Krasovskis, E.; Sutton, V.R.; Johnstone, R.W. The drug efflux protein, P-glycoprotein, additionally protects drug- resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 7024–7029. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Cretney, E.; Smyth, M.J. P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood 1999, 93, 1075–1085. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis avoidance in cancer. Curr. Opin. Oncol. 1999, 11, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.; Kitanaka, C.; Noguchi, K.; Mochizuki, T.; Nagashima, Y.; Shirouzu, M.; Fujita, H.; Yoshida, M.; Chen, W.; Asai, A.; et al. Oncogenic Ras triggers cell suicide through the activation of a caspase- independent cell death program in human cancer cells. Oncogene 1999, 18, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Chao, D.T.; Korsmeyer, S.J. BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA 1996, 93, 14559–14563. [Google Scholar] [CrossRef] [PubMed]
- Pinkoski, M.J.; Hobman, M.; Heibein, J.A.; Tomaselli, K.; Li, F.; Seth, P.; Froelich, C.J.; Bleackley, R.C. Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood 1998, 92, 1044–1054. [Google Scholar] [CrossRef]
- Beresford, P.J.; Xia, Z.; Greenberg, A.H.; Lieberman, J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 1999, 10, 585–594. [Google Scholar] [CrossRef]
- Onoda, T.; Ono, T.; Dhar, D.K.; Yamanoi, A.; Nagasue, N. Tetracycline analogues (doxycycline and COL-3) induce caspase-dependent and -independent apoptosis in human colon cancer cells. Int. J. Cancer 2006, 118, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Parreño, M.; Casanova, I.; Céspedes, M.V.; Vaqué, J.P.; Pavón, M.A.; Leon, J.; Mangues, R. Bobel-24 and derivatives induce caspase-independent death in pancreatic cancer regardless of apoptotic resistance. Cancer Res. 2008, 68, 6313–6323. [Google Scholar] [CrossRef]
- Wang, S.W.; Pan, S.L.; Huang, Y.C.; Guh, J.H.; Chiang, P.C.; Huang, D.Y.; Kuo, S.C.; Lee, K.H.; Teng, C.M. CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 350–360. [Google Scholar] [CrossRef]
- McCafferty-Grad, J.; Bahlis, N.J.; Krett, N.; Aguilar, T.M.; Reis, I.; Lee, K.P.; Boise, L.H. Arsenic trioxide uses caspase-dependent and caspase-independent death pathways in myeloma cells. Mol. Cancer Ther. 2003, 2, 1155–1164. [Google Scholar]
- Michel, L.; Dupuy, A.; Jean-Louis, F.; Sors, A.; Poupon, J.; Viguier, M.; Musette, P.; Dubertret, L.; Degos, L.; Dombret, H.; et al. Arsenic trioxide induces apoptosis of cutaneous Tcell lymphoma cells: Evidence for a partially caspase-independent pathway and potentiation by ascorbic acid (vitamin C). J. Investig. Dermatol. 2003, 121, 881–893. [Google Scholar] [CrossRef]
- Cho, Y.S. Receptor interacting protein 3 is required for arsenite mediated necroptosis. Int. J. Sci. Basic Appl. Res. 2020, 53, 51–65. [Google Scholar]
- Liu, G.; Zou, H.; Luo, T.; Long, M.; Bian, J.; Liu, X.; Gu, J.; Yuan, Y.; Song, R.; Wang, Y.; et al. Caspase-Dependent and Caspase-Independent Pathways Are Involved in Cadmium-Induced Apoptosis in Primary Rat Proximal Tubular Cell Culture. PLoS ONE 2016, 11, e0166823. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.-M.; Ko, W.-C.; Wu, J.-S.; Wei, Y.-H.; Wang, L.-F.; Chang, E.-E.; Lo, T.-Y.; Cheng, H.-H.; Chen, C.-T. Mediating of caspase-independent apoptosis by cadmium through the mitochondria-ROS pathway in MRC-5 fibroblasts. J. Cell. Biochem. 2004, 91, 384–397. [Google Scholar] [CrossRef]
- Mathiasen, I.S.; Lademann, U.; Jäättelä, M. Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res. 1999, 59, 4848–4856. [Google Scholar] [PubMed]
- Bidère, N.; Lorenzo, H.K.; Carmona, S.; Laforge, M.; Harper, F.; Dumont, C.; Senik, A. Cathepsin D triggers Bax activation, resulting in selective apoptosis inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J. Biol. Chem. 2003, 278, 31401–31411. [Google Scholar] [CrossRef]
- Johansson, A.C.; Steen, H.; Ollinger, K.; Roberg, K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003, 10, 1253–1259. [Google Scholar] [CrossRef]
- Saumet, A.; Slimane, M.B.; Lanotte, M.; Lawler, J.; Dubernard, V. Type 3 repeat/C-terminal domain of thrombospondin-1 triggers caspase-independent cell death through CD47/α vβ3 in promyelocytic leukemia NB4 cells. Blood 2005, 106, 658–667. [Google Scholar] [CrossRef]
- Rudolf, E.; Rudolf, K.; Cervinka, M. Selenium activates p53 and p38 pathways and induces caspase-independent cell death in cervical cancer cells. Cell Biol. Toxicol. 2008, 24, 123–141. [Google Scholar] [CrossRef]
- Selvakumar, E.; Hsieh, T.C. Regulation of cell cycle transition and induction of apoptosis in HL-60 leukemia cells by lipoic acid: Role in cancer prevention and therapy. J. Hematol. Oncol. 2008, 1, 4. [Google Scholar] [CrossRef]
- Chen, T.; Wong, Y.S. Selenocysteine induces caspase-independent apoptosis in MCF-7 human breast carcinoma cells with involvement of p53 phosphorylation and reactive oxygen species generation. Int. J. Biochem. Cell. Biol. 2009, 41, 666–676. [Google Scholar] [CrossRef]
- Pinto-Garcia, L.; Efferth, T.; Torres, A.; Hoheisel, J.D.; Youns, M. Berberine inhibits cell growth and mediates caspase-independent cell death in human pancreatic cancer cells. Planta Med. 2010, 76, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, L.; Shi, Y.; Cao, H.; Chaturvedi, R.; Calcutt, M.W.; Hu, T.; Ren, X.; Wilson, K.T.; Polk, D.B.; et al. Berberine induces caspase-independent cell death in colon tumor cells through activation of apoptosis inducing factor. PLoS ONE 2012, 7, e36418. [Google Scholar] [CrossRef] [PubMed]
- White, B.; Beckford, J.; Yadegaryni, S.; Ngo, N.; Lialiutsk, T.; Alarcao, M.d. Some natural flavonoids are competitive inhibitors of Caspase-1, -3 and -7 despite their cellular toxicity. Food Chem. 2012, 131, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Gao, Y.; Deng, Z.; Cao, H.; Zhang, W.; Wang, Q.; Zhang, B.; Song, G.; Zhan, Y.; et al. Matrine induces caspase-independent program cell death in hepatocellular carcinoma through bid-mediated nuclear translocation of apoptosis inducing factor. Mol. Cancer 2014, 13, 59–70. [Google Scholar] [CrossRef]
- Kim, S.P.; Nam, S.H.; Friedman, M. The Tomato Glycoalkaloid α-Tomatine Induces Caspase-Independent Cell Death in Mouse Colon Cancer CT-26 Cells and Transplanted Tumors in Mice. Agric. Food Chem. 2015, 63, 1142–1150. [Google Scholar] [CrossRef]
- Pozo-uisado, E.; Merino, J.M.; Mulero-Navarro, S.; LorenzoBenayas, M.J.; Centeno, F.; Alvarez-Barrientos, A.; Fernandez Salguero, P.M. Resveratrol-induced apoptosis in MCF-7 human breast cancer cells involves a caspase-independent mechanism with downregulation of Bcl-2 and NF-kappa B. Int. J. Cancer 2005, 116, 1004. [Google Scholar]
- Kumar, D.S.; Drishya, G.; Chandrasekharan, A.; Shaji, S.K.; Bose, C.; Jossart, J.; Perry, J.P.; Mishra, N.; Kumar, G.B.; Nair, B.G. Oxyresveratrol drives caspase-independent apoptosis-like cell death in MDA-MB-231 breast cancer cells through the induction of ROS. Biochem. Pharmacol. 2020, 173, 113724. [Google Scholar]
- Song, W.; Ren, Y.-J.; Liu, L.L.; Zhao, Y.-Y.; Li, Q.-F.; Yang, H.-B. Curcumin induced the cell death of immortalized human keratinocytes (HaCaT) through caspase-independent and caspase-dependent pathways. Food Funct. 2021, 12, 8669–8680. [Google Scholar] [CrossRef]
- Qa’Dan, M.; Ramsey, M.; Daniel, J.; Spyres, L.M.; Safifiejko-Mroczka, B.; Ortiz-Leduc, W.; Ballard, J.D. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell. Microbiol. 2002, 4, 425–434. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, K.W.; Kim, J.Y.; Jeong, I.Y.; Byun, M.W.; Park, J.E.; Yee, S.T.; Kim, K.H.; Rhim, J.S.; Yamada, K.; et al. Thiosulfinates from Allium tuberosum L. induce apoptosis via caspase-dependent and -independent pathways in PC-3 human prostate cancer cells. Bioorg. Med. Chem. Lett. 2008, 18, 199–204. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Lee, K.W.; Kim, M.; Lee, Y.; Yoo, J.; Hwangbo, C.; Park, K.H.; Kim, K.D. Chelidonine Induces Caspase-Dependent and Caspase-Independent Cell Death through G2/M Arrest in the T98G Human Glioblastoma Cell Line. Evid.-Based Complementary Altern. Med. Hindawi. 2019, 2019, 6318179. [Google Scholar] [CrossRef] [PubMed]
- Nedungadi, D.; Binoy, A.; Vinod, V.; Vanuopadath, M.; Nair, S.S.; Nair, B.G.; Mishra, N. Ginger extract activates caspase independent paraptosis in cancer cells via ER stress, mitochondrial dysfunction, AIF translocation and DNA damage. Nutr. Cancer 2021, 73, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Bruynzeel, A.M.E.; Hassan, M.A.A.; El Torun, E.; Bast, A.; van der Vijgh, W.J.F.; Kruyt, F.A.E. Caspase-dependent and -independent suppression of apoptosis by mono HER in Doxorubicin treated cells. Br. J. Cancer 2007, 96, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Tamasdan, C.; Miller, D.C.; Newcomb, E.W. Flavopiridol induces apoptosis in glioma cell lines independent of retinoblastoma and p53 tumour suppressor pathway alterations by a caspase-independent pathway. Mol. Cancer Therp. 2003, 2, 139–150. [Google Scholar]
- Newcomb, E.W.; Tamasdan, C.; Entzminger, Y.; Alonso, J.; Friedlander, D.; Crisan, D.; Miller, D.C.; Zagzag, D. Flavopiridol induces mitochondrial-mediated apoptosis in murine glioma GL261 cells via release of cytochrome c and apoptosis inducing factor. Cell Cycle 2003, 2, 243–250. [Google Scholar] [CrossRef][Green Version]
- Roberts, L.R.; Adjei, P.N.; Gores, G.J. Cathepsins as effector proteases in hepatocyte apoptosis. Cell Biochem. Biophys. 1999, 30, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Marzo, I.; Perez-Galan, P.; Giraldo, P.; Rubio-Felix, D.; Anel, A.; Naval, J. Cladribine induces apoptosis in human leukaemia cells by caspase-dependent and –independent pathways acting on mitochondria. Biochem. J. 2001, 359, 537–546. [Google Scholar] [CrossRef]
- Joseph, B.; Marchetti, P.; Formstecher, P.; Kromer, G.; Lewenshohn, R.; Zhivotovsky, B. Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene 2002, 21, 65–77. [Google Scholar] [CrossRef][Green Version]
- Ahn, H.J.; Kim, Y.S.; Kim, J.U.; Han, S.M.; Shin, J.W.; Yang, H.O. Mechanism of taxol induced apoptosis in human SKOV3 ovarian carcinoma cells. J. Cell. Biochem. 2004, 91, 1043–1052. [Google Scholar] [CrossRef]
- Kim, R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 2005, 103, 1551–1560. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, C.; Narayani, N.; Du, C.; Balaji, K.C. Nuclear translocation of apoptosis inducing factor is associated with cisplatin induced apoptosis in LNCaP prostate cancer cells. Cancer Lett. 2007, 255, 127–134. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Han, X.; Yang, J.; Qian, J.; Hong, S.; Samaniego, F.; Romaguera, J.; Yi, Q. Atiprimod inhibits the growth of mantle cell lymphoma in vitro and in vivo and induces apoptosis via activating the mitochondrial pathways. Blood 2007, 109, 5455–5461. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.A.; Baker-Lee, C.; Kennedy, J.; Lai, M.S.; de Vries, P.; Buhler, K.; Singer, J.W. In vitro and in vivo metabolism of paclitaxel poliglumex: Identification of metabolites and active proteases. Cancer Chemother. Pharmacol. 2007, 59, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Albain, K.S.; Belani, C.P.; Bonomi, P.; O’Byre, K.J.; Schiller, J.H.; Socinski, M. PIONEER: A phase III randomized trial of paclitaxel poliglumex versus paclitaxel in chemotherapy-naive women with advanced-stage non-small-cell lung cancer and performance status of 2. Clin. Lung Cancer 2006, 7, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Hüsemann, L.C.; Reese, A.; Radine, C.; Piekorz, R.P.; Budach, W.; Sohn, D.; Jänicke, R.U. The microtubule targeting agents eribulin and paclitaxel activate similar signaling pathways and induce cell death predominantly in a caspase-independent manner. Cell Cycle 2020, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Sung, E.-G.; Song, I.n.-H.; Lee, T.-J.; Kim, J.-Y. Metformin induces caspase-dependent and caspase-independent apoptosis in human bladder cancer T24 cells. Anti-Cancer Drugs 2020, 31, 655–662. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Classification | Name of the Compounds | System; Triggering Pathway | References |
|---|---|---|---|
| Intracellular molecular factors/extract | TNF-α, CD47, IMMUNEPOTENT C-reactive protein (I-CRP) | Human neutrophils, leukemic cells, Hela cells; induced caspase free cell death pathway. | [42,44,48] |
| Synthetic molecules | Tetracycline analogues: TCNAs, COL-3 (chemically modified tetracycline-3; 6-demethyl, 6-deoxy, 4-dedimethylamino tetracycline) and doxyclycline DOXY | Colon cancer cell line HT29; evaluated to be mitochondria-mediated apoptosis through both caspase-dependent and -independent pathways. | [57] |
| Bobel-24/AM-24 (2,4,6-triiodophenol) and derivatives | Human pancreatic carcinoma cell lines (NP18, NP9, NP31, and NP29), leukemia cell; follows caspase-independent pathways via production of ROS, mitochondrial depolarization, release of cytochrome c, AIF and release of lysosomal cathepsin. | [58] | |
| 6,7-substituted 2-phenyl-4-quinolone, CHM-1, (2′-fluoro-6,7-methylenedioxy-2-phenyl-4-quinolone) | HCC (hepatocellular carcinoma) HA22T, Hep3B, and HepG2; selective and potent anti-cancer agent and acts without activation of the caspase cascade. | [59] | |
| Inorganic compounds | Arsenic trioxide As2O3 | Myeloma cells and cutaneous T cell lymphoma viz. HuT-78, SeAx, Myla, and of peripheral blood mononuclear cells; follows both caspase-dependent and caspase-independent cell death pathways, caspase-independent cell death is triggered by ascorbic acid (vitamin C). | [60,61,62] |
| Cadmium | Rat proximal tubular cell, MRC-5 fibroblasts; Both caspase-dependent and caspase-independent pathways are caused that acts synergistically. BNIP-3 (Bcl-2/adenovirus E1B 19-kDa interacting protein 3) acts as an upstream factor inducing translocation of AIF and endonuclease G. It also induces mitochondria-ROS pathway and causes caspase-independent cell death. | [63,64] | |
| Molecular iodine | Breast carcinoma cell lines viz., MCF-7, MDA-MB-231, MDA-MB-453, ZR-75–1, and T-47D; caspase-independent apoptosis involving the mitochondria-mediated pathway. | [46] | |
| Natural compounds | Vit D | Breast cancer cell lines: MCF7, T47D; activates CIPCD by activating cathepsin D and inhibited by Bcl-2 but does not require p53. | [65] |
| Staurosporine | Human fibroblast; exhibits anti-cancer activity by Cathepsin D mediating cytochrome c release. Cathepsin D triggers Bax activation, resulting in relocation of AIF in T-lymphocytes, leading to apoptosis. | [66,67] | |
| Thrombospondin-1 | Promyelocytic leukemia NB4 cells and freshly isolated monocytes and monocyte-derived dendritic cells through Thrombospondin-1 membrane receptors CD47 and αvβ3, triggered caspase free cell death and characterized by the instantaneous permeability of plasma membrane, exposure of phosphatidylserine, decreased mitochondrial membrane potential and highly fragmented DNA. | [68] | |
| Selenite | Cervical cancer cell lines HeLa and Hep 2 cell lines; The Caspase free programmed cell death pathway involves activation of p53, accumulation of Bax and release of AIF and Smac/DIABLO, co-treatment with the caspase inhibitor Z-VAD-FMK, cell death was observed. | [69] | |
| Lipoic acid | HL-60 leukemia cells; activates CI-PCD via up-regulation of Bax, downregulating Bcl-2, release and translocation of AIF and cytochrome c to nucleus from mitochondria. | [70] | |
| Selenocystine | Breast cancer cells MCF7; CIPCD via translocation of AIF and phosphorylation of p53. | [71] | |
| Berberine | Human pancreatic cancer cells, BxPC-3, mouse immorto-Min colonic epithelial cells (IMCE), normal colon epithelial cells, namely young adult mouse colonic epithelium (YAMC) cells; stimulate caspase-independent cell death through production of ROS leading to the release of cathepsin B and activation of PARP dependent AIF translocation. | [72,73] | |
| Natural flavonoids: Quercetine, Myricetine, Apigenin | MDA-MB-231, an epithelial human breast cancer cells; induced cell death through a non-classical apoptosis pathway that is not dependent on caspase activity. Hence, they may be lead source for the rational drug design of caspase specific inhibitors. | [74] | |
| Matrine | HepG2 cells; induces parallelly caspase-dependent and caspase free cell death through Bid regulated AIF nuclear translocation pathway. | [75] | |
| α-Tomatine | Mouse colon cancer cells CT-26; induces both in vitro and in vivo caspase free cell death by expression of apoptosis-inducing protein (AIF) localizes from mitochondria to nucleus and down-regulation of surviving, an inhibitor of apoptosis. It also failed to express the active form of caspase-3, -8, and -9 produced by proteolytic cleavage. | [76] | |
| Resveratrol, derivative- Oxyresveratrol | MCF-7 breast cancer cells, MDA-MB-231 breast cancer cells; trigger the caspase-independent cell death through changes in mitochondrial membrane potential, downregulating Bcl-2, increased ROS and nitric oxide production and prevention of NF-kB. The derivative compound, induces apoptosis-like cell death by caspase-independent pathway through the induction of ROS, DNA fragmentation, Phosphatidyl serine externalization, PARP cleavage, decrease in mitochondrial membrane potential Δψm and nuclear translocation of AIF. | [77,78] | |
| Curcumin | Human keratinocytes (HaCaT); induced apoptosis of the cells through both caspase-dependent and caspase-independent pathways. | [79] | |
| Natural extracts | Clostridium diffificile toxin B (TcdB) | HeLa and MCF-7 cancer cell line; activates caspase-dependent and caspase-free apoptosis, respectively. | [80] |
| The thiosulfinates from Allium tuberosum L. extract | Prostate cancer cell line PC3; activate both CD-PCD and CI-PCD by increasing the expression of Bax and AIF and decreasing the expression of Bcl-2. | [81] | |
| Extract of Chelidonine majus L., containing isoquinoline alkaloids like sanquinarine, chelidonine, chelerythrine, berberine, protopine and coptisine, flavonoids and phenolic acids | T98G Human Glioblastoma Cell Line; reported to induce both caspase-dependent and caspase-independent cell death through G2/M arrest. | [82] | |
| The rhizome of ginger (Zingiber officinale) | Triple negative breast cancer (MDA-MB-231) and non-small lung (A549) cancer cells; known for its caspase-independent paraptosis via ER stress, mitochondrial dysfunction, AIF translocation and DNA damage. | [83] | |
| Repurposing drug molecules | Flavopiridol | Glioma cell lines; independent of retinoblastoma and p53 tumor suppressor pathway alterations by a caspase-independent pathway. | [85,86] |
| Doxorubicin | Human endothelial cell HUVECs and ovarian cancer cell line A2780, neonatal rat cardiac myocytes NeRCaMs; induced apoptosis through caspase-dependent and caspase-independent mechanisms, respectively. It was further confirmed that flavonoid mono hydroxy ethylrutoside, monoHER, a protective agent used against toxicity of Doxorubicin, at different concentrations, acts by suppressing the caspase-dependent or -independent cell death activation. | [84] | |
| Cladribine, camptothecin and cisplatin | Hepatocellular cancer cell line, Human SKOV3 ovarian carcinoma cells, LNCaP prostate cancer cells; capable of causing cell death through caspase-independent programmed cell death pathway involving translocation of AIF from mitochondrion to nucleus. | [87,88,89,90,91,92] | |
| Atiprimod | Mantle cell lymphoma MCL; inducing cell apoptosis mainly via activation of AIF pathway. | [93] | |
| Paclitaxel poliglumex | Prostate, ovarian cancer and non-small cell lung carcinoma; is a phase II clinical trial drug, metabolized via cathepsin B to paclitaxel in the cancer cells. In addition, it also induced caspase-independent apoptosis via apoptosis inducing factor AIF. | [94,95] | |
| Eribulin, Paclitaxel | MCF-7 breast carcinoma; predominantly causes cell death without activation of caspase. On the molecular level, both the compounds to a similar extent activate the key proteins involved in apoptosis such as p53, Plk1, caspase-2, and Bim as well as MAPK pathway mediated by ERK and JNK. | [96] | |
| Metformin | Human bladder cancer cell line T24; induces apoptosis by both caspase-dependent and caspase-independent signaling pathways through the stimulation of AIF signaling pathway and increasing c-FLIPL protein (FADD like interleukin-1β-converting enzyme inhibitory protein) instability. | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadra, K. A Mini Review on Molecules Inducing Caspase-Independent Cell Death: A New Route to Cancer Therapy. Molecules 2022, 27, 6401. https://doi.org/10.3390/molecules27196401
Bhadra K. A Mini Review on Molecules Inducing Caspase-Independent Cell Death: A New Route to Cancer Therapy. Molecules. 2022; 27(19):6401. https://doi.org/10.3390/molecules27196401
Chicago/Turabian StyleBhadra, Kakali. 2022. "A Mini Review on Molecules Inducing Caspase-Independent Cell Death: A New Route to Cancer Therapy" Molecules 27, no. 19: 6401. https://doi.org/10.3390/molecules27196401
APA StyleBhadra, K. (2022). A Mini Review on Molecules Inducing Caspase-Independent Cell Death: A New Route to Cancer Therapy. Molecules, 27(19), 6401. https://doi.org/10.3390/molecules27196401