
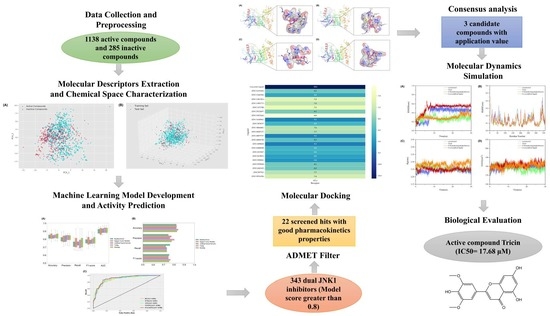
Discovery of Novel c-Jun N-Terminal Kinase 1 Inhibitors from Natural Products: Integrating Artificial Intelligence with Structure-Based Virtual Screening and Biological Evaluation


Abstract
:
1. Introduction
2. Results
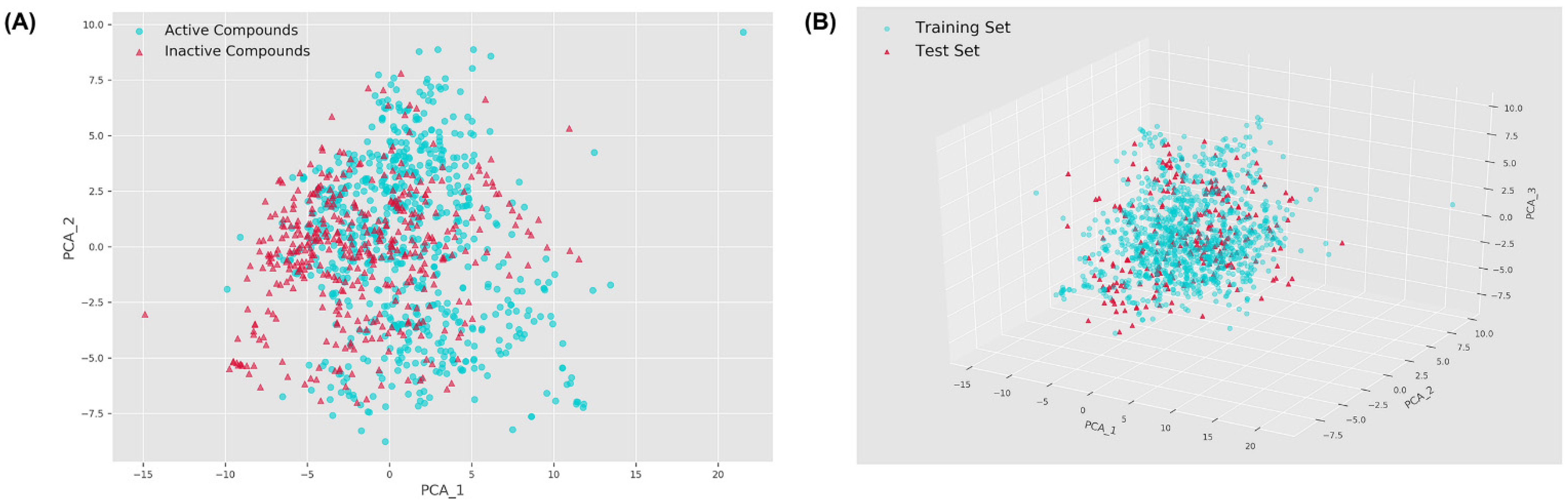
2.1. Chemical Space Distribution
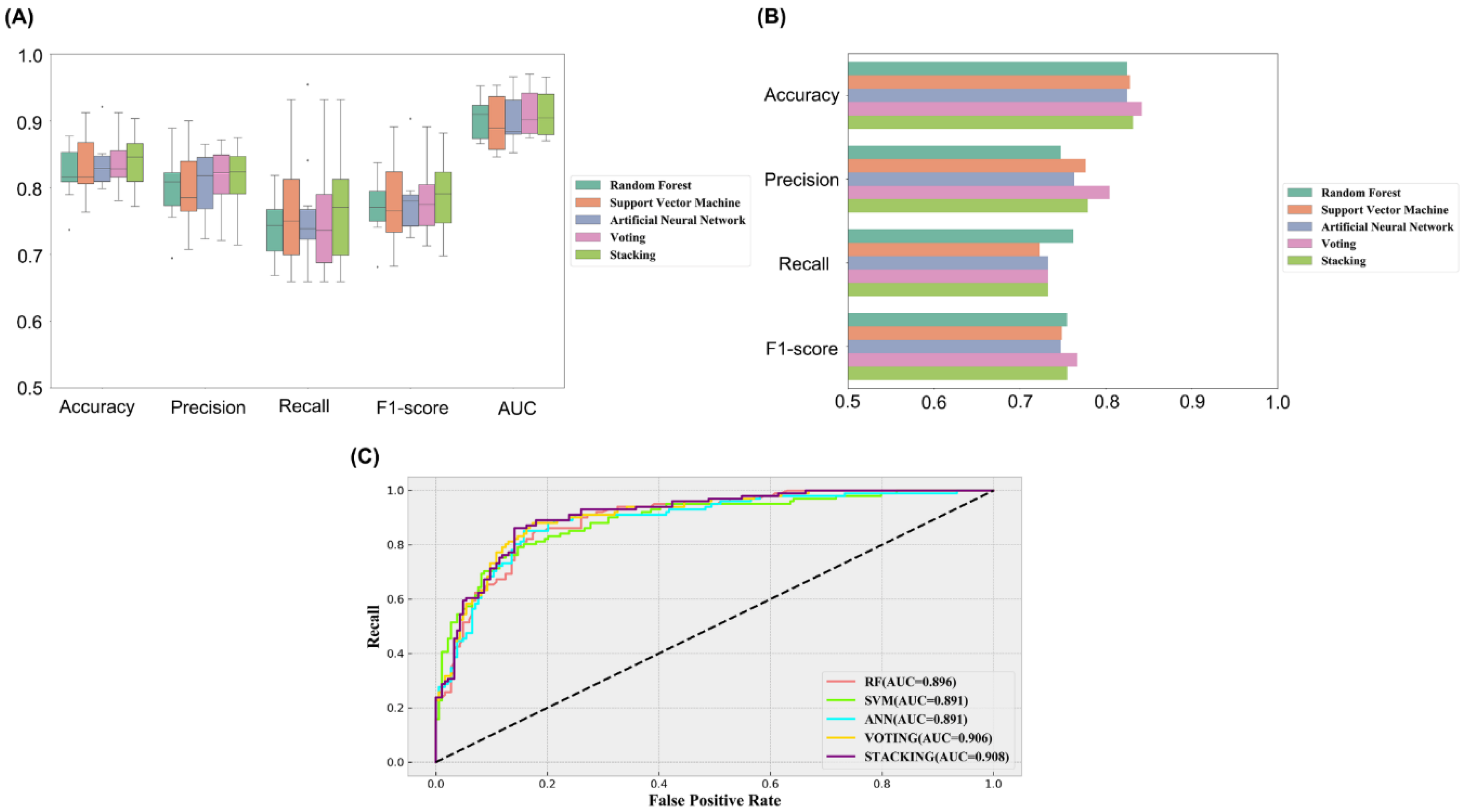
2.2. Establishment and Comparison of Machine Learning Models
2.3. Feature Importance Ranking
2.4. Integrated Model-Based Activity Prediction and Drug-Likeness Filter
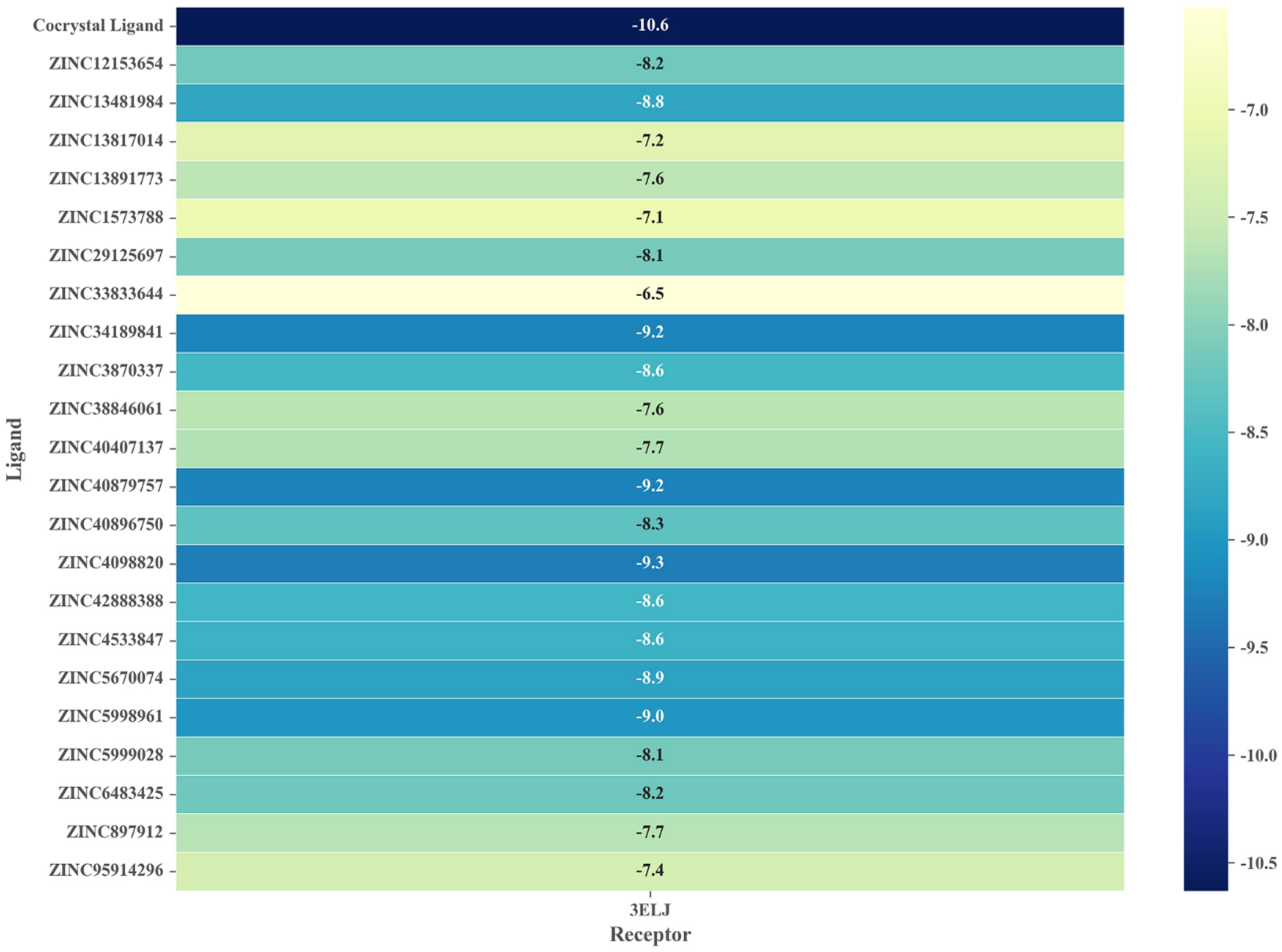
2.5. Virtual Screening Based on Molecular Docking
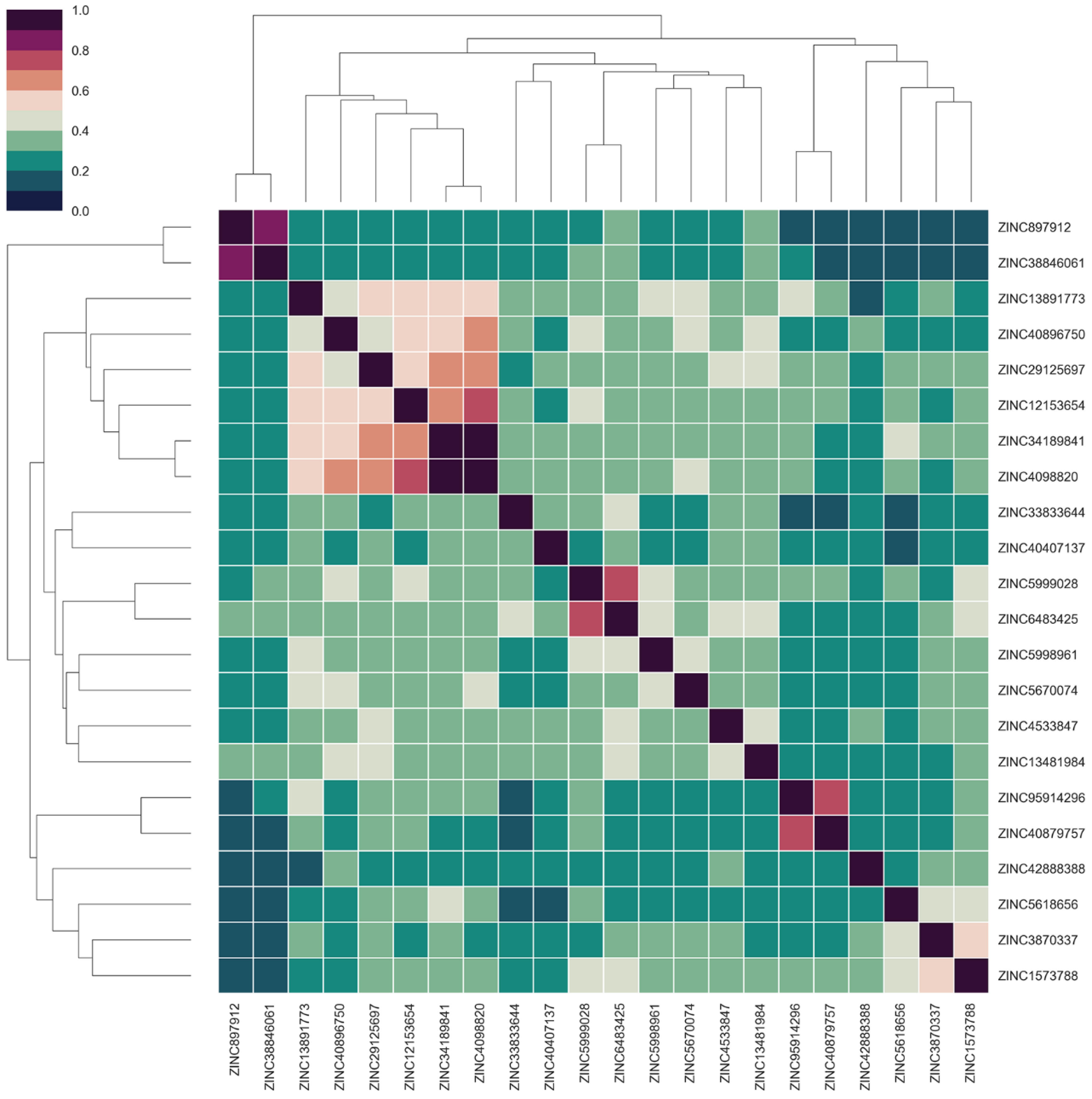
2.6. Identification of Candidate Compounds
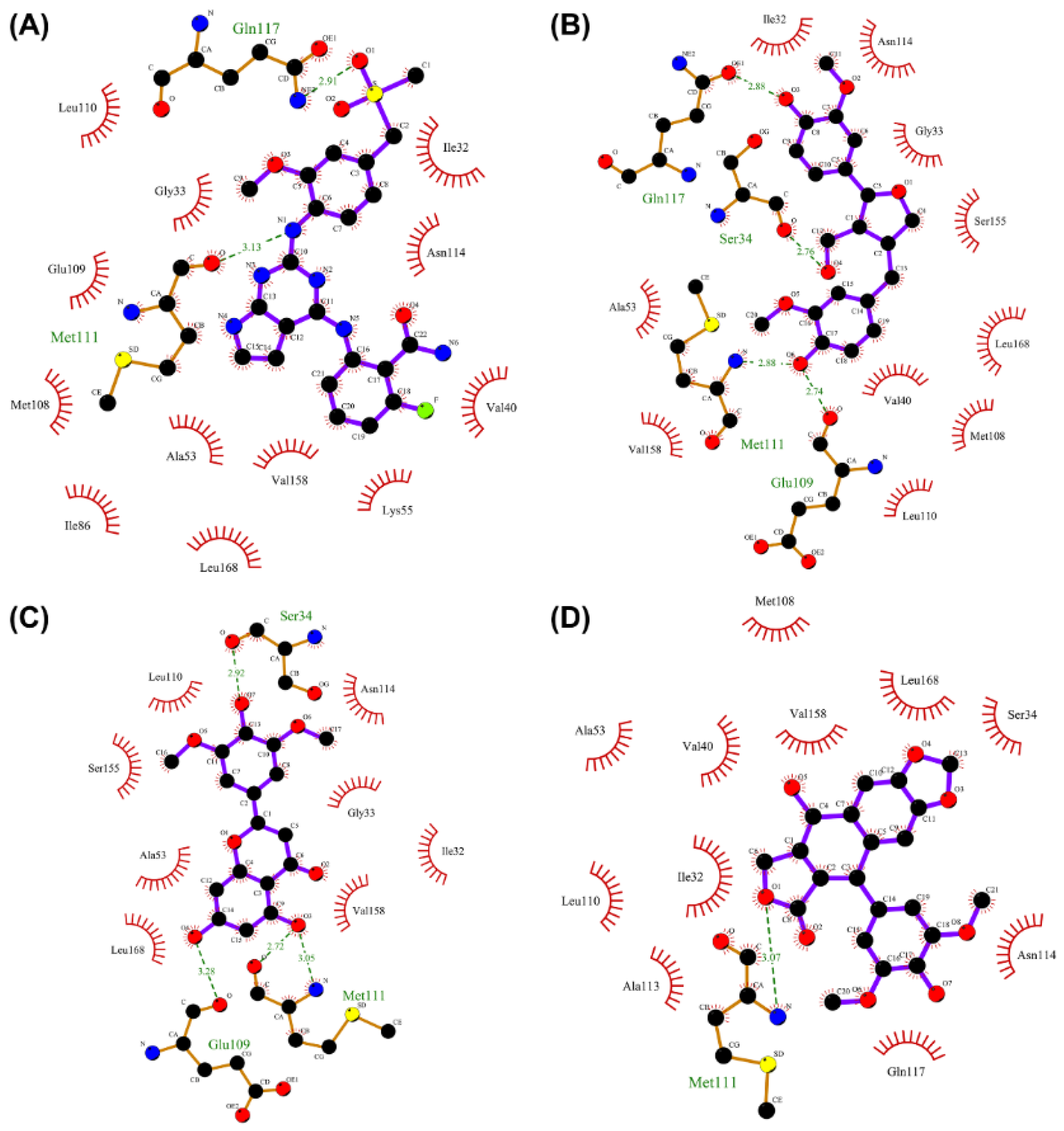
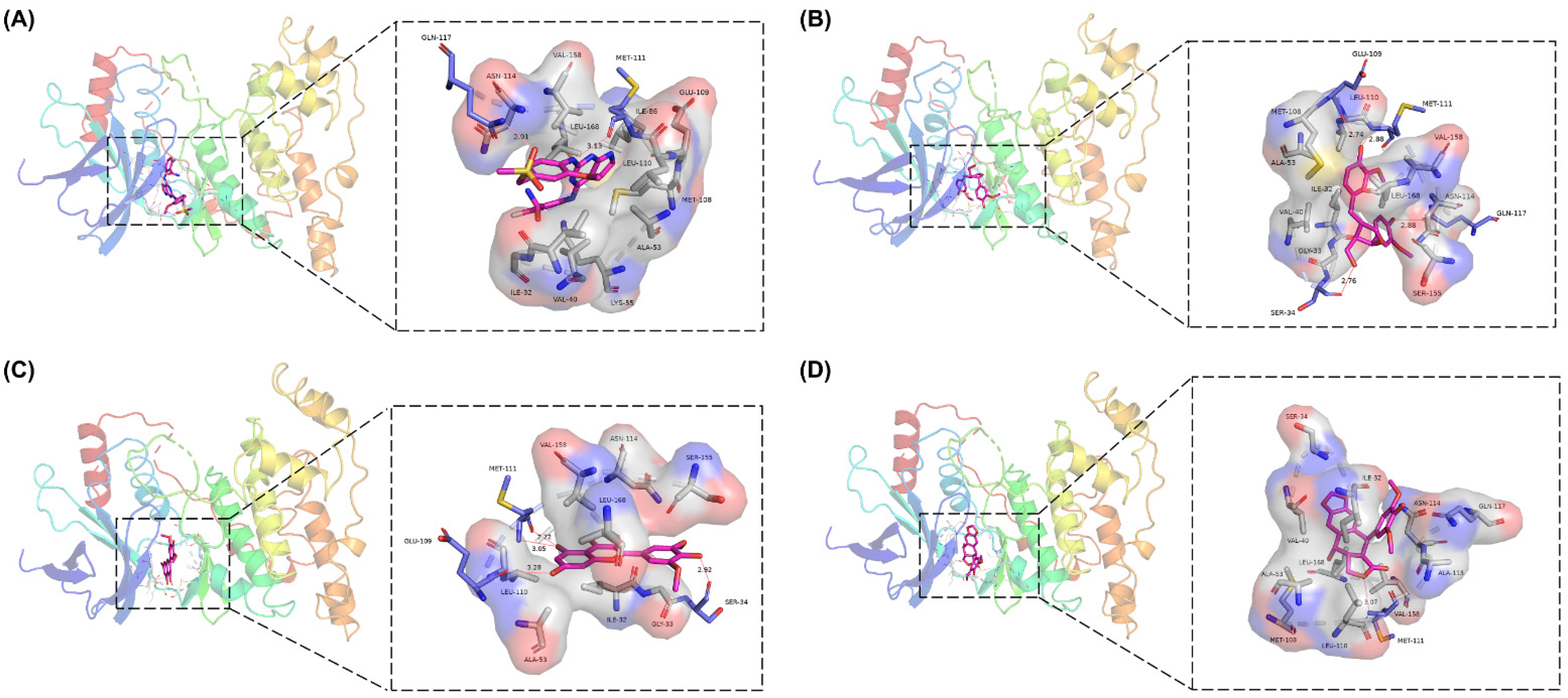
2.7. Analysis of Drug–Protein Interaction
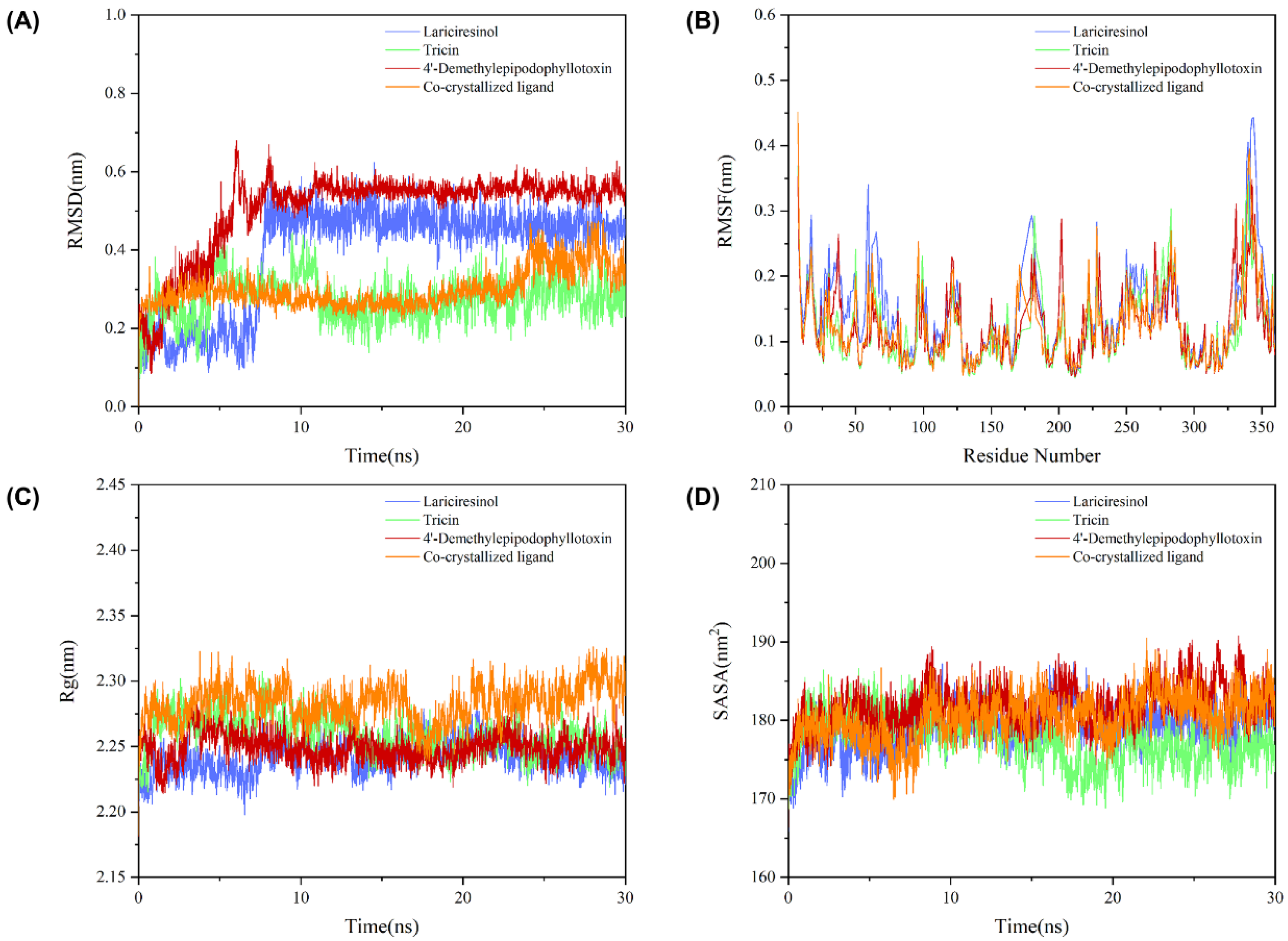
2.8. Molecular Dynamics Simulation
2.9. Biological Evaluation
3. Discussion
4. Materials and Methods
4.1. Machine Learning Dataset
4.1.1. Data Preparation
4.1.2. Descriptors Pruning
4.2. Machine Learning Algorithms
4.2.1. Classifier Construction
4.2.2. Classifier Evaluation
4.3. ADMET Studies
4.4. Molecular Docking
4.5. Consensus Analysis
4.6. Molecular Dynamics Simulation
4.7. Kinase Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chen, F. JNK-Induced Apoptosis, Compensatory Growth, and Cancer Stem Cells. Cancer Res. 2012, 72, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse Roles of JNK and MKK Pathways in the Brain. J. Signal Transduct. 2012, 2012, 459265. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.; Eyck, L.F.T.; Ashford, V.A.; Xuong, N.-H.; Taylor, S.S.; Sowadski, J.M. Crystal Structure of the Catalytic Subunit of Cyclic Adenosine Monophosphate-Dependent Protein Kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Bennett, B.L. JNK: A new therapeutic target for diabetes. Curr. Opin. Pharmacol. 2003, 3, 420–425. [Google Scholar] [CrossRef]
- Rayan, A.; Raiyn, J.; Falah, M. Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS ONE 2017, 12, e0187925. [Google Scholar] [CrossRef]
- Bradley, S.A.; Zhang, J.; Jensen, M.K. Deploying Microbial Synthesis for Halogenating and Diversifying Medicinal Alkaloid Scaffolds. Front. Bioeng. Biotechnol. 2020, 8, 594126. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Li, R.E. Emerging Methods for Ensemble-Based Virtual Screening. Curr. Top. Med. Chem. 2010, 10, 3–13. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, Y.; Wang, M.; Li, K.; Xu, L.; Chen, Y.; Cai, Y.; Jin, J. Integrating Machine Learning-Based Virtual Screening with Multiple Protein Structures and Bio-Assay Evaluation for Discovery of Novel GSK3β Inhibitors. Front. Pharmacol. 2020, 11, 566058. [Google Scholar] [CrossRef]
- Che, J.; Feng, R.; Gao, J.; Yu, H.; Weng, Q.; He, Q.; Dong, X.; Wu, J.; Yang, B. Evaluation of Artificial Intelligence in Participating Structure-Based Virtual Screening for Identifying Novel Interleukin-1 Receptor Associated Kinase-1 Inhibitors. Front. Oncol. 2020, 10, 1769. [Google Scholar] [CrossRef] [PubMed]
- Nyamundanda, G.; Brennan, L.; Gormley, I.C. Probabilistic principal component analysis for metabolomic data. BMC Bioinform. 2010, 11, 571. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Z.; Yao, X.; Li, Y.; Lei, T.; Wang, E.; Xu, L.; Zhu, F.; Li, D.; Hou, T. Comprehensive assessment of nine docking programs on type II kinase inhibitors: Prediction accuracy of sampling power, scoring power and screening power. Briefings Bioinform. 2018, 21, 282–297. [Google Scholar] [CrossRef]
- Chung, D.-J.; Wang, C.-J.; Yeh, C.-W.; Tseng, T.-H. Inhibition of the Proliferation and Invasion of C6 Glioma Cells by Tricin via the Upregulation of Focal-Adhesion-Kinase-Targeting MicroRNA-7. J. Agric. Food Chem. 2018, 66, 6708–6716. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Cho, J.; Hwang, I.-S.; Jin, H.-G.; Woo, E.-R.; Lee, D.G. Antifungal activity of lariciresinol derived from Sambucus williamsii and their membrane-active mechanisms in Candida albicans. Biochem. Biophys. Res. Commun. 2011, 410, 489–493. [Google Scholar] [CrossRef]
- Tang, Y.-J.; Zhao, W.; Li, H.-M. Novel Tandem Biotransformation Process for the Biosynthesis of a Novel Compound, 4-(2,3,5,6-Tetramethylpyrazine-1)-4′-Demethylepipodophyllotoxin. Appl. Environ. Microbiol. 2011, 77, 3023–3034. [Google Scholar] [CrossRef]
- Duong, M.T.H.; Lee, J.-H.; Ahn, H.-C. C-Jun N-terminal kinase inhibitors: Structural insight into kinase-inhibitor complexes. Comput. Struct. Biotechnol. J. 2020, 18, 1440–1457. [Google Scholar] [CrossRef]
- Liu, M.; Xin, Z.; Clampit, J.E.; Wang, S.; Gum, R.J.; Haasch, D.L.; Trevillyan, J.M.; Abad-Zapatero, C.; Fry, E.H.; Sham, H.L.; et al. Synthesis and SAR of 1,9-dihydro-9-hydroxypyrazolo [3,4-b]quinolin-4-ones as novel, selective c-Jun N-terminal kinase inhibitors. Bioorganic Med. Chem. Lett. 2006, 16, 2590–2594. [Google Scholar] [CrossRef]
- Gong, L.; Han, X.; Silva, T.; Tan, Y.-C.; Goyal, B.; Tivitmahaisoon, P.; Trejo, A.; Palmer, W.; Hogg, H.; Jahagir, A.; et al. Development of indole/indazole-aminopyrimidines as inhibitors of c-Jun N-terminal kinase (JNK): Optimization for JNK potency and physicochemical properties. Bioorganic Med. Chem. Lett. 2013, 23, 3565–3569. [Google Scholar] [CrossRef]
- Joshi, T.; Joshi, T.; Pundir, H.; Sharma, P.; Mathpal, S.; Chandra, S. Predictive modeling by deep learning, virtual screening and molecular dynamics study of natural compounds against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2021, 39, 6728–6746. [Google Scholar] [CrossRef]
- Esmaeilzadeh, P. Use of AI-based tools for healthcare purposes: A survey study from consumers’ perspectives. BMC Med. Inform. Decis. Mak. 2020, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Chen, Y.-P.P. Machine learning on adverse drug reactions for pharmacovigilance. Drug Discov. Today 2019, 24, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Mercader, A.G.; Duchowicz, P.R.; Fernández, F.M.; Castro, E.A. Modified and enhanced replacement method for the selection of molecular descriptors in QSAR and QSPR theories. Chemom. Intell. Lab. Syst. 2008, 92, 138–144. [Google Scholar] [CrossRef]
- Vergara, J.R.; Estévez, P.A. A review of feature selection methods based on mutual information. Neural Comput. Appl. 2014, 24, 175–186. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep Learning in Neural Networks: An Overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kongsted, J.; Ryde, U. An improved method to predict the entropy term with the MM/PBSA approach. J. Comput. Mol. Des. 2009, 23, 63–71. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Accuracy | Precision | Recall | F1-Score | AUC | |
|---|---|---|---|---|---|---|
| Training Set | RF | 0.821 | 0.801 | 0.713 | 0.752 | 0.905 |
| SVM | 0.832 | 0.795 | 0.763 | 0.777 | 0.896 | |
| ANN | 0.835 | 0.806 | 0.759 | 0.779 | 0.901 | |
| Voting | 0.836 | 0.811 | 0.752 | 0.778 | 0.913 | |
| Stacking | 0.840 | 0.810 | 0.768 | 0.786 | 0.912 | |
| Test Set | RF | 0.825 | 0.748 | 0.762 | 0.755 | 0.896 |
| SVM | 0.828 | 0.777 | 0.723 | 0.749 | 0.891 | |
| ANN | 0.825 | 0.763 | 0.733 | 0.748 | 0.891 | |
| Voting | 0.842 | 0.804 | 0.733 | 0.767 | 0.906 | |
| Stacking | 0.832 | 0.779 | 0.733 | 0.755 | 0.908 |
| Name | CAS Number | Structure | Binding Free Energy (kcal/mol) | S(Molecular Docking) | Confidence Level of Voting Model | Confidence Level of Stacking Model | S(Machine Learning) | S(JNK1) |
|---|---|---|---|---|---|---|---|---|
| Lariciresinol | 27003-73-2 | 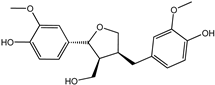 | −9.282 | 0.873 | 0.855 | 0.861 | 0.858 | 0.866 |
| Nigracin | 18463-25-7 |  | −9.229 | 0.868 | 0.829 | 0.838 | 0.834 | 0.851 |
| Tricin | 520-32-1 | 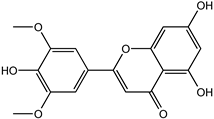 | −9.007 | 0.847 | 0.830 | 0.836 | 0.833 | 0.840 |
| 4′-Demethylepipodophyllotoxin | 6559-91-7 | 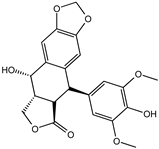 | −8.856 | 0.833 | 0.826 | 0.859 | 0.842 | 0.838 |
| Ophiopogonanone E | 588706-66-5 | 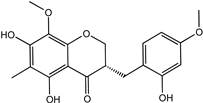 | −8.802 | 0.828 | 0.844 | 0.846 | 0.845 | 0.837 |
| Compound | van der Waals Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Total Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| Lariciresinol | −147.82 ± 9.72 | −39.20 ± 7.35 | 119.25 ± 8.46 | −16.74 ± 0.82 | −84.53 ± 9.51 |
| Tricin | −142.25 ± 10.38 | −18.34 ± 7.86 | 97.03 ± 12.46 | −16.55 ± 1.00 | −80.22 ± 10.08 |
| 4′-Demethylepipodophyllotoxin | −127.96 ± 9.74 | −34.25 ± 8.06 | 98.59 ± 14.58 | −14.48 ± 0.84 | −78.18 ± 10.67 |
| Co-crystallized ligand | −172.17 ± 18.60 | −98.91 ± 30.84 | 169.53 ± 18.81 | −16.80 ± 1.30 | −118.34 ± 29.11 |
| Compound | 50 μM | 25 μM | 12.5 μM | 6.25 μM | 3.125 μM | IC50 |
|---|---|---|---|---|---|---|
| Lariciresinol | 15.1% | 11.6% | 9.0% | 7.2% | 5.6% | >50 μM |
| Tricin | 75.4% | 61.8% | 47.2% | 34.8% | 25.9% | 17.68 μM |
| 4′-Demethylepipodophyllotoxin | 10.2% | 7.9% | 5.7% | 3.3% | 1.9% | >50 μM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, R.; Zhao, G.; Yan, B. Discovery of Novel c-Jun N-Terminal Kinase 1 Inhibitors from Natural Products: Integrating Artificial Intelligence with Structure-Based Virtual Screening and Biological Evaluation. Molecules 2022, 27, 6249. https://doi.org/10.3390/molecules27196249
Yang R, Zhao G, Yan B. Discovery of Novel c-Jun N-Terminal Kinase 1 Inhibitors from Natural Products: Integrating Artificial Intelligence with Structure-Based Virtual Screening and Biological Evaluation. Molecules. 2022; 27(19):6249. https://doi.org/10.3390/molecules27196249
Chicago/Turabian StyleYang, Ruoqi, Guiping Zhao, and Bin Yan. 2022. "Discovery of Novel c-Jun N-Terminal Kinase 1 Inhibitors from Natural Products: Integrating Artificial Intelligence with Structure-Based Virtual Screening and Biological Evaluation" Molecules 27, no. 19: 6249. https://doi.org/10.3390/molecules27196249
APA StyleYang, R., Zhao, G., & Yan, B. (2022). Discovery of Novel c-Jun N-Terminal Kinase 1 Inhibitors from Natural Products: Integrating Artificial Intelligence with Structure-Based Virtual Screening and Biological Evaluation. Molecules, 27(19), 6249. https://doi.org/10.3390/molecules27196249