
Identification of Novel Dopamine D2 Receptor Ligands—A Combined In Silico/In Vitro Approach


Abstract
:1. Introduction
2. Results
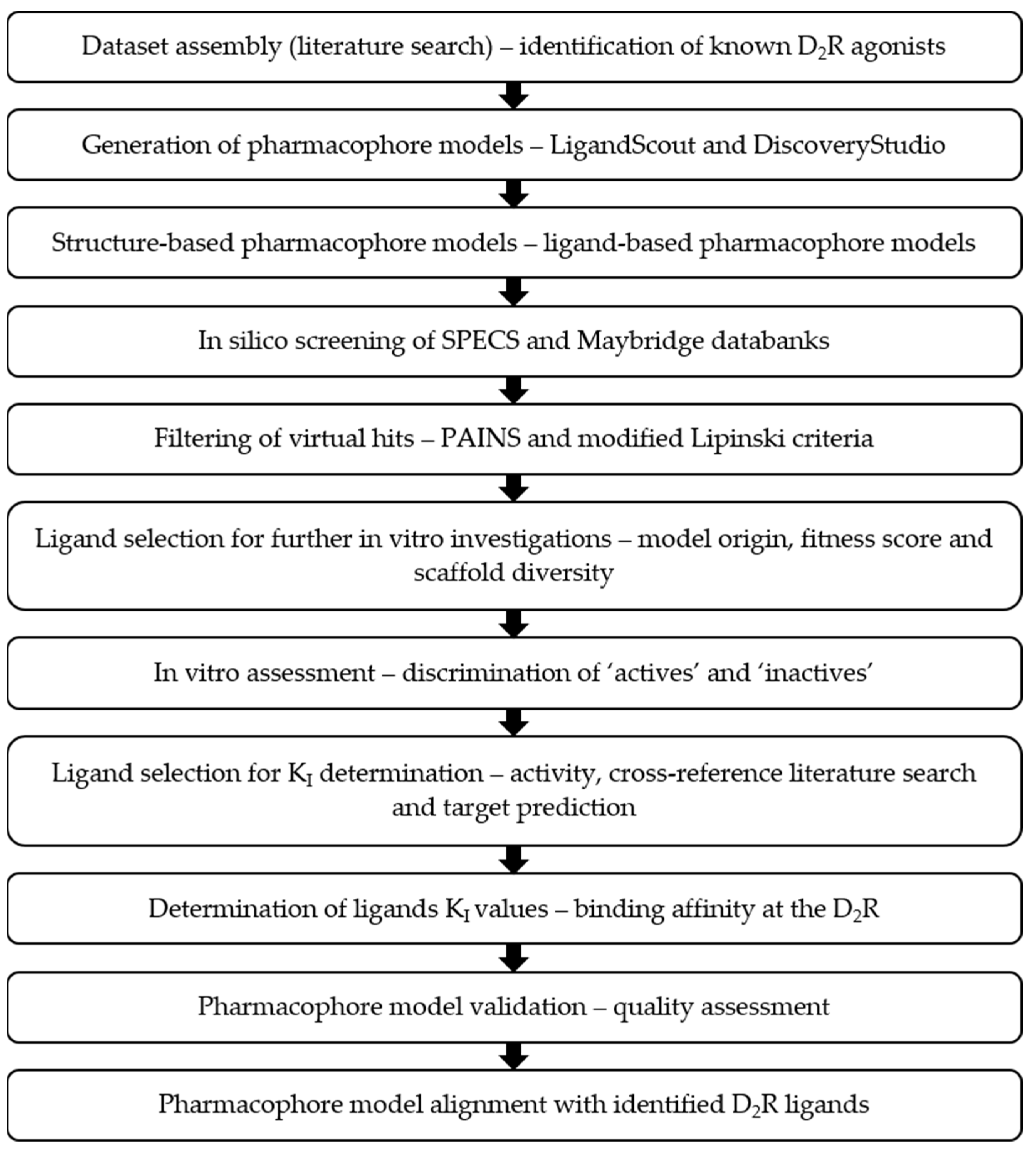
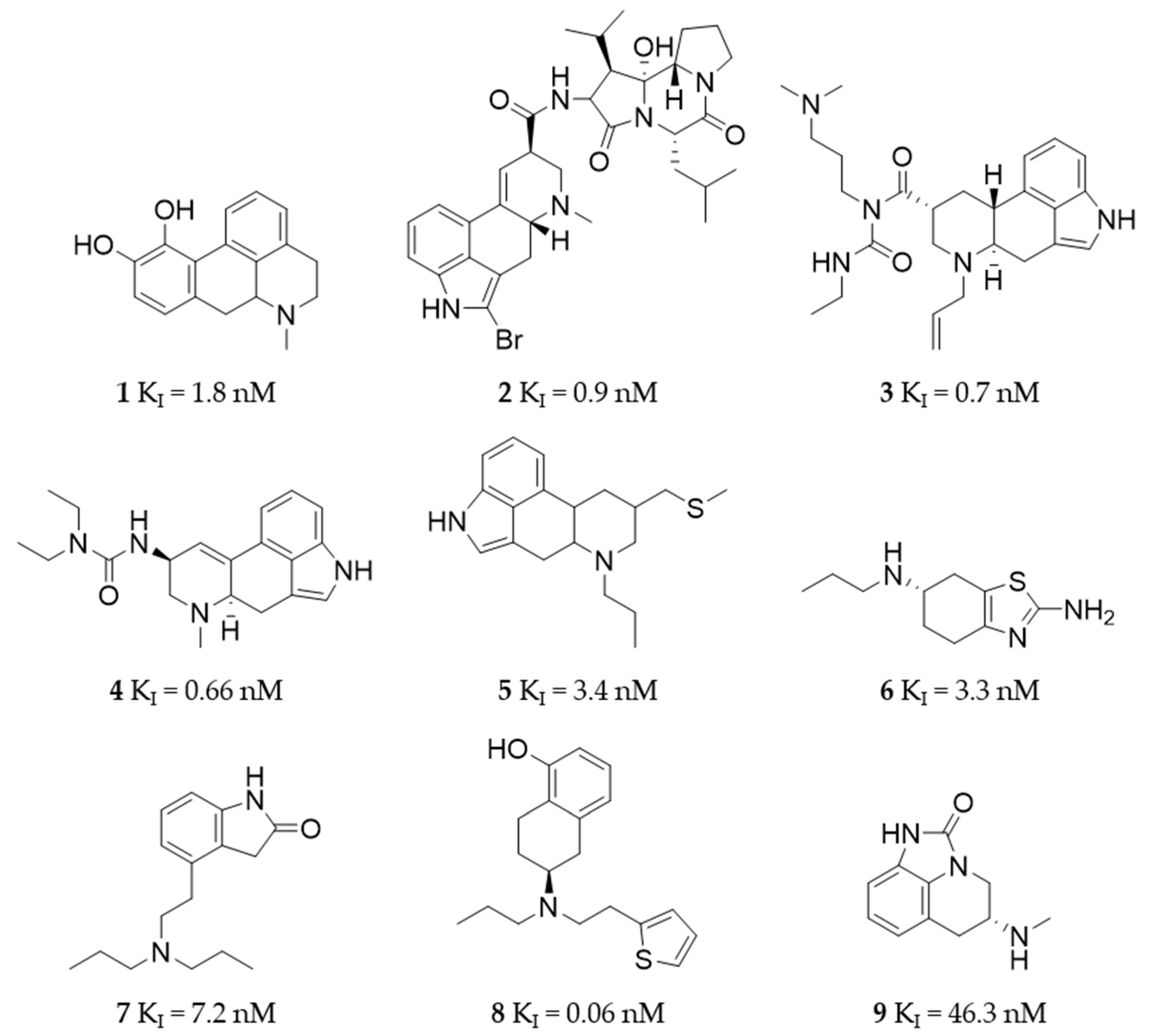
2.1. Dataset Assembly
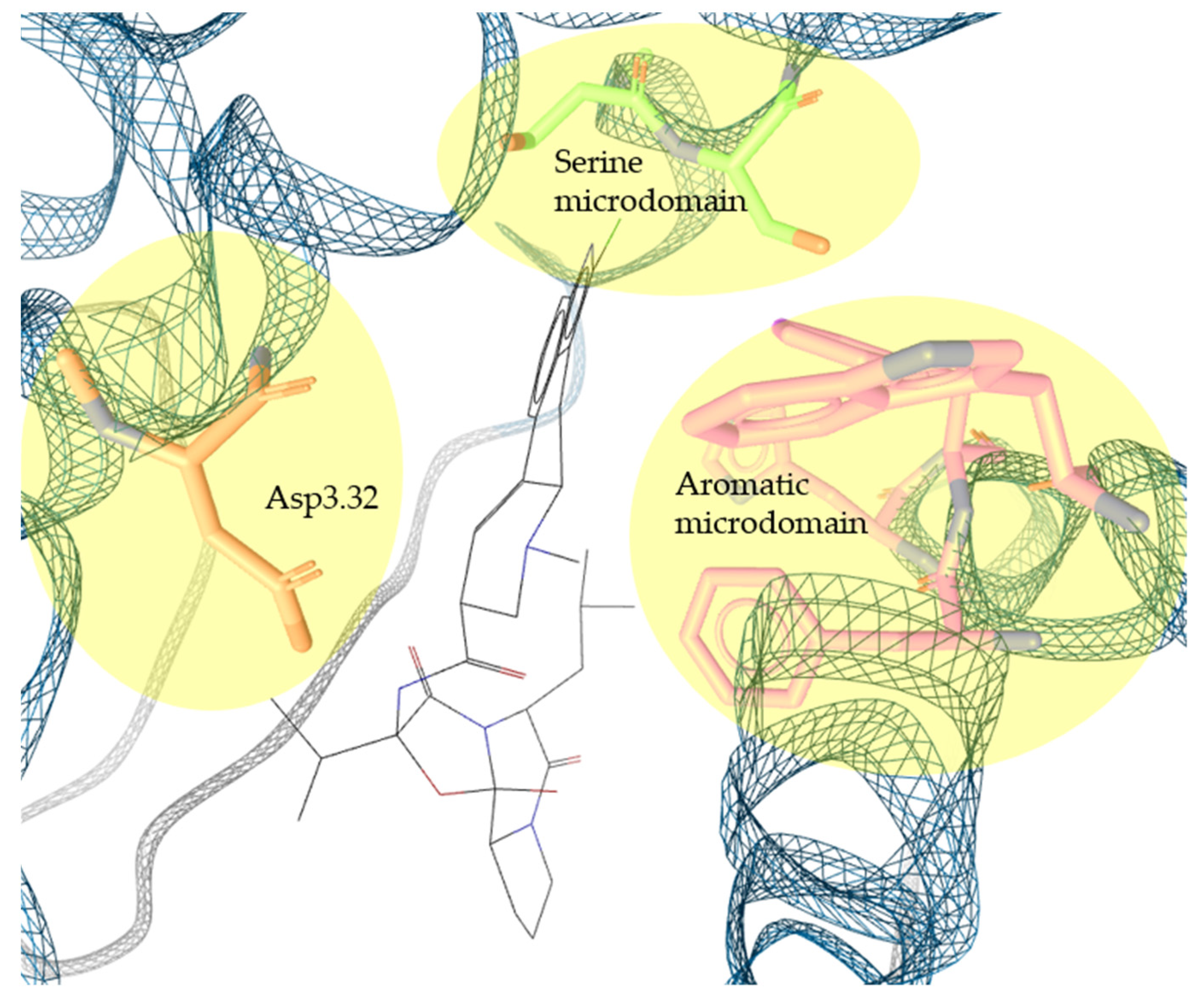
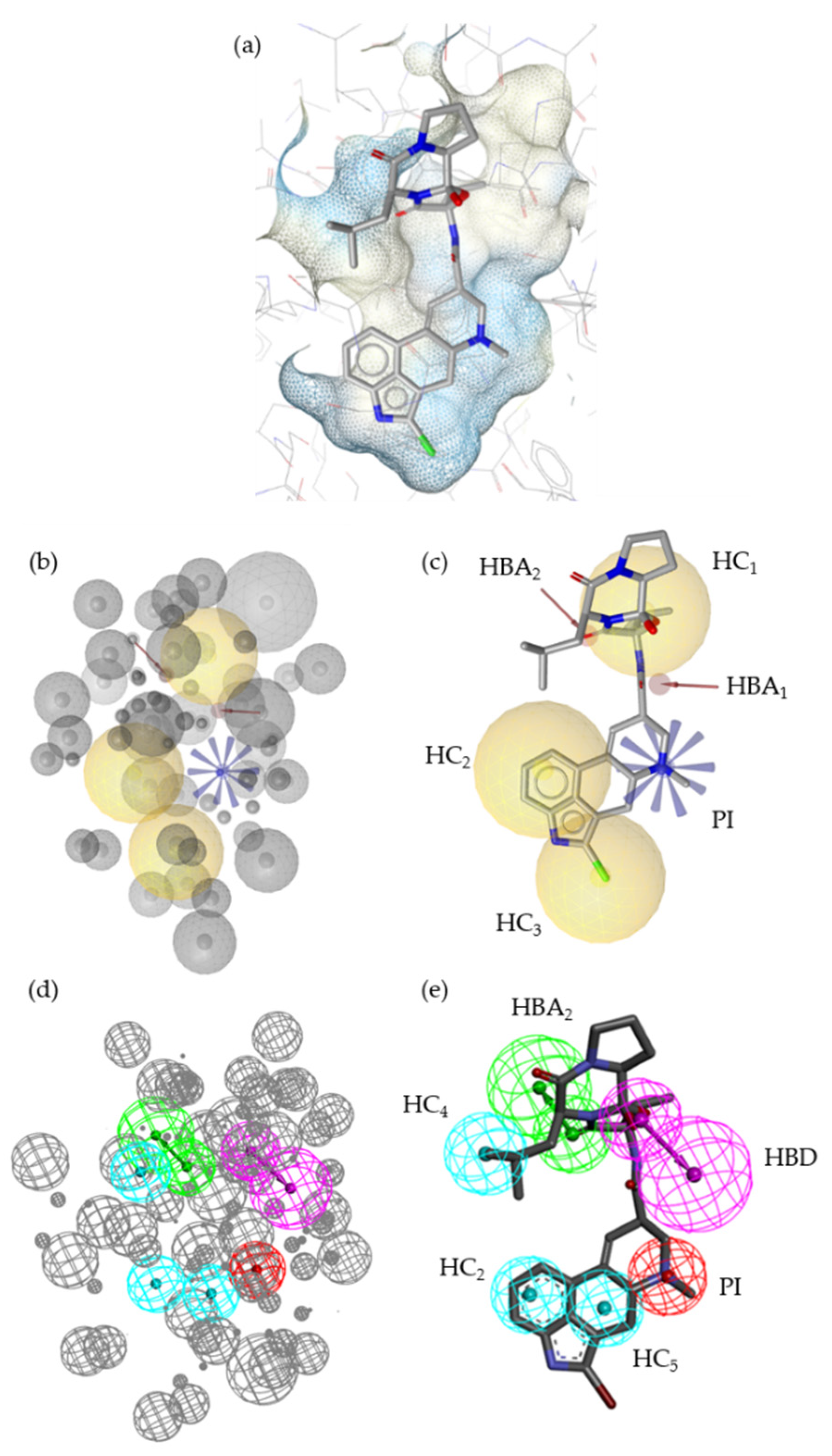
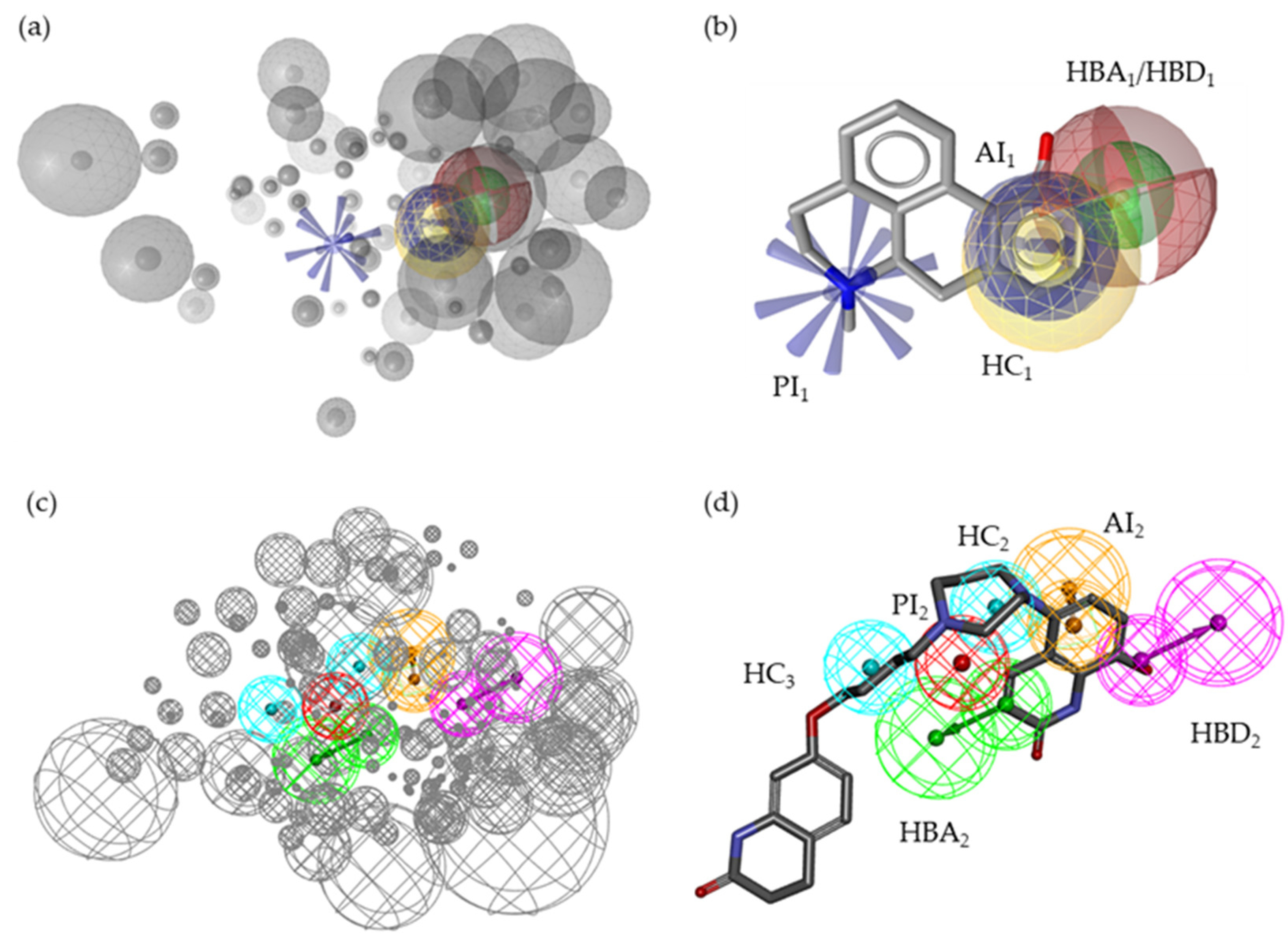
2.2. Pharmacophore Models—LB and SB Approaches in LigandScout and DiscoveryStudio
2.3. Screening of the SPECS and Maybridge Databases
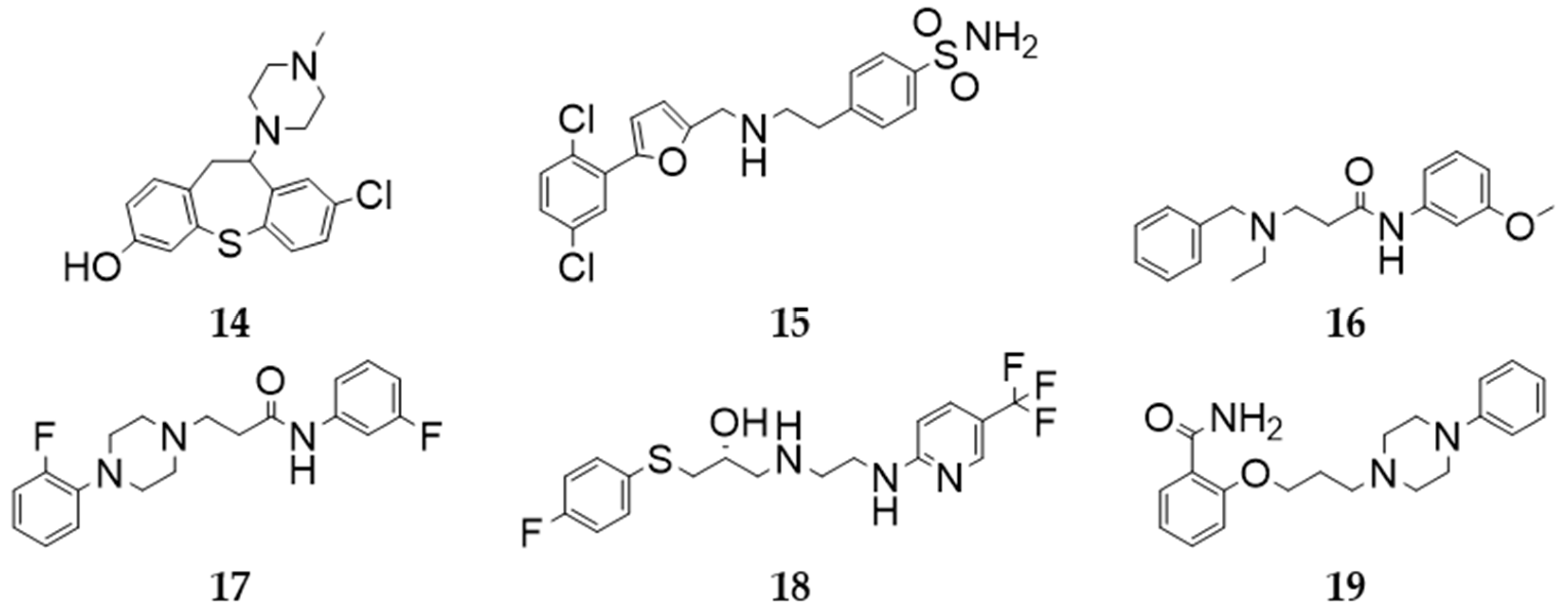
2.4. In Vitro Compound Screening
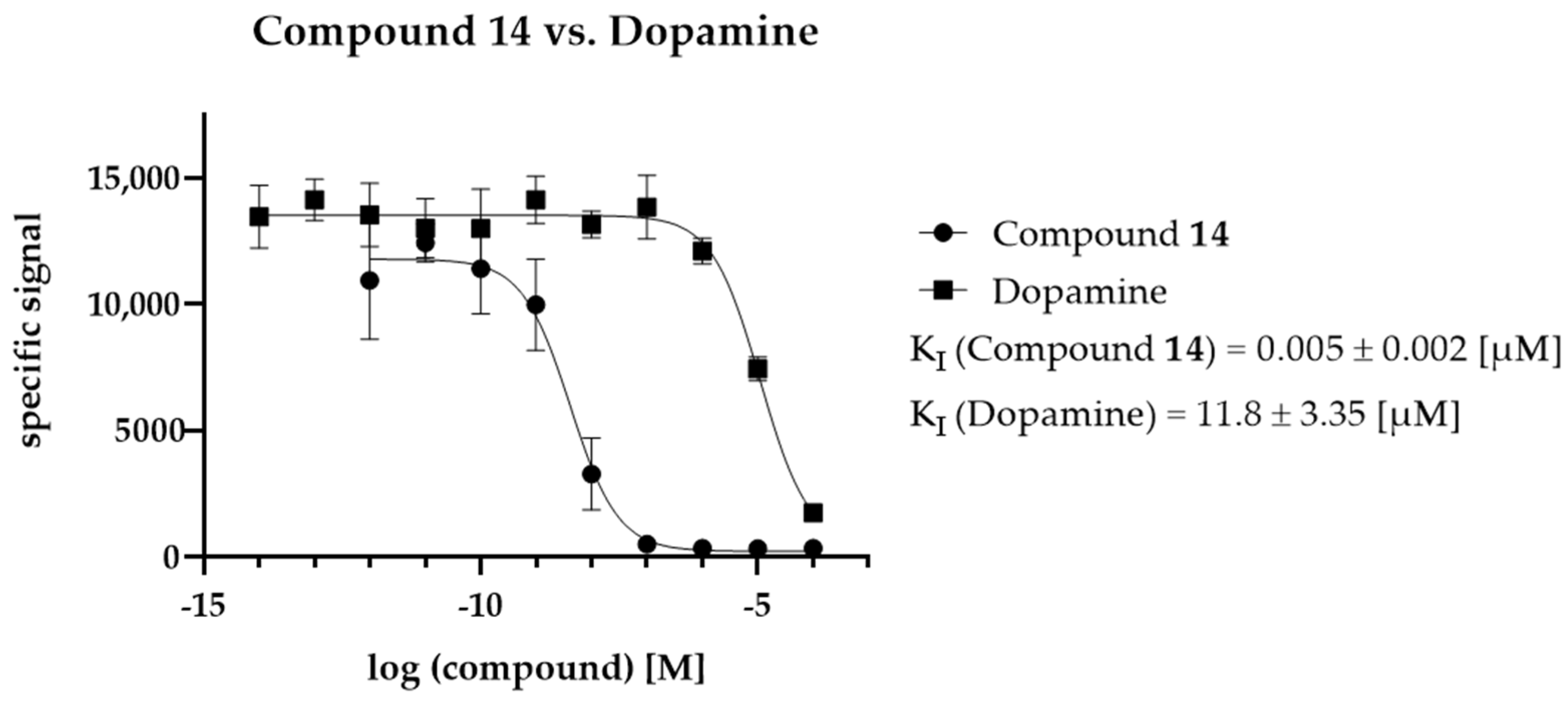
2.5. KI Determination—D2R Binding Affinity of the Selected Compounds
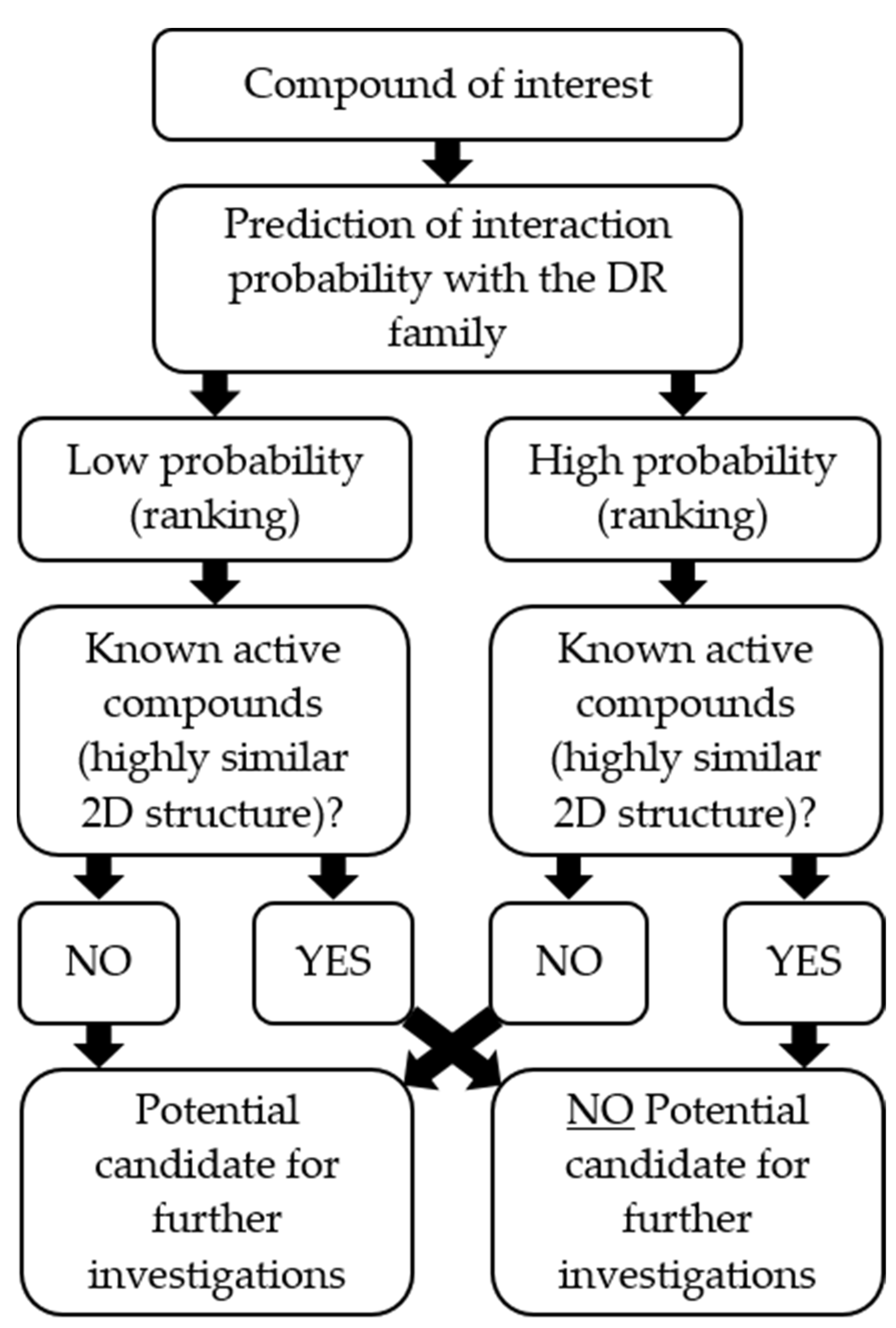
2.6. In Silico Model Evaluation
2.7. Alignment of the Pharmacophore Models with Novel D2R Ligands
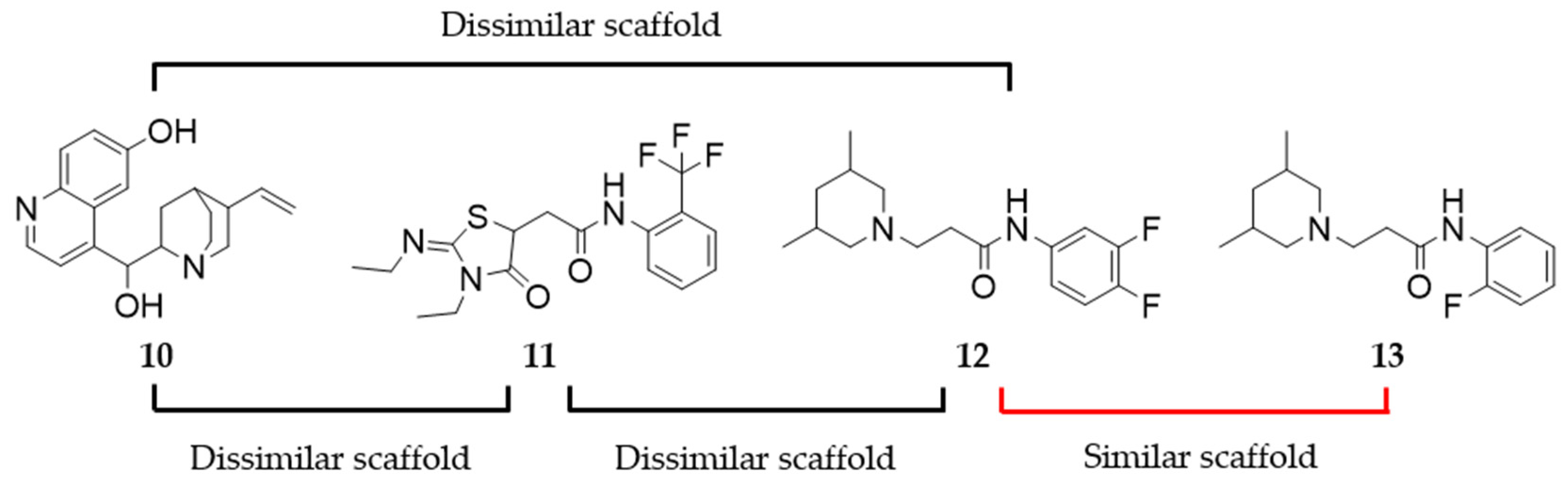
2.8. Assessing the Scaffold Novelty of the Identified Ligands Compared to Known D2R Ligands in ChEMBL
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Dataset Assembly
4.3. Generation of Pharmacophore Models
4.3.1. SB Pharmacophore
4.3.2. LB Pharmacophore
4.3.3. Pharmacophore Model Validation Metrics
4.4. Predictive Screening—SPECS and Maybridge
4.5. Settings for an HTRF-Compatible Tecan Spark Plate Reader
4.6. Characterization of D2R Carrier Cells—KD Determination
4.7. Compound Selection for Assessing In Vitro Activity
4.8. In Vitro Assessment of Compound Activity
4.9. Ligand Selection for Detailed In Vitro Investigation (KI Determination)
4.10. KI Determination of the Selected Ligands
4.11. Pharmacophore Model Evaluation
4.12. Assessing Scaffold Similarity/Dissimilarity
4.13. Data Processing, Representation, and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Haque, M.E.; Jakaria, M.; Jo, S.H.; Kim, I.S.; Choi, D.K. G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits. Cells 2020, 9, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Wu, X.; Acheampong, K.; Liu, A. Dopamine and Dopamine Receptors in Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Stocchi, F.; Torti, M.; Fossati, C. Advances in dopamine receptor agonists for the treatment of Parkinson’s disease. Expert Opin. Pharmacother. 2016, 17, 1889–1902. [Google Scholar] [CrossRef]
- Li, P.; Snyder, G.L.; Vanover, K.E. Dopamine Targeting Drugs for the Treatment of Schizophrenia: Past, Present and Future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [Green Version]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Carbone, F.; Djamshidian, A.; Seppi, K.; Poewe, W. Apomorphine for Parkinson’s Disease: Efficacy and Safety of Current and New Formulations. CNS Drugs 2019, 33, 905–918. [Google Scholar] [CrossRef] [Green Version]
- Smith, Y.; Wichmann, T.; Factor, S.A.; DeLong, M.R. Parkinson’s disease therapeutics: New developments and challenges since the introduction of levodopa. Neuropsychopharmacology 2012, 37, 213–246. [Google Scholar] [CrossRef]
- Kuo, J.; Hwu, H.G. Aripiprazole and haloperidol: Beneficial combination antipsychotic therapy for a schizophrenic patient. Clin. Neuropharmacol. 2008, 31, 173–175. [Google Scholar] [CrossRef]
- Thanvi, B.R.; Lo, T.C. Long term motor complications of levodopa: Clinical features, mechanisms, and management strategies. Postgrad Med. J. 2004, 80, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 2018, 16, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.A.; Moore, H.; Stott, L.; Holliday, N.; Javitch, J.A.; Lane, J.R.; Charlton, S.J. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat. Commun. 2017, 8, 763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, B.D. Neuroleptic malignant syndrome: A review for neurohospitalists. Neurohospitalist 2011, 1, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Mach, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef] [Green Version]
- Butini, S.; Nikolic, K.; Kassel, S.; Bruckmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Prasad, M.E.; Hung, S.Y. Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals 2021, 14, 717. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef]
- Fan, L.; Tan, L.; Chen, Z.; Qi, J.; Nie, F.; Luo, Z.; Cheng, J.; Wang, S. Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands. Nat. Commun. 2020, 11, 1074. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chen, K.M.; Clark, M.J.; Hijazi, M.; Kumari, P.; Bai, X.C.; Sunahara, R.K.; Barth, P.; Rosenbaum, D.M. Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature 2020, 584, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.P. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e18. [Google Scholar] [CrossRef]
- Lemos, A.; Melo, R.; Preto, A.J.; Almeida, J.G.; Moreira, I.S.; Dias Soeiro Cordeiro, M.N. In Silico Studies Targeting G-protein Coupled Receptors for Drug Research Against Parkinson’s Disease. Curr. Neuropharmacol. 2018, 16, 786–848. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.W.S.; Kumar, H. Structure- and ligand-based drug design: Concepts, approaches, and challenges. In Chemoinformatics and Bioinformatics in the Pharmaceutical Sciences; Sharma, H.N.O., Raghav, P.W., Goyal, R.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 27–53. [Google Scholar]
- Lian, P.; Xu, L.; Geng, C.; Qian, Y.; Li, W.; Zhen, X.; Fu, W. A computational perspective on drug discovery and signal transduction mechanism of dopamine and serotonin receptors in the treatment of schizophrenia. Curr. Pharm. Biotechnol. 2014, 15, 916–926. [Google Scholar] [CrossRef]
- Ishiki, H.M.; Filho, J.M.B.; da Silva, M.S.; Scotti, M.T.; Scotti, L. Computer-aided Drug Design Applied to Parkinson Targets. Curr. Neuropharmacol. 2018, 16, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Ostopovici-Halip, L.; Rad-Curpan, R. Modeling of ligand binding to the dopamine D2 receptor. J. Serbian Chem. Soc. 2014, 79, 175–183. [Google Scholar] [CrossRef]
- Malo, M.; Brive, L.; Luthman, K.; Svensson, P. Investigation of D(2) receptor-agonist interactions using a combination of pharmacophore and receptor homology modeling. ChemMedChem 2012, 7, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Penjisevic, J.Z.; Sukalovic, V.V.; Andric, D.B.; Roglic, G.M.; Novakovic, I.T.; Soskic, V.; Kostic-Rajacic, S.V. Synthesis, biological evaluation and docking analysis of substituted piperidines and (2-methoxyphenyl)piperazines. J. Serbian Chem. Soc. 2016, 81, 347–356. [Google Scholar] [CrossRef]
- Penjisevic, J.Z.; Sukalovic, V.V.; Andric, D.B.; Roglic, G.M.; Soskic, V.; Kostic-Rajacic, S.V. Synthesis, Biological, and Computational Evaluation of Substituted 1-(2-Methoxyphenyl)-4-(1-phenethylpiperidin-4-yl)piperazines and 1-(2-Methoxyphenyl)-4-[(1-phenethylpiperidin-4-yl)methyl]piperazines as Dopaminergic Ligands. Arch. der Pharm. 2016, 349, 614–626. [Google Scholar] [CrossRef]
- Platania, C.B.; Salomone, S.; Leggio, G.M.; Drago, F.; Bucolo, C. Homology modeling of dopamine D2 and D3 receptors: Molecular dynamics refinement and docking evaluation. PLoS ONE 2012, 7, e44316. [Google Scholar] [CrossRef] [PubMed]
- Ortore, G.; Tuccinardi, T.; Bertini, S.; Martinelli, A. A theoretical study to investigate D2DAR/D4DAR selectivity: Receptor modeling and molecular docking of dopaminergic ligands. J. Med. Chem. 2006, 49, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Sukalovic, V.; Andric, D.; Roglic, G.; Kostic-Rajacic, S.; Schrattenholz, A.; Soskic, V. Synthesis, dopamine D2 receptor binding studies and docking analysis of 5-[3-(4-arylpiperazin-1-yl)propyl]-1H-benzimidazole, 5-[2-(4-arylpiperazin-1-yl)ethoxy]-1H-benzimidazole and their analogs. Eur. J. Med. Chem. 2005, 40, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Sukalovic, V.; Soskic, V.; Andric, D.; Roglic, G.; Kostic-Rajacic, S. Modeling key interactions between the second extracellular loop of the dopamine D2 receptor and arylpiperazine ligands. J. Serb. Chem. Soc. 2012, 77, 259–277. [Google Scholar] [CrossRef]
- Zhu, J.N.; Liang, L.; Stevens, C.L.K. A CoMFA study of dopamine D2 receptor agonists and X-ray crystal structure of quinelorane dihydrochloride dihydrate, R(-)-apomorphine hydrochloride and R(-)-N-n-propylnorapomorphine hydrochloride. Struct. Chem. 2004, 15, 553–565. [Google Scholar] [CrossRef]
- Modi, G.; Sharma, H.; Kharkar, P.S.; Dutta, A.K. Understanding the Structural Requirements of Hybrid (S)-6-((2-(4-Phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthal en-1-ol and its Analogs as D2/D3 Receptor Ligands: A Three-Dimensional Quantitative Structure-Activity Relationship (3D QSAR) Investigation. Medchemcomm 2014, 5, 1384–1399. [Google Scholar]
- Malo, M.; Brive, L.; Luthman, K.; Svensson, P. Selective pharmacophore models of dopamine D(1) and D(2) full agonists based on extended pharmacophore features. ChemMedChem 2010, 5, 232–246. [Google Scholar] [CrossRef]
- Floresca, Z.C.; Schetz, J.A. Dopamine receptor microdomains involved in molecular recognition and the regulation of drug affinity and function. J. Recept. Signal Transduct. Res. 2004, 24, 207–239. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Recept. Mol. Biol. 1995, 25, 366–428. [Google Scholar]
- Moreira, I.S.S.; Shi, L.; Freyberg, Z.; Ericksen, S.S.; Weinstein, H.; Javitch, J.A. Structural Basis of Dopamine Receptor Activation. In The Dopamine Receptors; Neve, K.A., Ed.; Humana Press/Springer: Berlin, Germany, 2010. [Google Scholar]
- Salmas, R.E.; Yurtsever, M.; Stein, M.; Durdagi, S. Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions. Mol. Divers. 2015, 19, 321–332. [Google Scholar] [CrossRef]
- Bueschbell, B.; Barreto, C.A.V.; Preto, A.J.; Schiedel, A.C.; Moreira, I.S. A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods. Molecules 2019, 24, 1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschammer, N.; Dorfler, M.; Hubner, H.; Gmeiner, P. Engineering a GPCR-ligand pair that simulates the activation of D(2L) by Dopamine. ACS Chem. Neurosci. 2010, 1, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kling, R.C.; Tschammer, N.; Lanig, H.; Clark, T.; Gmeiner, P. Active-state model of a dopamine D2 receptor-Galphai complex stabilized by aripiprazole-type partial agonists. PLoS ONE 2014, 9, e100069. [Google Scholar]
- Kalani, M.Y.; Vaidehi, N.; Hall, S.E.; Trabanino, R.J.; Freddolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.; Goddard, W.A. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 3815–3820. [Google Scholar] [CrossRef] [Green Version]
- Holst, B.; Nygaard, R.; Valentin-Hansen, L.; Bach, A.; Engelstoft, M.S.; Petersen, P.S.; Frimurer, T.M.; Schwartz, T.W. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 2010, 285, 3973–3985. [Google Scholar] [CrossRef] [Green Version]
- Degorce, F.; Card, A.; Soh, S.; Trinquet, E.; Knapik, G.P.; Xie, B. HTRF: A technology tailored for drug discovery—A review of theoretical aspects and recent applications. Curr. Chem. Genom. 2009, 3, 22–32. [Google Scholar] [CrossRef]
- Yasi, E.A.; Kruyer, N.S.; Peralta-Yahya, P. Advances in G protein-coupled receptor high-throughput screening. Curr. Opin. Biotechnol. 2020, 64, 210–217. [Google Scholar] [CrossRef]
- Durdagi, S.; Salmas, R.E.; Stein, M.; Yurtsever, M.; Seeman, P. Binding Interactions of Dopamine and Apomorphine in D2High and D2Low States of Human Dopamine D2 Receptor Using Computational and Experimental Techniques. ACS Chem. Neurosci. 2015, 7, 185–195. [Google Scholar] [CrossRef]
- Seeman, P. Antiparkinson therapeutic potencies correlate with their affinities at dopamine D2(High) receptors. Synapse 2007, 61, 1013–1018. [Google Scholar] [CrossRef]
- Tadori, Y.; Forbes, R.A.; McQuade, R.D.; Kikuchi, T. Functional potencies of dopamine agonists and antagonists at human dopamine D(2) and D(3) receptors. Eur. J. Pharmacol. 2011, 666, 43–52. [Google Scholar] [CrossRef]
- Krogsgaard-Larsen, N.; Jensen, A.A.; Schroder, T.J.; Christoffersen, C.T.; Kehler, J. Novel aza-analogous ergoline derived scaffolds as potent serotonin 5-HT(6) and dopamine D(2) receptor ligands. J. Med. Chem. 2014, 57, 5823–5828. [Google Scholar] [CrossRef] [PubMed]
- De Keyser, J.D.B.; De Backer, J.P.; Wilczak, N.; Herroelen, L. Dopamine agonists used in the treatment of Parkinson’s disease and their selectivity for the D1, D2, and D3 dopamine receptors in human striatum. Prog. Neuro-Psychopharmacol. Biol. Psychiat. 1995, 19, 1147–1154. [Google Scholar] [CrossRef]
- Mierau, J.; Schneider, F.J.; Ensinger, H.A.; Chio, C.L.; Lajiness, M.E.; Huff, R.M. Pramipexole binding and activation of cloned and expressed dopamine D2, D3 and D4 receptors. Eur. J. Pharmacol. Mol. Pharmacol. 1995, 290, 29–36. [Google Scholar] [CrossRef]
- Heier, R.F.; Dolak, L.A.; Duncan, J.N.; Hyslop, D.K.; Lipton, M.F.; Martin, I.J.; Mauragis, M.A.; Piercey, M.F.; Nichols, N.F.; Schreur, P.J.; et al. Synthesis and biological activities of (R)-5,6-dihydro-N,N-dimethyl-4H-imidazo [4,5,1-ij]quinolin-5-amine and its metabolites. J. Med. Chem. 1997, 40, 639–646. [Google Scholar] [CrossRef]
- Dijkstra, D.; Rodenhuis, N.; Vermeulen, E.S.; Pugsley, T.A.; Wise, L.D.; Wikstrom, H.V. Further characterization of structural requirements for ligands at the dopamine D(2) and D(3) receptor: Exploring the thiophene moiety. J. Med. Chem. 2002, 45, 3022–3031. [Google Scholar] [CrossRef]
- Bonifazi, A.; Yano, H.; Ellenberger, M.P.; Muller, L.; Kumar, V.; Zou, M.F.; Cai, N.S.; Guerrero, A.M.; Woods, A.S.; Shi, L. Novel Bivalent Ligands Based on the Sumanirole Pharmacophore Reveal Dopamine D2 Receptor (D2R) Biased Agonism. J. Med. Chem. 2017, 60, 2890–2907. [Google Scholar] [CrossRef] [PubMed]
- van Wieringen, J.P.; Shalgunov, V.; Janssen, H.M.; Fransen, P.M.; Janssen, A.G.; Michel, M.C.; Booij, J.; Elsinga, P.H. Synthesis and characterization of a novel series of agonist compounds as potential radiopharmaceuticals for imaging dopamine D(2)/(3) receptors in their high-affinity state. J. Med. Chem. 2014, 57, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Kuhhorn, J.; Gotz, A.; Hubner, H.; Thompson, D.; Whistler, J.; Gmeiner, P. Development of a bivalent dopamine D(2) receptor agonist. J. Med. Chem. 2011, 54, 7911–7919. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sassano, M.F.; Zheng, L.; Setola, V.; Chen, M.; Bai, X.; Frye, S.V.; Wetsel, W.C.; Roth, B.L.; Jin, J. Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J. Med. Chem. 2012, 55, 7141–7153. [Google Scholar] [CrossRef] [Green Version]
- Weichert, D.; Banerjee, A.; Hiller, C.; Kling, R.C.; Hubner, H.; Gmeiner, P. Molecular determinants of biased agonism at the dopamine D(2) receptor. J. Med. Chem. 2015, 58, 2703–2717. [Google Scholar] [CrossRef]
- Hiller, C.; Kling, R.C.; Heinemann, F.W.; Meyer, K.; Hubner, H.; Gmeiner, P. Functionally selective dopamine D2/D3 receptor agonists comprising an enyne moiety. J. Med. Chem. 2013, 56, 5130–5141. [Google Scholar] [CrossRef] [PubMed]
- Burris, K.D.; Pacheco, M.A.; Filtz, T.M.; Kung, M.P.; Kung, H.F.; Molinoff, P.B. Lack of discrimination by agonists for D2 and D3 dopamine receptors. Neuropsychopharmacology 1995, 12, 335–345. [Google Scholar] [CrossRef]
- Ghosh, B.; Antonio, T.; Zhen, J.; Kharkar, P.; Reith, M.E.; Dutta, A.K. Development of (S)-N6-(2-(4-(isoquinolin-1-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydro benzo[d]-thiazole-2,6-diamine and its analogue as a D3 receptor preferring agonist: Potent in vivo activity in Parkinson’s disease animal models. J. Med. Chem. 2010, 53, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Szabo, M.; Klein Herenbrink, C.; Christopoulos, A.; Lane, J.R.; Capuano, B. Structure-activity relationships of privileged structures lead to the discovery of novel biased ligands at the dopamine D(2) receptor. J. Med. Chem. 2014, 57, 4924–4939. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Downing, D.M.; Feng, M.R.; Hayes, R.N.; Heffner, T.G.; MacKenzie, R.G.; Meltzer, L.T.; Pugsley, T.A.; Wise, L.D. Identification, characterization and pharmacological profile of three metabolites of (R)-(+)-1,2,3,6-tetrahydro-4-phenyl-1-[(3-phenylcyclohexen-1- yl)methyl]pyridine (CI-1007), a dopamine autoreceptor agonist and potential antipsychotic agent. J. Med. Chem. 1995, 38, 5007–5014. [Google Scholar] [CrossRef] [PubMed]
- Mannel, B.; Dengler, D.; Shonberg, J.; Hubner, H.; Moller, D.; Gmeiner, P. Hydroxy-Substituted Heteroarylpiperazines: Novel Scaffolds for beta-Arrestin-Biased D2R Agonists. J. Med. Chem. 2017, 60, 4693–4713. [Google Scholar] [CrossRef]
- Ghosh, B.A.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl)piperazin-1-yl)quinolin-8-ol and Its Analogues as Highly Potent Dopamine D2/D3 Agonists and as Iron Chelator: In Vivo Activity Indicates Potential Application in Symptomatic and Neuroprotective Therapy for Parkinson’s Disease. J. Med. Chem. 2010, 53, 2114–2125. [Google Scholar]
- Zou, M.F.; Keck, T.M.; Kumar, V.; Donthamsetti, P.; Michino, M.; Burzynski, C.; Schweppe, C.; Bonifazi, A.; Free, R.B.; Sibley, D.R.; et al. Novel Analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo [4,5,1-ij]quinolin-2(1H)-one (Sumanirole) Provide Clues to Dopamine D2/D3 Receptor Agonist Selectivity. J. Med. Chem. 2016, 59, 2973–2988. [Google Scholar] [CrossRef]
- Linz, S.; Muller, J.; Hubner, H.; Gmeiner, P.; Troschutz, R. Design, synthesis and dopamine D4 receptor binding activities of new N-heteroaromatic 5/6-ring Mannich bases. Bioorg. Med. Chem. 2009, 17, 4448–4458. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Cai, W.; Yu, L.; Zhen, X.; Zhang, A. ‘Click’ D(1) receptor agonists with a 5-HT(1A) receptor pharmacophore producing D(2) receptor activity. Bioorg. Med. Chem. 2009, 17, 4873–4880. [Google Scholar] [CrossRef]
- Cao, Y.; Min, C.; Acharya, S.; Kim, K.M.; Cheon, S.H. Design, synthesis and evaluation of bitopic arylpiperazinephenyl-1,2,4-oxadiazoles as preferential dopamine D3 receptor ligands. Bioorg. Med. Chem. 2016, 24, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Suckling, C.J.; Murphy, J.A.; Khalaf, A.I.; Zhou, S.Z.; Lizos, D.E.; van Nhien, A.N.; Yasumatsu, H.; McVie, A.; Young, L.C.; McCraw, C.; et al. M4 agonists/5HT7 antagonists with potential as antischizophrenic drugs: Serominic compounds. Bioorg. Med. Chem. Lett. 2007, 17, 2649–2655. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.; Wiecek, M.; Lazewska, D.; Kurczab, R.; Jastrzebska-Wiesek, M.; Satala, G.; Kucwaj-Brysz, K.; Lubelska, A.; Gluch-Lutwin, M.; Mordyl, B.; et al. Synthesis and computer-aided SAR studies for derivatives of phenoxyalkyl-1,3,5-triazine as the new potent ligands for serotonin receptors 5-HT6. Eur. J. Med. Chem. 2019, 178, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Löber, S.; Aboul-Fadl, T.; Hübner, H.; Gmeiner, P. Di- and trisubstituted pyrazolo [1,5-a]pyridine derivatives: Synthesis, dopamine receptor binding and ligand efficacy. Bioorg. Med. Chem. Lett. 2002, 12, 633–636. [Google Scholar] [CrossRef]
- Haubmann, C.; Hübner, H.; Gmeiner, P. 2,2-dicyanovinyl as a nonaromatic aryl bioisostere: Synthesis, binding experiments and SAR studies of highly selective dopamine D4 receptor ligands. Bioorg. Med. Chem. Lett. 1999, 9, 1969–1972. [Google Scholar] [CrossRef]
- Cao, J.; Slack, R.D.; Bakare, O.M.; Burzynski, C.; Rais, R.; Slusher, B.S.; Kopajtic, T.; Bonifazi, A.; Ellenberger, M.P.; Yano, H.; et al. Novel and High Affinity 2-[(Diphenylmethyl)sulfinyl]acetamide (Modafinil) Analogues as Atypical Dopamine Transporter Inhibitors. J. Med. Chem. 2016, 59, 10676–10691. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Moritz, A.E.; Keck, T.M.; Bonifazi, A.; Ellenberger, M.P.; Sibley, C.D.; Free, R.B.; Shi, L.; Lane, J.R.; Sibley, D.R.; et al. Synthesis and Pharmacological Characterization of Novel trans-Cyclopropylmethyl-Linked Bivalent Ligands That Exhibit Selectivity and Allosteric Pharmacology at the Dopamine D3 Receptor (D3R). J. Med. Chem. 2017, 60, 1478–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, S.W.; Riad, A.A.; Hsieh, C.J.; Sahlholm, K.; Jacome, D.A.; Griffin, S.; Taylor, M.; Weng, C.C.; Xu, K.; Kirschner, N.; et al. Leveraging a Low-Affinity Diazaspiro Orthosteric Fragment to Reduce Dopamine D3 Receptor (D3R) Ligand Promiscuity across Highly Conserved Aminergic G-Protein-Coupled Receptors (GPCRs). J. Med. Chem. 2019, 62, 5132–5147. [Google Scholar] [CrossRef]
- Battiti, F.O.; Cemaj, S.L.; Guerrero, A.M.; Shaik, A.B.; Lam, J.; Rais, R.; Slusher, B.S.; Deschamps, J.R.; Imler, G.H.; Newman, A.H.; et al. The Significance of Chirality in Drug Design and Synthesis of Bitopic Ligands as D3 Receptor (D3R) Selective Agonists. J. Med. Chem. 2019, 62, 6287–6314. [Google Scholar] [CrossRef]
- Neumeyer, J.L.; Baindur, N.; Niznik, H.B.; Guan, H.C.; Seeman, P. (+/−)-3-Allyl-6-bromo-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3- benzazepin, a new high-affinity D1 dopamine receptor ligand: Synthesis and structure-activity relationship. J. Med. Chem. 1991, 34, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Elsner, J.; Boeckler, F.; Heinemann, F.W.; Hubner, H.; Gmeiner, P. Pharmacophore-guided drug discovery investigations leading to bioactive 5-aminotetrahydropyrazolopyridines. Implications for the binding mode of heterocyclic dopamine D3 receptor agonists. J. Med. Chem. 2005, 48, 5771–5779. [Google Scholar] [CrossRef] [PubMed]
- Enguehard-Gueiffier, C.; Hubner, H.; El Hakmaoui, A.; Allouchi, H.; Gmeiner, P.; Argiolas, A.; Melis, M.R.; Gueiffier, A. 2-[(4-phenylpiperazin-1-yl)methyl]imidazo(di)azines as selective D4-ligands. Induction of penile erection by 2-[4-(2-methoxyphenyl)piperazin-1-ylmethyl]imidazo [1,2-a]pyridine (PIP3EA), a potent and selective D4 partial agonist. J. Med. Chem. 2006, 49, 3938–3947. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, S.; Hubner, H.; Heinemann, F.W.; Meyer, K.; Gmeiner, P. Novel pyridylmethylamines as highly selective 5-HT(1A) superagonists. J. Med. Chem. 2010, 53, 7167–7179. [Google Scholar] [CrossRef]
- Tschammer, N.; Elsner, J.; Goetz, A.; Ehrlich, K.; Schuster, S.; Ruberg, M.; Kuhhorn, J.; Thompson, D.; Whistler, J.; Hubner, H.; et al. Highly potent 5-aminotetrahydropyrazolopyridines: Enantioselective dopamine D3 receptor binding, functional selectivity, and analysis of receptor-ligand interactions. J. Med. Chem. 2011, 54, 2477–2491. [Google Scholar] [CrossRef] [PubMed]
- Prante, O.; Tietze, R.; Hocke, C.; Lober, S.; Hubner, H.; Kuwert, T.; Gmeiner, P. Synthesis, radiofluorination, and in vitro evaluation of pyrazolo [1,5-a]pyridine-based dopamine D4 receptor ligands: Discovery of an inverse agonist radioligand for PET. J. Med. Chem. 2008, 51, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Dorfler, M.; Tschammer, N.; Hamperl, K.; Hubner, H.; Gmeiner, P. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: Synthesis, bioactivity, and mutagenesis studies. J. Med. Chem. 2008, 51, 6829–6838. [Google Scholar] [CrossRef]
- Colabufo, N.A.; Berardi, F.; Perrone, R.; Rapposelli, S.; Digiacomo, M.; Vanni, M.; Balsamo, A. 2-[(3-Methoxyphenylethyl)phenoxy]-based ABCB1 inhibitors: Effect of different basic side-chains on their biological properties. J. Med. Chem. 2008, 51, 7602–7613. [Google Scholar] [CrossRef]
- Berry, C.B.; Bubser, M.; Jones, C.K.; Hayes, J.P.; Wepy, J.A.; Locuson, C.W.; Daniels, J.S.; Lindsley, C.W.; Hopkins, C.R. Discovery and Characterization of ML398, a Potent and Selective Antagonist of the D4 Receptor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1060–1064. [Google Scholar] [CrossRef]
- Keck, T.M.; Free, R.B.; Day, M.M.; Brown, S.L.; Maddaluna, M.S.; Fountain, G.; Cooper, C.; Fallon, B.; Holmes, M.; Stang, C.T. Dopamine D4 Receptor-Selective Compounds Reveal Structure-Activity Relationships that Engender Agonist Efficacy. J. Med. Chem. 2019, 62, 3722–3740. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.W.; Griffin, S.; Taylor, M.; Sahlholm, K.; Weng, C.C.; Xu, K.; Jacome, D.A.; Luedtke, R.R.; Mach, R.H. Highly Selective Dopamine D3 Receptor Antagonists with Arylated Diazaspiro Alkane Cores. J. Med. Chem. 2017, 60, 9905–9910. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. Dassault Systèmes, BIOVIA Discovery Studio, Release 2018; Dassault Systems: San Diego, CA, USA, 2018. [Google Scholar]
- Temml, V.; Kaserer, T.; Kutil, Z.; Landa, P.; Vanek, T.; Schuster, D. Pharmacophore modeling for COX-1 and -2 inhibitors with LigandScout in comparison to Discovery Studio. Fut. Med. Chem. 2014, 6, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, T.T. An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: Armonk, NY, USA, 1958. [Google Scholar]
- Vazquez, J.; Lopez, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Smith, J.L.; Stein, D.A.; Shum, D.; Fischer, M.A.; Radu, C.; Bhinder, B.; Djaballah, H.; Nelson, J.A.; Fruh, K.; Hirsch, A.J. Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J. Virol. 2014, 88, 5533–5542. [Google Scholar] [CrossRef] [Green Version]
- Campiani, G.; Nacci, V.; Bechelli, S.; Ciani, S.M.; Garofalo, A.; Fiorini, I.; Wikstrom, H.; de Boer, P.; Liao, Y.; Tepper, P.G.; et al. New antipsychotic agents with serotonin and dopamine antagonist properties based on a pyrrolo [2,1-b][1,3]benzothiazepine structure. J. Med. Chem. 1998, 41, 3763–3772. [Google Scholar] [CrossRef]
- Naporra, F.; Gobleder, S.; Wittmann, H.J.; Spindler, J.; Bodensteiner, M.; Bernhardt, G.; Hubner, H.; Gmeiner, P.; Elz, S.; Strasser, A. Dibenzo[b,f][1,4]oxazepines and dibenzo[b,e]oxepines: Influence of the chlorine substitution pattern on the pharmacology at the H1R, H4R, 5-HT2AR and other selected GPCRs. Pharmacol. Res. 2016, 113, 610–625. [Google Scholar] [CrossRef]
- Ginex, T.; Munoz-Muriedas, J.; Herrero, E.; Gibert, E.; Cozzini, P.; Luque, F.J. Development and validation of hydrophobic molecular fields derived from the quantum mechanical IEF/PCM-MST solvation models in 3D-QSAR. J. Comput. Chem. 2016, 37, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.M.; Ejendal, K.F.; Avramova, L.V.; Garland-Kuntz, E.E.; Giraldo-Calderon, G.I.; Brust, T.F.; Watts, V.J.; Hill, C.A. A “genome-to-lead” approach for insecticide discovery: Pharmacological characterization and screening of Aedes aegypti D(1)-like dopamine receptors. PLoS Negl. Trop. Dis. 2012, 6, e1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogeso, K.P.; Liljefors, T.; Arnt, J.; Hyttel, J.; Pedersen, H. Octoclothepin enantiomers. A reinvestigation of their biochemical and pharmacological activity in relation to a new receptor-interaction model for dopamine D-2 receptor antagonists. J. Med. Chem. 1991, 34, 2023–2030. [Google Scholar] [CrossRef]
- Staron, J.; Kurczab, R.; Warszycki, D.; Satala, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A.; et al. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2020, 185, 111857. [Google Scholar] [CrossRef] [PubMed]
- Perrone, R.; Berardi, F.; Colabufo, N.A.; Leopoldo, M.; Tortorella, V. A structure-affinity relationship study on derivatives of N-[2-[4-(4-Chlorophenyl)piperazin-1-yl]ethyl]-3-methoxybenzamide, a high-affinity and selective D(4) receptor ligand. J. Med. Chem. 2000, 43, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Peprah, K.; Zhu, X.Y.; Eyunni, S.V.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Multi-receptor drug design: Haloperidol as a scaffold for the design and synthesis of atypical antipsychotic agents. Bioorg. Med. Chem. 2012, 20, 1291–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoemaker, H.; Claustre, Y.; Fage, D.; Rouquier, L.; Chergui, K.; Curet, O.; Oblin, A.; Gonon, F.; Carter, C.; Benavides, J.; et al. Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity. J. Pharmacol. Exp. Ther. 1997, 280, 83–97. [Google Scholar]
- Poli, G.; Seidel, T.; Langer, T. Conformational Sampling of Small Molecules With iCon: Performance Assessment in Comparison With OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Glen, R.C.; Bender, A.; Arnby, C.H.; Carlsson, L.; Boyer, S.; Smith, J. Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME (vol 9, pg 199, 2006). Idrugs 2006, 9, 311. [Google Scholar]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
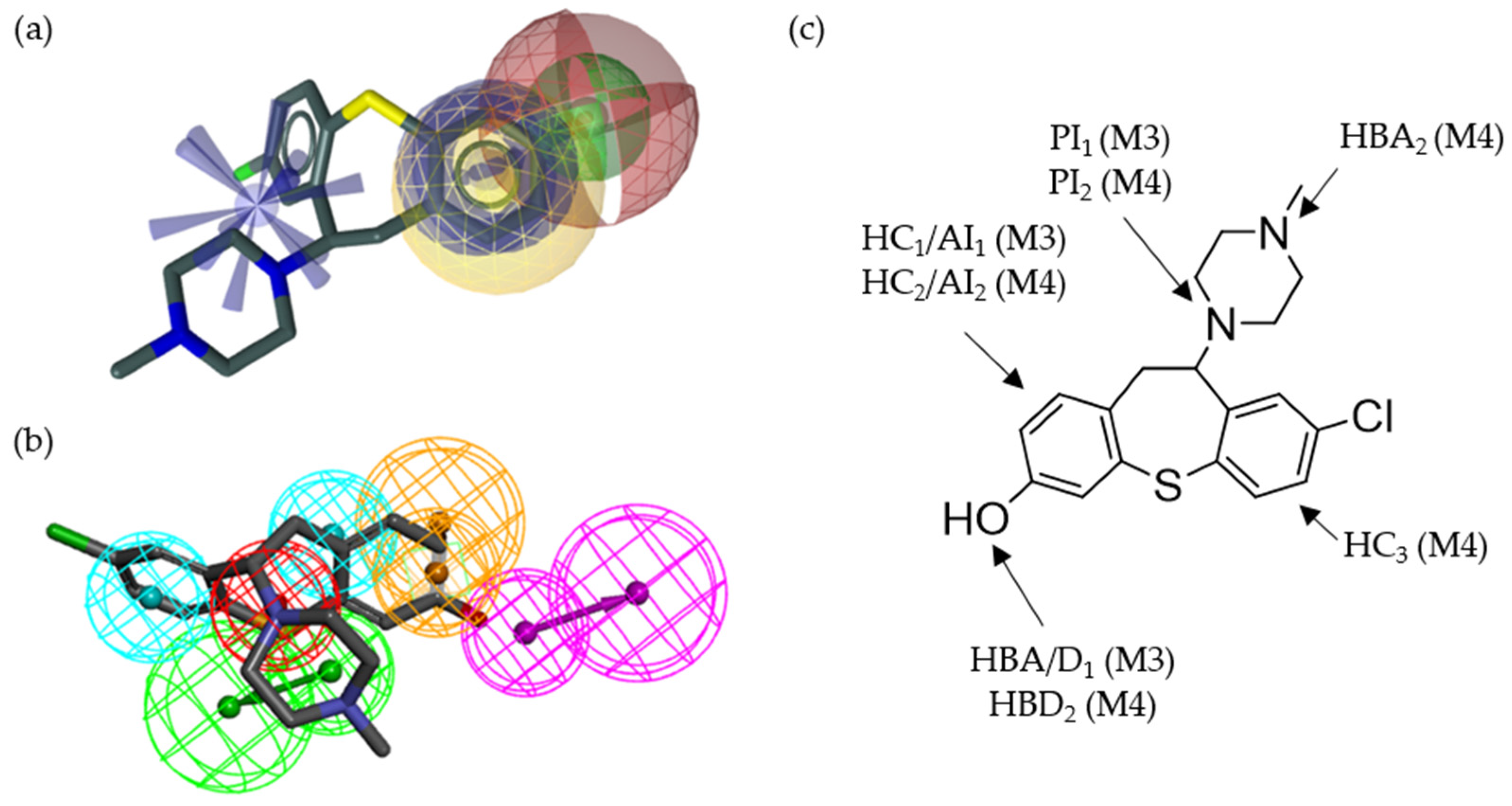

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M1 | M2 | M3 | M4 | All Models | |
|---|---|---|---|---|---|
| Active hits (n = 68) | 9 | 16 | 26 | 30 | 51 |
| Inactive hits (n = 68) | 1 | 0 | 1 | 0 | 2 |
| Decoy hits (n = 3752) | 35 | 36 | 9 | 27 | 108 |
| TPs | 9 | 16 | 26 | 30 | 51 |
| FPs | 36 | 36 | 10 | 27 | 110 |
| TNs | 3784 | 3783 | 3810 | 3792 | 3710 |
| Specificity | 0.99 | 0.99 | 1.00 | 0.99 | 0.97 |
| Sensitivity | 0.13 | 0.24 | 0.38 | 0.44 | 0.75 |
| Accuracy | 0.98 | 0.98 | 0.99 | 0.98 | 0.97 |
| EF | 11.44 | 17.26 | 41.29 | 29.57 | 18.11 |
| YoA | 0.20 | 0.30 | 0.74 | 0.52 | 0.32 |
| Number of Hits | ||||
|---|---|---|---|---|
| Pharmacophore Model | SPECS | Maybridge | Combined | |
| Single hits | M1 | 236 | 118 | 354 |
| M2 | 595 | 76 | 671 | |
| M3 | 157 | 17 | 174 | |
| M4 | 762 | 78 | 840 | |
| Consensus hits (n = 2) | M2 + M4 | 122 | 11 | 133 |
| M1 + M3 | 0 | 0 | 0 | |
| M4 + M1/M3 | 47 | 6 | 53 | |
| M2 + M1/M3 | 10 | 1 | 11 | |
| Consensus hits (n = 3) | M2 + M4 + M1/M3 | 4 | 0 | 4 |
| Compound | Normalized Decrease in Fluorescence ± SD | Compound | Normalized Decrease in Fluorescence ± SD |
|---|---|---|---|
| Control | 1 | SC160 | 2.41 ± 2.01 |
| 1 | 9.44 ± 5.97 | 15 | 3.99 ± 2.58 |
| Dopamine | 1.50 ± 0.88 | SC175 | 2.98 ± 1.77 |
| Sulpiride | 30.65 ± 6.79 | 16 | 15.74 ± 18.15 |
| 2 | 39.10 ± 1.31 | 17 | 8.18 ± 3.62 |
| Droperidol | 38.69 ± 2.56 | SC191 | 1.83 ± 0.83 |
| Haloperidol | 39.18 ± 0.83 | 18 | 10.85 ± 4.93 |
| Piribedil | 9.21 ± 3.80 | SC198 | 2.43 ± 1.39 |
| 14 | 40.40 ± 1.39 | SC201 | 1.99 ± 0.85 |
| SC149 | 2.04 ± 1.34 | 19 | 22.08 ± 6.62 |
| SC155 | 1.68 ± 0.90 | SC207 | 22.89 ± 8.41 |
| Compound No. (ID) | Fold Difference | KI (µM) | 95% CI (µM) |
|---|---|---|---|
| 14 | 2700 | 0.004 | 0.003–0.006 |
| 15 | 2.6 | 4.31 | 2.01–19.58 |
| 16 | 1.1 | 10.04 | 5.69–17.43 |
| 17 | 9.0 | 1.24 | 0.77–2.05 |
| 18 | 4.2 | 2.63 | 1.62–4.28 |
| 19 | 34.7 | 0.32 | 0.23–0.45 |
| Dopamine | 1 | 11.12 | 7.77–15.84 |
| D2R Ligands (Identified by Models) | Tested Virtual Hits | Active Virtual Hits | Number of Active Hits | Hit Rate (%) |
|---|---|---|---|---|
| M1 | 22 | 17 | 1 | 4.5 |
| M2 | 37 | 15, 16, 17, 18 | 4 | 10.8 |
| M3 | 25 | 14, 15, 19 | 3 | 12 |
| M4 | 45 | 14, 16, 19 | 3 | 6.7 |
| Parameter | Value |
|---|---|
| Molecular weight (MW) (g/mol) | 600 ≥ x ≥ 200 |
| cLogP | 6.0 ≥ x ≥ 1.5 |
| HB acceptors | 7 ≥ x ≥ 0 |
| HB donors | 3 ≥ x ≥ 0 |
| Rotatable bonds | 18 ≥ x ≥ 0 |
| Rings | 5 ≥ x ≥ 2 |
| Aromatic rings | 3 ≥ x ≥ 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zell, L.; Lainer, C.; Kollár, J.; Temml, V.; Schuster, D. Identification of Novel Dopamine D2 Receptor Ligands—A Combined In Silico/In Vitro Approach. Molecules 2022, 27, 4435. https://doi.org/10.3390/molecules27144435
Zell L, Lainer C, Kollár J, Temml V, Schuster D. Identification of Novel Dopamine D2 Receptor Ligands—A Combined In Silico/In Vitro Approach. Molecules. 2022; 27(14):4435. https://doi.org/10.3390/molecules27144435
Chicago/Turabian StyleZell, Lukas, Constanze Lainer, Jakub Kollár, Veronika Temml, and Daniela Schuster. 2022. "Identification of Novel Dopamine D2 Receptor Ligands—A Combined In Silico/In Vitro Approach" Molecules 27, no. 14: 4435. https://doi.org/10.3390/molecules27144435
APA StyleZell, L., Lainer, C., Kollár, J., Temml, V., & Schuster, D. (2022). Identification of Novel Dopamine D2 Receptor Ligands—A Combined In Silico/In Vitro Approach. Molecules, 27(14), 4435. https://doi.org/10.3390/molecules27144435