
Pure and Hybrid SCAN, rSCAN, and r2SCAN: Which One Is Preferred in KS- and HF-DFT Calculations, and How Does D4 Dispersion Correction Affect This Ranking?



Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
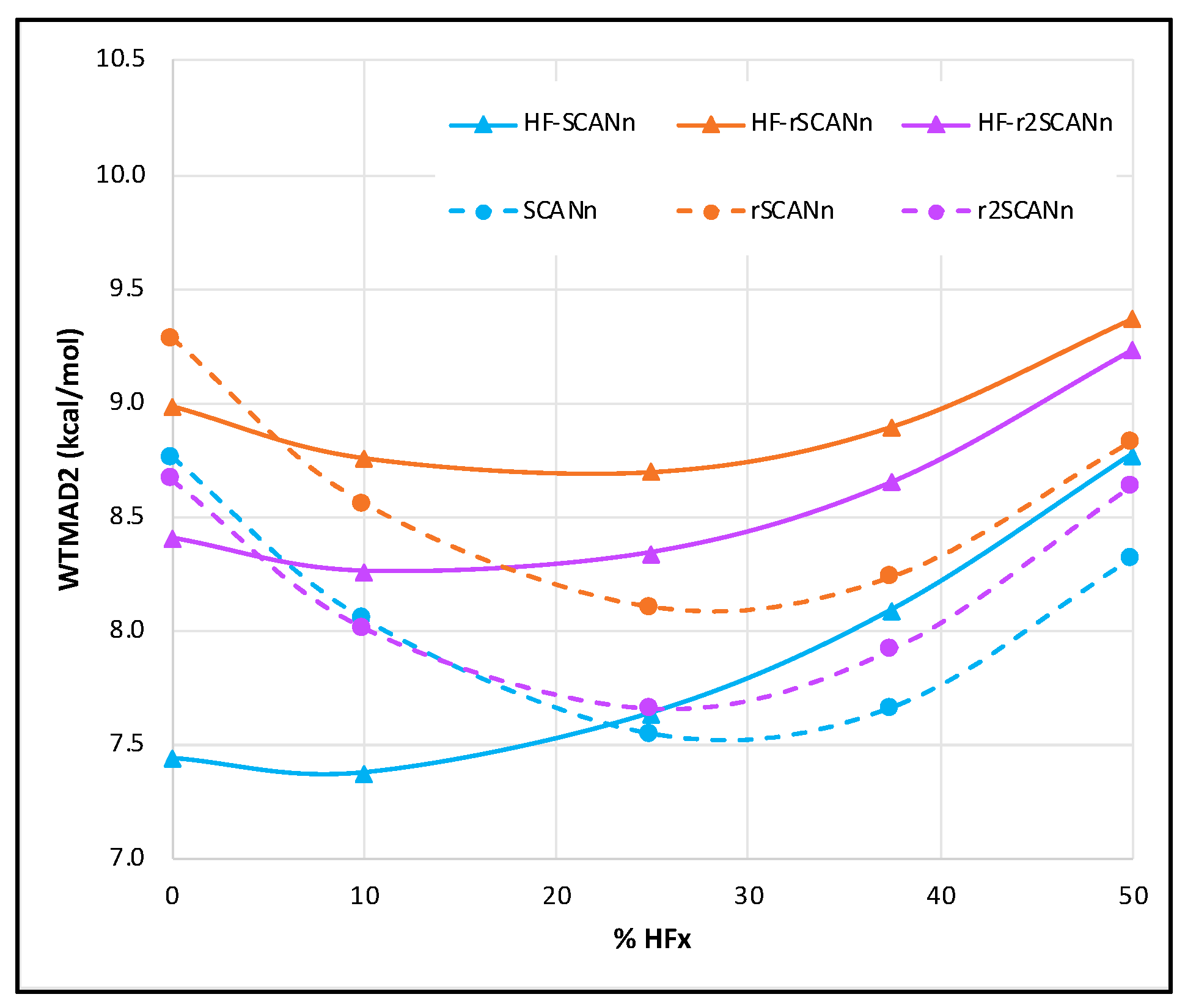
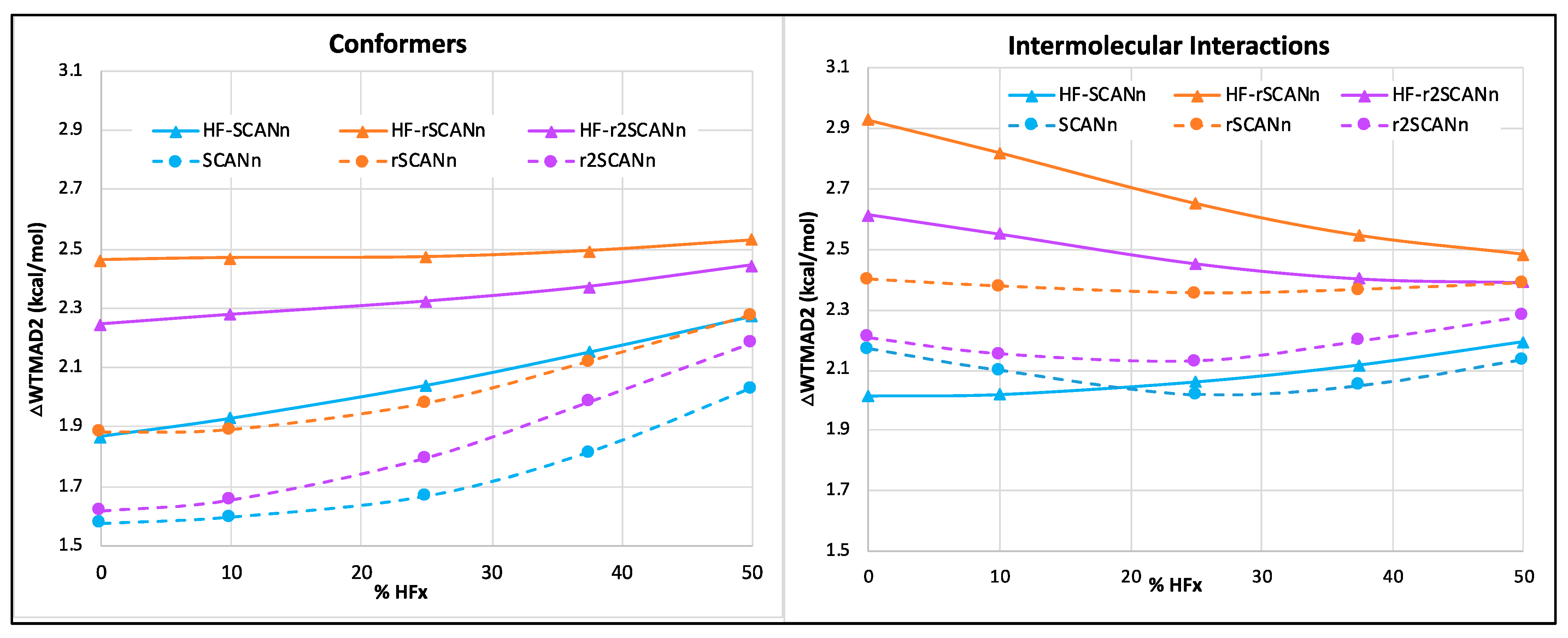
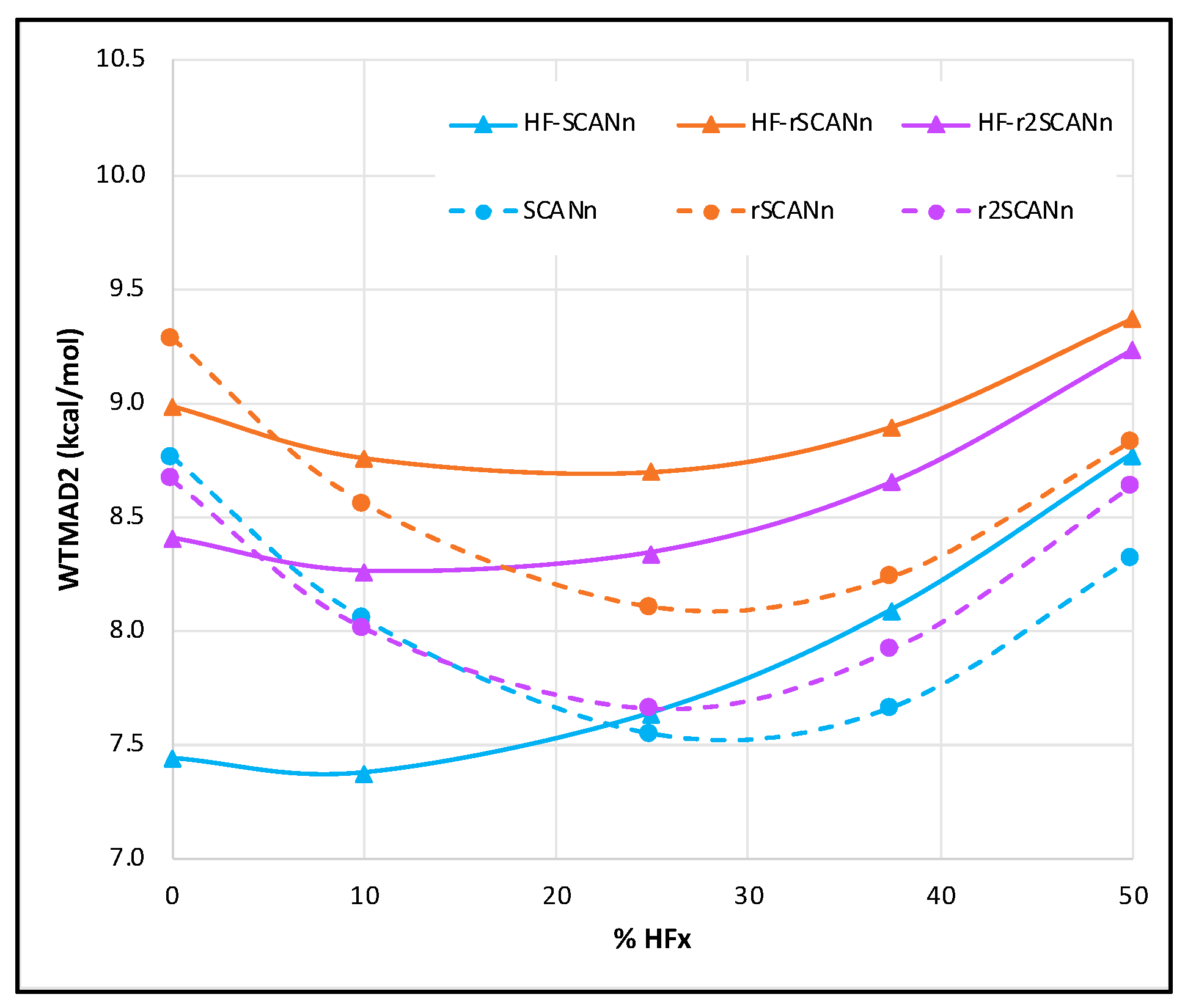
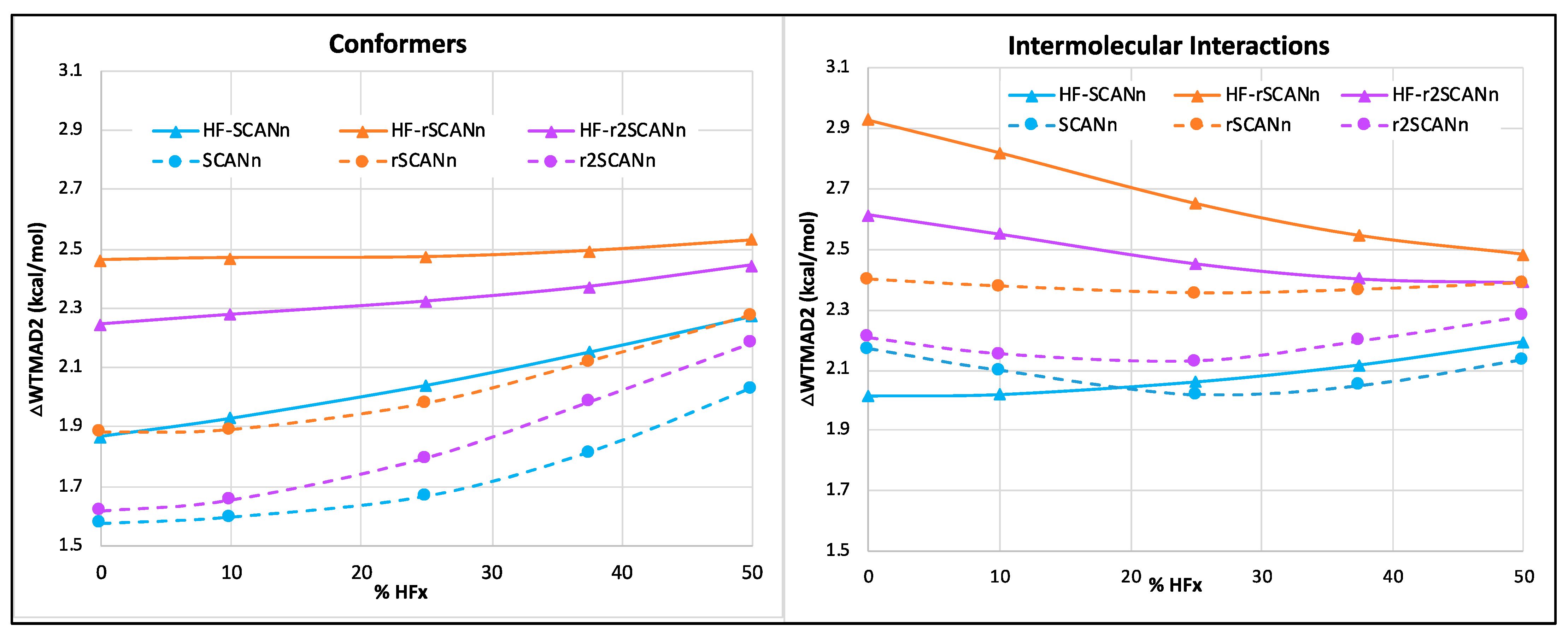
3.1. Without Dispersion Correction
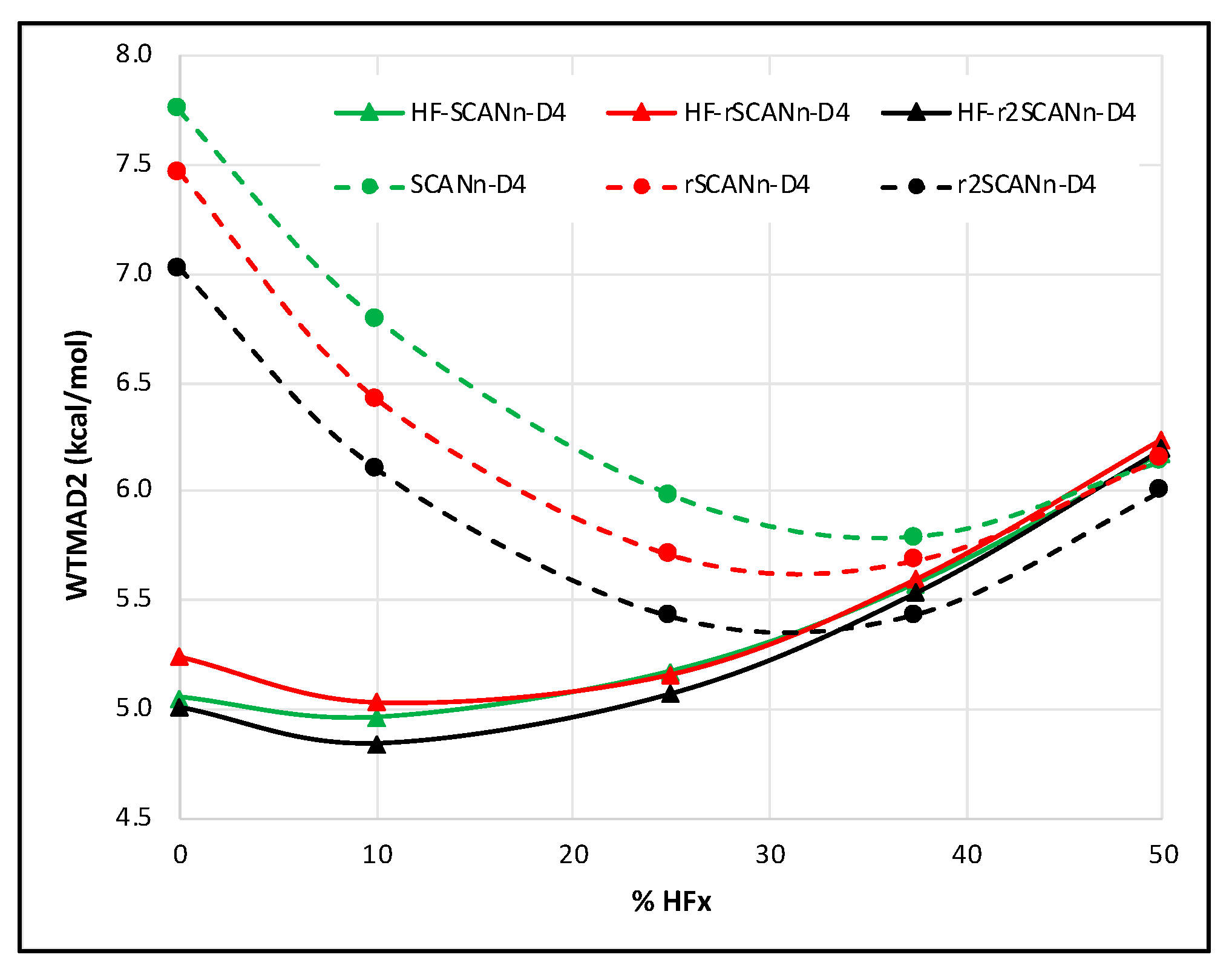
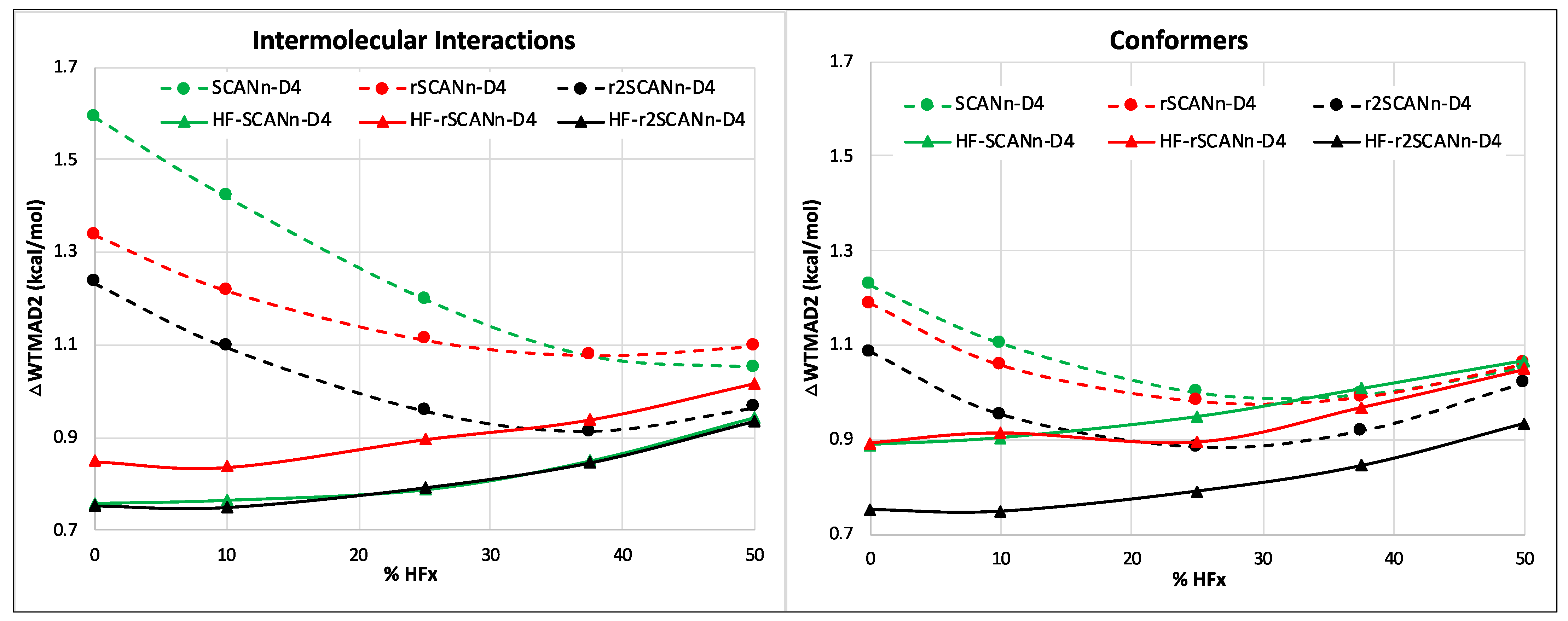
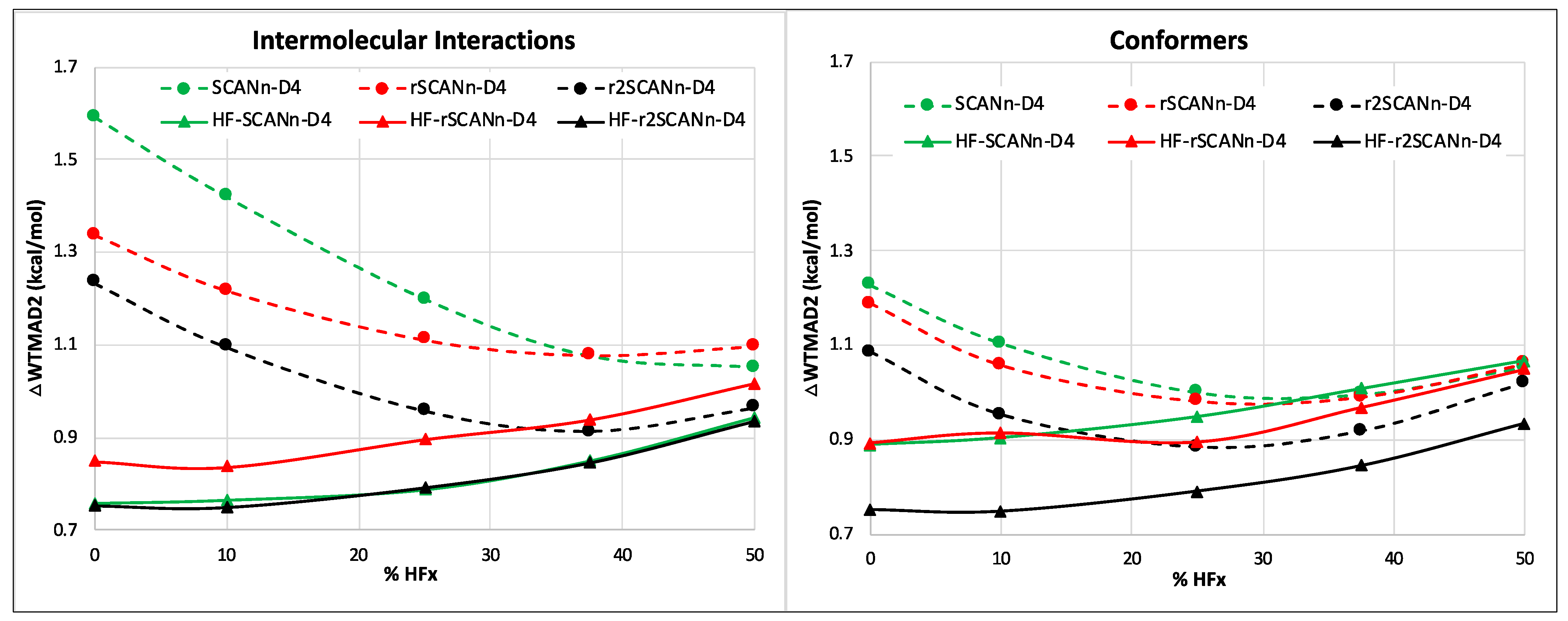
3.2. Impact of Introducing D4 Dispersion Correction
4. Conclusions
- Both for self-consistent and for HF-DFT series, the WTMAD2 global minimum is the same for all three SCAN variants. The only exception is HF-rSCANn, where the overall minimum is near 25% HF exchange instead of near 10% for the other two. Among all the functionals tested, the pure mGGA form is a low-cost alternative for all three SCAN variants.
- The choice of SCAN variant can significantly influence the performance of the dispersion-uncorrected HF-DFT series. However, upon introducing D4 dispersion, the WTMAD2 gaps between different SCAN variants almost vanish. At lower percentages of HF exchange, self-consistent SCANn and r2SCANn hybrids perform similarly. However, with D4 correction, r2SCANn-D4 outperforms rSCANn-D4 and SCANn-D4.
- For the small-molecule thermochemistry and barrier height subsets, different SCAN variants perform comparably in the pure and hybrid self-consistent and HF-DFT series.
- Irrespective of the choice of SCAN variant, the use of ROHF and ROKS densities are clearly beneficial for hybrid HF-DFT and self-consistent functionals.
- Among all the functionals tested, HF-r2SCAN10-D4 offers the lowest WTMAD2 (4.85 kcal/mol), just below HF-SCAN10-D4 (WTMAD2 = 4.96 kcal/mol) and without the latter’s grid convergence issues. The same remark applies concerning HF-DFT, with HF-r2SCAN-D4 (WTMAD2 = 5.01 kcal/mol) slightly outperforming HF-SCAN-D4 (WTMAD2 = 5.05 kcal/mol).
- Overall, and taking into account the reduced grid sensitivity resulting from its regularization, we find that r2SCAN’s superiority over SCAN is also retained for hybrids and for HF-DFT.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [Google Scholar] [CrossRef]
- Von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case: I. J. Phys. C Solid State Phys. 1972, 5, 1629–1642. [Google Scholar] [CrossRef]
- Perdew, J.P.; Yue, W. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 1986, 33, 8800–8802. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249, Erratum in Phys. Rev. B 2018, 98, 079904. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Sun, J.; Furness, J.W.; Zhang, Y. Density functional theory. In Mathematical Physics in Theoretical Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 119–159. ISBN 9780128136515. [Google Scholar]
- Perdew, J.P. Climbing the ladder of density functional approximations. MRS Bull. 2013, 38, 743–750. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Ruzsinszky, A.; Perdew, J. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Remsing, R.C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; et al. Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional. Nat. Chem. 2016, 8, 831–836. [Google Scholar] [CrossRef]
- Sai Gautam, G.; Carter, E.A. Evaluating transition metal oxides within DFT-SCAN and SCAN+U frameworks for solar thermochemical applications. Phys. Rev. Mater. 2018, 2, 095401. [Google Scholar] [CrossRef]
- Lane, C.; Zhang, Y.; Furness, J.W.; Markiewicz, R.S.; Barbiellini, B.; Sun, J.; Bansil, A. First-principles calculation of spin and orbital contributions to magnetically ordered moments in Sr2IrO4. Phys. Rev. B 2020, 101, 155110. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Ko, H.Y.; Remsing, R.C.; Calegari Andrade, M.F.; Santra, B.; Sun, Z.; Selloni, A.; Car, R.; Klein, M.L.; Perdew, J.P.; et al. Ab initio theory and modeling of water. Proc. Natl. Acad. Sci. USA 2017, 114, 10846–10851. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Peng, H.; Sun, J.; Perdew, J.P. More-Realistic Band Gaps from Meta-Generalized Gradient Approximations: Only in a Generalized Kohn-Sham Scheme. Phys. Rev. B 2016, 93, 205205. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Diaz, C.M.; Basurto, L.; Jackson, K.A.; Baruah, T.; Zope, R.R. Fermi-Löwdin orbital self-interaction correction using the strongly constrained and appropriately normed meta-GGA functional. J. Chem. Phys. 2019, 151, 154105. [Google Scholar] [CrossRef]
- Bartók, A.P.; Yates, J.R. Regularized SCAN functional. J. Chem. Phys. 2019, 150, 161101. [Google Scholar] [CrossRef]
- Mejía-Rodríguez, D.; Trickey, S.B. Comment on “Regularized SCAN functional” [J. Chem. Phys. 150, 161101 (2019)]. J. Chem. Phys. 2019, 151, 207101. [Google Scholar] [CrossRef]
- Bartók, A.P.; Yates, J.R. Response to “Comment on ‘Regularized SCAN functional’” [J. Chem. Phys. 151, 207101 (2019)]. J. Chem. Phys. 2019, 151, 207102. [Google Scholar] [CrossRef]
- Furness, J.W.; Kaplan, A.D.; Ning, J.; Perdew, J.P.; Sun, J. Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation. J. Phys. Chem. Lett. 2020, 11, 8208–8215, Erratum in J. Phys. Chem. Lett. 2020, 11, 9248–9248. [Google Scholar] [CrossRef]
- Furness, J.W.; Kaplan, A.D.; Ning, J.; Perdew, J.P.; Sun, J. Construction of meta-GGA functionals through restoration of exact constraint adherence to regularized SCAN functionals. J. Chem. Phys. 2021, 1–49. [Google Scholar] [CrossRef]
- Kim, M.-C.; Sim, E.; Burke, K. Understanding and reducing errors in density functional calculations. Phys. Rev. Lett. 2012, 111, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Song, S.; Sim, E.; Burke, K. Halogen and Chalcogen Binding Dominated by Density-Driven Errors. J. Phys. Chem. Lett. 2019, 10, 295–301. [Google Scholar] [CrossRef]
- Kim, M.-C.; Park, H.; Son, S.; Sim, E.; Burke, K. Improved DFT Potential Energy Surfaces via Improved Densities. J. Phys. Chem. Lett. 2015, 6, 3802–3807. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, A.; Nafziger, J.; Jiang, K.; Kim, M.-C.; Sim, E.; Burke, K. The Importance of Being Inconsistent. Annu. Rev. Phys. Chem. 2017, 68, 555–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Kim, M.-C.; Sim, E.; Benali, A.; Heinonen, O.; Burke, K. Benchmarks and Reliable DFT Results for Spin Gaps of Small Ligand Fe(II) Complexes. J. Chem. Theory Comput. 2018, 14, 2304–2311. [Google Scholar] [CrossRef]
- Song, S.; Vuckovic, S.; Sim, E.; Burke, K. Density Sensitivity of Empirical Functionals. J. Phys. Chem. Lett. 2021, 12, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Vuckovic, S.; Sim, E.; Burke, K. Density-corrected DFT explained: Questions and answers. arXiv 2021, arXiv:2110.07849. [Google Scholar]
- Santra, G.; Martin, J.M.L. What Types of Chemical Problems Benefit from Density-Corrected DFT? A Probe Using an Extensive and Chemically Diverse Test Suite. J. Chem. Theory Comput. 2021, 17, 1368–1379. [Google Scholar] [CrossRef]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef] [Green Version]
- Caldeweyher, E.J. Development and Application of London Dispersion Corrections for Electronic Structure Methods. Ph.D. Thesis, Rhenish Friedrich Wilhelm University of Bonn, Bonn, Germany, 2020. [Google Scholar]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Axilrod, B.M.; Teller, E. Interaction of the van der Waals type between three atoms. J. Chem. Phys. 1943, 11, 299–300. [Google Scholar] [CrossRef]
- Muto, Y. Force between nonpolar molecules. Proc. Physico-Math. Soc. Jpn. 1943, 17, 629–631. [Google Scholar] [CrossRef]
- Ehlert, S.; Huniar, U.; Ning, J.; Furness, J.W.; Sun, J.; Kaplan, A.D.; Perdew, J.P.; Brandenburg, J.G. r2SCAN-D4: Dispersion corrected meta-generalized gradient approximation for general chemical applications. J. Chem. Phys. 2021, 154, 061101. [Google Scholar] [CrossRef]
- Powell, M. The BOBYQA Algorithm for Bound Constrained Optimization without Derivatives (DAMPT Report 2009/NA06); University of Cambridge: Cambridge, UK, 2009. [Google Scholar]
- Santra, G.; Sylvetsky, N.; Martin, J.M.L. Minimally Empirical Double-Hybrid Functionals Trained against the GMTKN55 Database: revDSD-PBEP86-D4, revDOD-PBE-D4, and DOD-SCAN-D4. J. Phys. Chem. A 2019, 123, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, V.I.; Laikov, D.N. A quadrature formula for the sphere of the 131st algebraic order of accuracy. Dokl. Math. 1999, 59, 477–481. [Google Scholar]
- Murray, C.W.; Handy, N.C.; Laming, G.J. Quadrature schemes for integrals of density functional theory. Mol. Phys. 1993, 78, 997–1014. [Google Scholar] [CrossRef]
- Gill, P.M.; Johnson, B.G.; Pople, J.A. A standard grid for density functional calculations. Chem. Phys. Lett. 1993, 209, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Krack, M.; Köster, A.M. An adaptive numerical integrator for molecular integrals. J. Chem. Phys. 1998, 108, 3226–3234. [Google Scholar] [CrossRef]
- Řezáč, J.; Riley, K.E.; Hobza, P. S66: A well-balanced database of benchmark interaction energies relevant to biomolecular structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, M.K.; Karton, A.; Sylvetsky, N.; Martin, J.M.L. The S66 Non-Covalent Interactions Benchmark Reconsidered Using Explicitly Correlated Methods Near the Basis Set Limit. Aust. J. Chem. 2018, 71, 238–248. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functionals | MAD (kcal/mol) | MSD (kcal/mol) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H-Bonds | π-Stack | London | Mixed-Influence | Full S66 | H-Bonds | π-Stack | London | Mixed Influence | Full S66 | |
| HF-SCAN-D4 | 0.21 | 0.57 | 0.47 | 0.23 | 0.32 | 0.09 | 0.57 | −0.45 | 0.02 | 0.03 |
| HF-SCAN10-D4 | 0.31 | 0.44 | 0.44 | 0.24 | 0.33 | 0.24 | 0.44 | −0.42 | 0.07 | 0.09 |
| HF-SCAN0-D4 | 0.45 | 0.26 | 0.41 | 0.25 | 0.35 | 0.42 | 0.26 | −0.41 | 0.11 | 0.14 |
| HF-SCAN38-D4 | 0.59 | 0.15 | 0.37 | 0.28 | 0.39 | 0.58 | 0.15 | −0.37 | 0.16 | 0.2 |
| HF-SCAN50-D4 | 0.79 | 0.13 | 0.28 | 0.32 | 0.45 | 0.79 | 0.13 | −0.26 | 0.26 | 0.32 |
| SCAN-D4 | 0.73 | 0.1 | 0.34 | 0.23 | 0.41 | 0.73 | −0.03 | −0.34 | 0.01 | 0.19 |
| SCAN10-D4 | 0.79 | 0.08 | 0.23 | 0.2 | 0.39 | 0.79 | −0.01 | −0.22 | 0.1 | 0.26 |
| SCAN0-D4 | 0.84 | 0.08 | 0.16 | 0.23 | 0.41 | 0.84 | −0.02 | −0.14 | 0.18 | 0.32 |
| SCAN38-D4 | 0.89 | 0.1 | 0.14 | 0.27 | 0.43 | 0.89 | −0.05 | −0.11 | 0.22 | 0.35 |
| SCAN50-D4 | 0.98 | 0.1 | 0.11 | 0.32 | 0.48 | 0.98 | −0.03 | −0.06 | 0.29 | 0.42 |
| HF-rSCAN-D4 | 0.11 | 0.52 | 0.26 | 0.16 | 0.21 | 0.03 | 0.52 | −0.24 | 0.01 | 0.05 |
| HF-rSCAN10-D4 | 0.18 | 0.38 | 0.28 | 0.15 | 0.22 | 0.15 | 0.38 | −0.27 | 0.03 | 0.07 |
| HF-rSCAN0-D4 | 0.43 | 0.33 | 0.22 | 0.22 | 0.31 | 0.43 | 0.33 | −0.21 | 0.15 | 0.20 |
| HF-rSCAN38-D4 | 0.59 | 0.16 | 0.24 | 0.24 | 0.35 | 0.59 | 0.16 | −0.24 | 0.17 | 0.23 |
| HF-rSCAN50-D4 | 0.80 | 0.11 | 0.19 | 0.30 | 0.42 | 0.80 | 0.10 | −0.18 | 0.26 | 0.34 |
| rSCAN-D4 | 0.57 | 0.19 | 0.13 | 0.15 | 0.30 | 0.56 | −0.17 | −0.13 | −0.04 | 0.13 |
| rSCAN10-D4 | 0.63 | 0.14 | 0.08 | 0.13 | 0.30 | 0.63 | −0.10 | −0.07 | 0.04 | 0.20 |
| rSCAN0-D4 | 0.74 | 0.10 | 0.06 | 0.16 | 0.33 | 0.74 | −0.04 | −0.01 | 0.14 | 0.29 |
| rSCAN38-D4 | 0.84 | 0.11 | 0.07 | 0.22 | 0.39 | 0.84 | −0.04 | 0.00 | 0.21 | 0.35 |
| rSCAN50-D4 | 0.97 | 0.13 | 0.07 | 0.29 | 0.46 | 0.97 | −0.04 | 0.00 | 0.28 | 0.42 |
| HF-r2SCAN-D4 | 0.14 | 0.77 | 0.40 | 0.25 | 0.32 | −0.09 | 0.77 | −0.35 | 0.06 | 0.03 |
| HF-r2SCAN10-D4 | 0.16 | 0.75 | 0.33 | 0.28 | 0.32 | 0.13 | 0.75 | −0.27 | 0.16 | 0.15 |
| HF-r2SCAN0-D4 | 0.37 | 0.58 | 0.28 | 0.30 | 0.36 | 0.36 | 0.58 | −0.23 | 0.22 | 0.24 |
| HF-r2SCAN38-D4 | 0.52 | 0.34 | 0.30 | 0.30 | 0.38 | 0.52 | 0.34 | −0.28 | 0.22 | 0.24 |
| HF-r2SCAN50-D4 | 0.72 | 0.21 | 0.26 | 0.33 | 0.43 | 0.72 | 0.21 | −0.25 | 0.27 | 0.32 |
| r2SCAN-D4 | 0.47 | 0.09 | 0.33 | 0.20 | 0.30 | 0.44 | −0.04 | −0.33 | −0.05 | 0.07 |
| r2SCAN10-D4 | 0.51 | 0.09 | 0.27 | 0.19 | 0.30 | 0.50 | −0.03 | −0.27 | 0.01 | 0.12 |
| r2SCAN0-D4 | 0.62 | 0.08 | 0.20 | 0.24 | 0.34 | 0.62 | 0.00 | −0.19 | 0.17 | 0.23 |
| r2SCAN38-D4 | 0.73 | 0.08 | 0.16 | 0.24 | 0.37 | 0.73 | 0.00 | −0.14 | 0.18 | 0.28 |
| r2SCAN50-D4 | 0.88 | 0.08 | 0.12 | 0.29 | 0.43 | 0.88 | 0.02 | -0.09 | 0.27 | 0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santra, G.; Martin, J.M.L. Pure and Hybrid SCAN, rSCAN, and r2SCAN: Which One Is Preferred in KS- and HF-DFT Calculations, and How Does D4 Dispersion Correction Affect This Ranking? Molecules 2022, 27, 141. https://doi.org/10.3390/molecules27010141
Santra G, Martin JML. Pure and Hybrid SCAN, rSCAN, and r2SCAN: Which One Is Preferred in KS- and HF-DFT Calculations, and How Does D4 Dispersion Correction Affect This Ranking? Molecules. 2022; 27(1):141. https://doi.org/10.3390/molecules27010141
Chicago/Turabian StyleSantra, Golokesh, and Jan M. L. Martin. 2022. "Pure and Hybrid SCAN, rSCAN, and r2SCAN: Which One Is Preferred in KS- and HF-DFT Calculations, and How Does D4 Dispersion Correction Affect This Ranking?" Molecules 27, no. 1: 141. https://doi.org/10.3390/molecules27010141
APA StyleSantra, G., & Martin, J. M. L. (2022). Pure and Hybrid SCAN, rSCAN, and r2SCAN: Which One Is Preferred in KS- and HF-DFT Calculations, and How Does D4 Dispersion Correction Affect This Ranking? Molecules, 27(1), 141. https://doi.org/10.3390/molecules27010141