
Hydrogen Delocalization in an Asymmetric Biomolecule: The Curious Case of Alpha-Fenchol



Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Techniques
2.2. Computational Techniques
3. Results and Discussion
3.1. Spectra of Monomeric -Fenchol
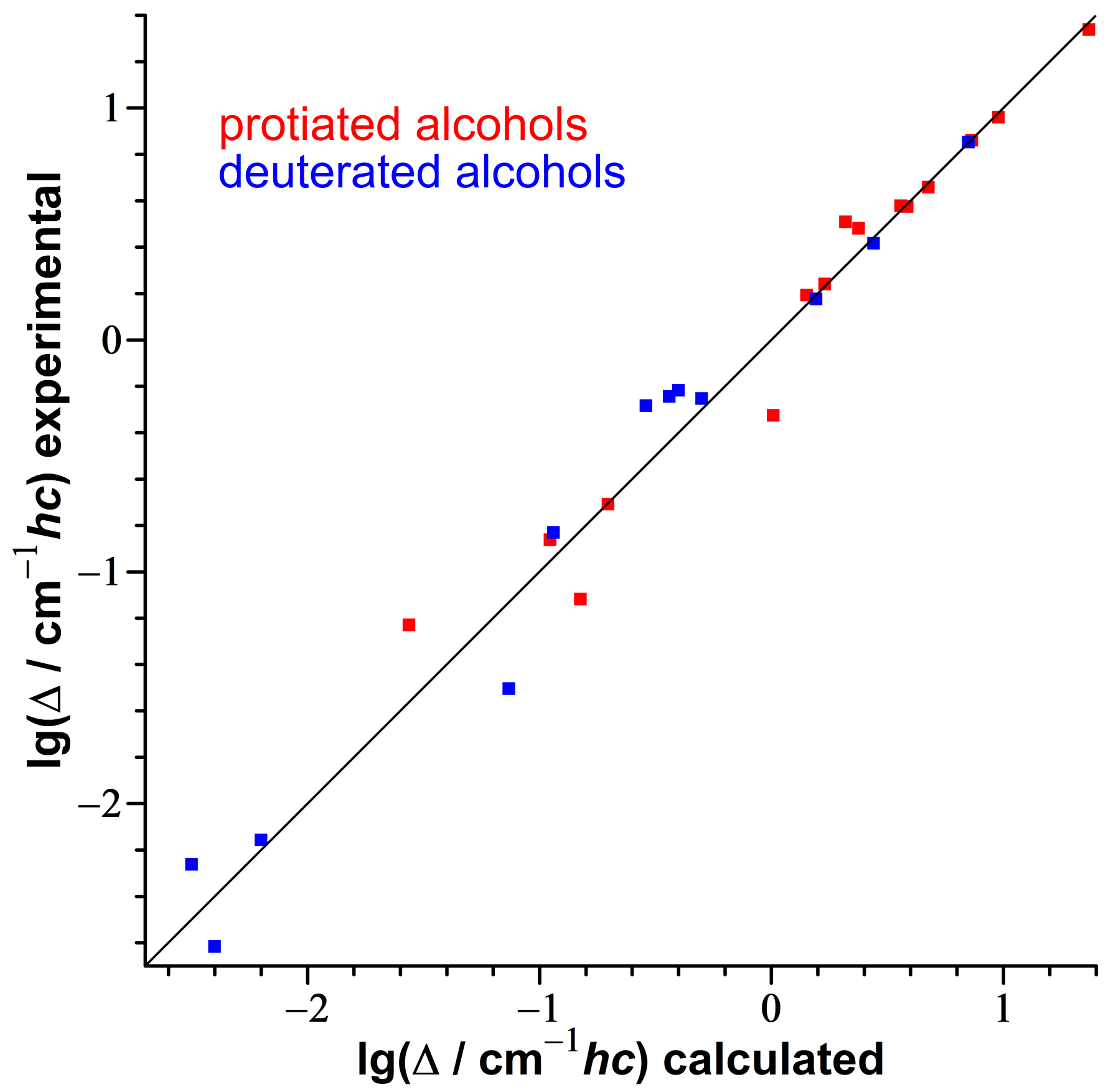
3.2. Consistency Checks for Isotope Effects
3.3. Theoretical Investigation of Torsional States of -Fenchol
3.4. Experimental Support from Monomeric Propargyl Alcohol
- Propargyl alcohol has a relatively high volatility (normal boiling point 115 ), which enables to utilize high vapor pressures without heating, which is helpful to retain signal intensity in mild expansions with low carrier gas pressure.
- The t conformer of propargyl alcohol, diastereomeric to the two enantiomeric g conformers interacting through tunneling, is calculated to be very high in energy (about 7 kJ mol) and therefore does not perturb the spectrum of the g conformers.
- The presence of the -system leads to both hydroxy groups donating a hydrogen bond each in dimers of propargyl alcohol [117,118]. This shifts their OH stretching fundamentals out of the monomer spectral region, so that the latter can be analyzed without the risk of contamination from signals of dangling hydroxy groups of dimers. This conclusion for propargyl alcohol will be reconfirmed in Section 3.5.
3.5. Computational Results for Propargyl Alcohol Dimers
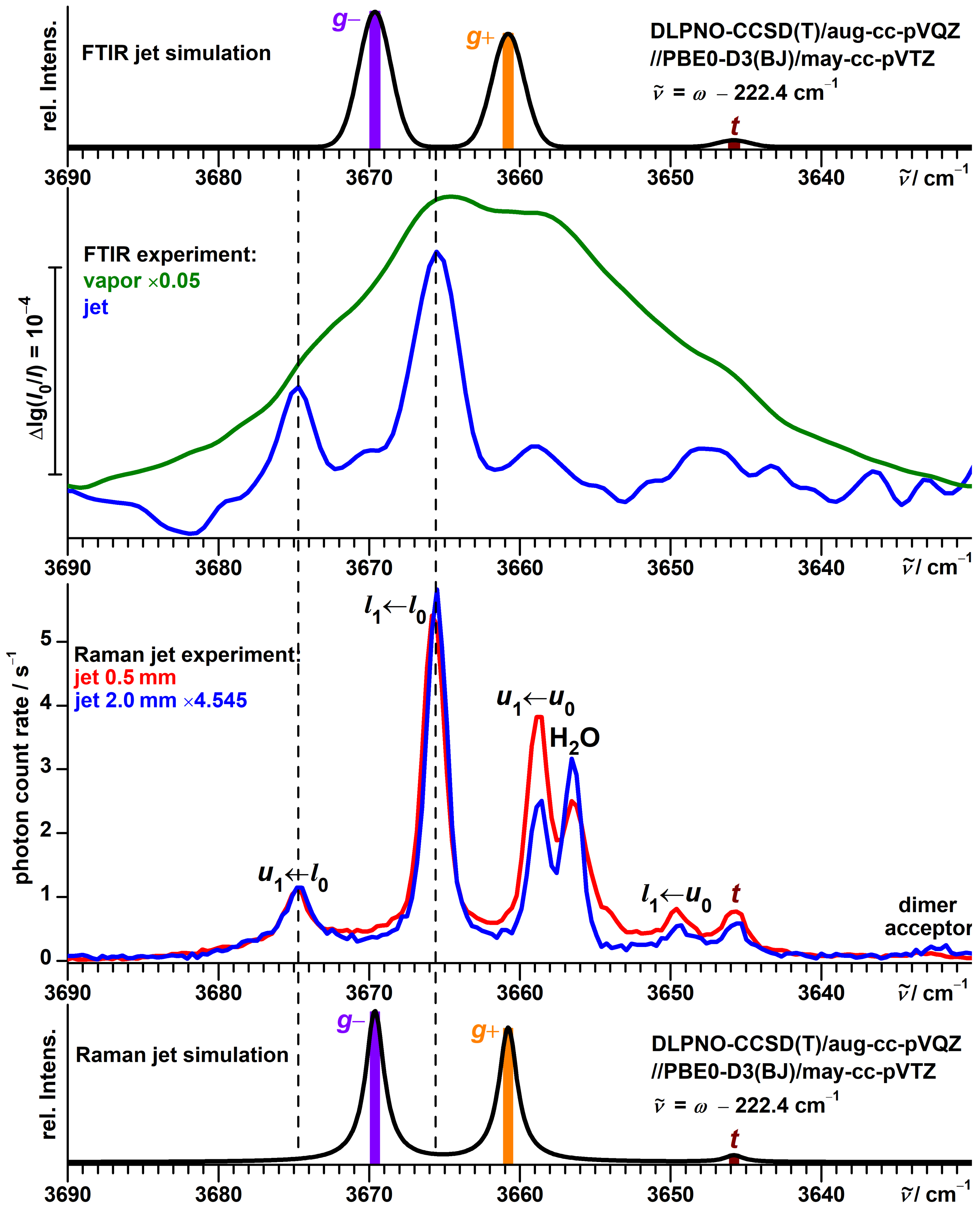
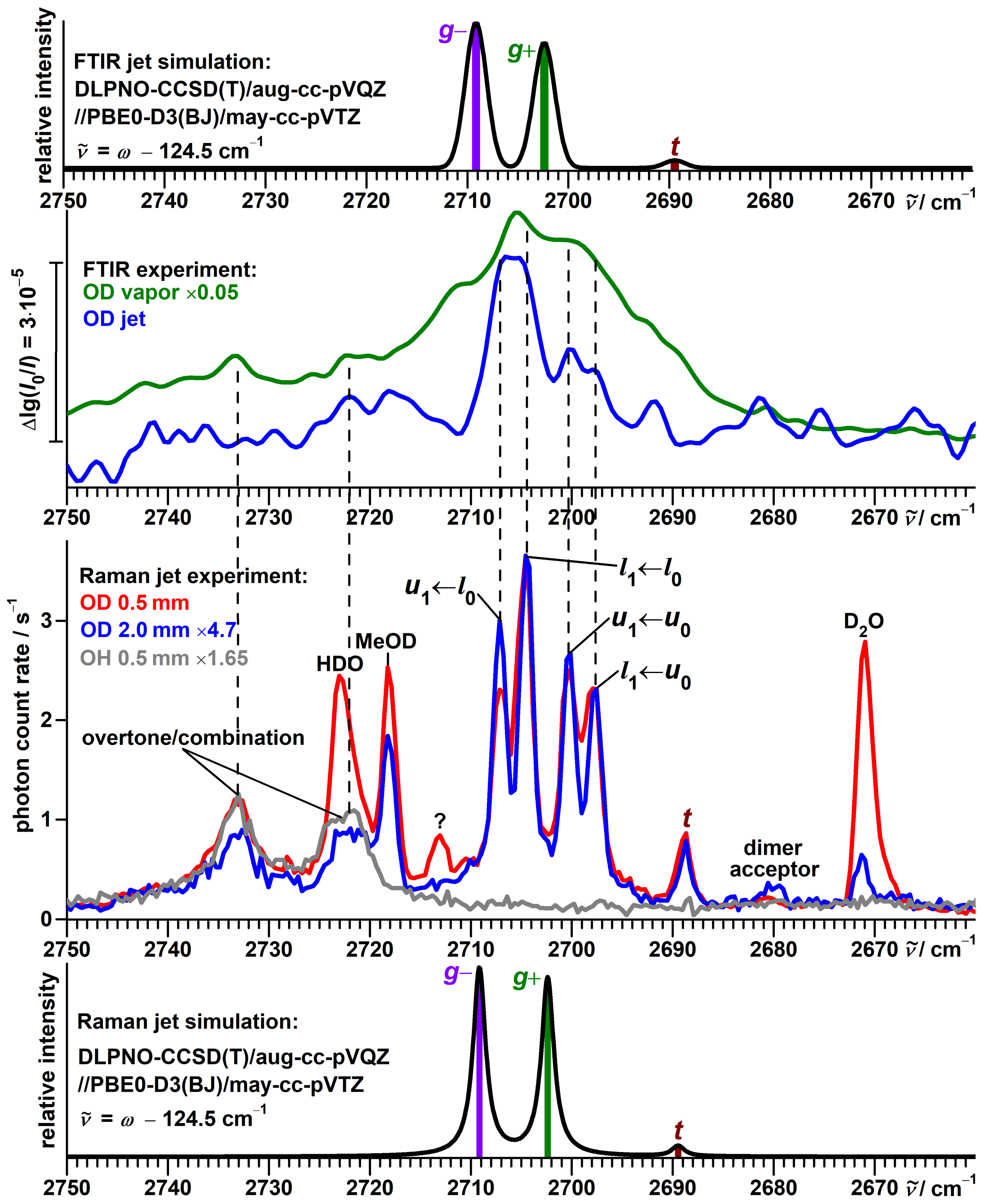
3.6. Vibrational Spectra of Propargyl Alcohol Dimers
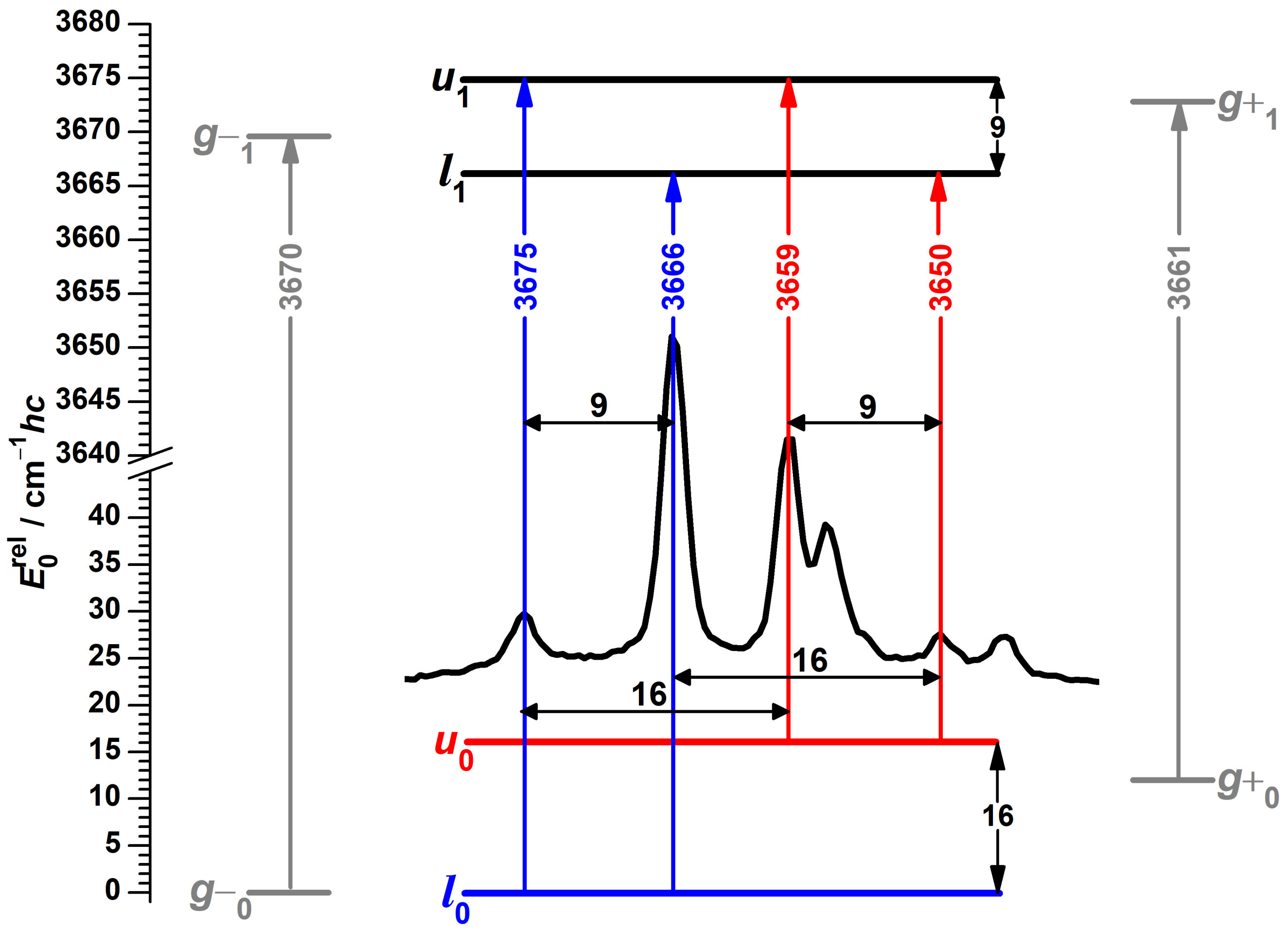
3.7. Vibrational Spectra of --Fenchol Dimers
3.8. Computational Results for --Fenchol Dimers
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schreiner, P.R. Quantum Mechanical Tunneling Is Essential to Understanding Chemical Reactivity. Trends Chem. 2020, 2, 980–989. [Google Scholar] [CrossRef]
- Quack, M.; Seyfang, G. Atomic and Molecular Tunneling Processes in Chemistry. In Molecular Spectroscopy and Quantum Dynamics; Elsevier: St. Louis, MO, USA, 2021; pp. 231–282. [Google Scholar] [CrossRef]
- Rossetti, R.; Rayford, R.; Haddon, R.C.; Brus, L.E. Proton localization in an asymmetric double-minimum potential: 2-methyl-9-hydroxyphenalenone. J. Am. Chem. Soc. 1981, 103, 4303–4307. [Google Scholar] [CrossRef]
- Baughcum, S.L.; Duerst, R.W.; Rowe, W.F.; Smith, Z.; Wilson, E.B. Microwave spectroscopic study of malonaldehyde (3-hydroxy-2-propenal). 2. Structure, dipole moment, and tunneling. J. Am. Chem. Soc. 1981, 103, 6296–6303. [Google Scholar] [CrossRef]
- Redington, R.L.; Redington, T.E.; Hunter, M.A.; Field, R.W. Ã1B2–X˜1A126v0 transitions of 18O-enriched tropolone. J. Chem. Phys. 1990, 92, 6456–6462. [Google Scholar] [CrossRef]
- Busch, J.H.; De la Vega, J.R. Symmetry and tunneling in proton transfer reactions. Proton exchange between methyloxonium ion and methyl alcohol, methyl alcohol and methoxide ion, hydronium ion and water, and water and hydroxyl ion. J. Am. Chem. Soc. 1977, 99, 2397–2406. [Google Scholar] [CrossRef]
- Bosch, E.; Moreno, M.; Lluch, J.M. Tunneling dynamics in isotopically substituted malonaldehyde. Comparison between symmetric and asymmetric species. J. Am. Chem. Soc. 1992, 114, 2072–2076. [Google Scholar] [CrossRef]
- Cordes, J.; Das, A. Tunnelling in asymmetric double-well potentials: Varying initial states. Superlattices Microstruct. 2001, 29, 121–132. [Google Scholar] [CrossRef]
- Song, D.Y. Tunneling and energy splitting in an asymmetric double-well potential. Ann. Phys. 2008, 323, 2991–2999. [Google Scholar] [CrossRef]
- Rastelli, G. Semiclassical formula for quantum tunneling in asymmetric double-well potentials. Phys. Rev. A 2012, 86, 012106. [Google Scholar] [CrossRef]
- Song, D.Y. Localization or tunneling in asymmetric double-well potentials. Ann. Phys. 2015, 362, 609–620. [Google Scholar] [CrossRef]
- Bredtmann, T.; Manz, J.; Zhao, J.M. Concerted Electronic and Nuclear Fluxes During Coherent Tunnelling in Asymmetric Double-Well Potentials. J. Phys. Chem. A 2016, 120, 3142–3154. [Google Scholar] [CrossRef]
- Halataei, S.M.H.; Leggett, A.J. Tunnel Splitting in Asymmetric Double Well Potentials: An Improved WKB Calculation. arXiv 2017, arXiv:1703.05758. [Google Scholar]
- Jahr, E.; Laude, G.; Richardson, J.O. Instanton theory of tunneling in molecules with asymmetric isotopic substitutions. J. Chem. Phys. 2020, 153, 094101. [Google Scholar] [CrossRef]
- Medel, R.; Suhm, M.A. Understanding benzyl alcohol aggregation by chiral modification: The pairing step. Phys. Chem. Chem. Phys. 2020, 22, 25538–25551. [Google Scholar] [CrossRef] [PubMed]
- De la Vega, J.R. Role of symmetry in the tunneling of the proton in double-minimum potentials. Acc. Chem. Res. 1982, 15, 185–191. [Google Scholar] [CrossRef]
- Pejlovas, A.M.; Lin, W.; Kukolich, S.G. Microwave spectra and structure of the cyclopropanecarboxylic acid-formic acid dimer. J. Chem. Phys. 2015, 143, 124311. [Google Scholar] [CrossRef]
- Evangelisti, L.; Caminati, W. Modeling the internal rotation tunnelling in benzyl alcohol by ring fluorination: The rotational spectrum of 3,5-difluorobenzyl alcohol. Chem. Phys. Lett. X 2019, 1, 100004. [Google Scholar] [CrossRef]
- Thomas, J.; Carrillo, M.J.; Serrato, A.; Xie, F.; Jäger, W.; Xu, Y.; Lin, W. Microwave spectrum of the complex of 3,3,3-trifluoro-2-(trifluoromethyl)propanoic acid and formic acid. Mol. Phys. 2019, 117, 1193–1199. [Google Scholar] [CrossRef]
- Kakar, R.K.; Quade, C.R. Microwave rotational spectrum and internal rotation in gauche ethyl alcohol. J. Chem. Phys. 1980, 72, 4300–4307. [Google Scholar] [CrossRef]
- Sekiya, H.; Takesue, H.; Nishimura, Y.; Li, Z.H.; Mori, A.; Takeshita, H. Electronic spectra of jet-cooled isopropyltropolones in the S1–S0 region. Proton tunneling in the S1 state. J. Chem. Phys. 1990, 92, 2790–2796. [Google Scholar] [CrossRef]
- Farrell, J.T.; Suhm, M.A.; Nesbitt, D.J. Breaking symmetry with hydrogen bonds: Vibrational predissociation and isomerization dynamics in HF–DF and DF–HF isotopomers. J. Chem. Phys. 1996, 104, 9313–9331. [Google Scholar] [CrossRef]
- Su, C.F.; Quade, C. Microwave Spectra of gauche CH2DCH2OH Including Excited States of the –OH Torsion. J. Mol. Spectrosc. 1998, 188, 1–8. [Google Scholar] [CrossRef]
- Lesarri, A.; Shipman, S.T.; Neill, J.L.; Brown, G.G.; Suenram, R.D.; Kang, L.; Caminati, W.; Pate, B.H. Interplay of Phenol and Isopropyl Isomerism in Propofol from Broadband Chirped-Pulse Microwave Spectroscopy. J. Am. Chem. Soc. 2010, 132, 13417–13424. [Google Scholar] [CrossRef]
- Albert, S.; Lerch, P.; Prentner, R.; Quack, M. Tunneling and Tunneling Switching Dynamics in Phenol and Its Isotopomers from High-Resolution FTIR Spectroscopy with Synchrotron Radiation. Angew. Chem. Int. Ed. 2013, 52, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Chen, Z.; Fábri, C.; Lerch, P.; Prentner, R.; Quack, M. A combined Gigahertz and Terahertz (FTIR) spectroscopic investigation of meta-D-phenol: Observation of tunnelling switching. Mol. Phys. 2016, 114, 2751–2768. [Google Scholar] [CrossRef]
- Saragi, R.T.; Juanes, M.; Caminati, W.; Lesarri, A.; Enríquez, L.; Jaraíz, M. Rotational Spectrum, Tunneling Motions, and Intramolecular Potential Barriers in Benzyl Mercaptan. J. Phys. Chem. A 2019, 123, 8435–8440. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tikhonov, D.; Schnell, M. Double proton transfer across a table: The formic acid dimer–fluorobenzene complex. Angew. Chem. 2021, 133, 26203. [Google Scholar] [CrossRef]
- Ensminger, F.A.; Plassard, J.; Zwier, T.S.; Hardinger, S. Vibronically state-selective photoisomerization in 5-hydroxytropolone. J. Chem. Phys. 1993, 99, 8341–8344. [Google Scholar] [CrossRef]
- Barclay, A.J.; Pietropolli Charmet, A.; Michaelian, K.H.; McKellar, A.R.W.; Moazzen-Ahmadi, N. Micro-solvation of CO in water: Infrared spectra and structural calculations for (D2O)2–CO and (D2O)3–CO. Phys. Chem. Chem. Phys. 2019, 21, 26564–26568. [Google Scholar] [CrossRef]
- Wassermann, T.N.; Luckhaus, D.; Coussan, S.; Suhm, M.A. Proton tunneling estimates for malonaldehyde vibrations from supersonic jet and matrix quenching experiments. Phys. Chem. Chem. Phys. 2006, 8, 2344–2348. [Google Scholar] [CrossRef]
- Lüttschwager, N.O.; Wassermann, T.N.; Coussan, S.; Suhm, M.A. Vibrational tuning of the Hydrogen transfer in malonaldehyde—A combined FTIR and Raman jet study. Mol. Phys. 2013, 111, 2211–2227. [Google Scholar] [CrossRef]
- Rambaud, C.; Trommsdorff, H. Cooperative proton transfer and tunneling in dye doped benzoic acid crystals. Chem. Phys. Lett. 1999, 306, 124–132. [Google Scholar] [CrossRef]
- Yamada, K.M.; Ross, S.C. Isomerisation: Don’t forget the possibility of enhanced tunnelling! A simple two-state model for the on-resonance and near-resonance behaviour of enhanced tunnelling. J. Mol. Struct. 2006, 795, 84–92. [Google Scholar] [CrossRef]
- Schuder, M.D.; Lovejoy, C.M.; Lascola, R.; Nesbitt, D.J. High resolution, jet-cooled infrared spectroscopy of (HCl)2: Analysis of ν1 and ν2 HCl stretching fundamentals, interconversion tunneling, and mode-specific predissociation lifetimes. J. Chem. Phys. 1993, 99, 4346–4362. [Google Scholar] [CrossRef]
- Schuder, M.D.; Nelson, D.D.; Nesbitt, D.J. Slit-jet near-infrared diode laser spectroscopy of (DCl)2: ν1, ν2 DCl stretching fundamentals, tunneling dynamics, and the influence of large amplitude “geared” intermolecular rotation. J. Chem. Phys. 1993, 99, 5045–5060. [Google Scholar] [CrossRef]
- Gil, M.; Jasny, J.; Vogel, E.; Waluk, J. Ground and excited state tautomerization in 9-acetoxy-2,7,12,17-tetra-n-propylporphycene. Chem. Phys. Lett. 2000, 323, 534–541. [Google Scholar] [CrossRef]
- Vdovin, A.; Sepioł, J.; Urbańska, N.; Pietraszkiewicz, M.; Mordziński, A.; Waluk, J. Evidence for Two Forms, Double Hydrogen Tunneling, and Proximity of Excited States in Bridge-Substituted Porphycenes: Supersonic Jet Studies. J. Am. Chem. Soc. 2006, 128, 2577–2586. [Google Scholar] [CrossRef]
- Fita, P.; Garbacz, P.; Nejbauer, M.; Radzewicz, C.; Waluk, J. Ground and Excited State Double Hydrogen Transfer in Symmetric and Asymmetric Potentials: Comparison of 2,7,12,17-Tetran-propylporphycene with 9-Acetoxy-2,7,12,17-tetran-propylporphycene. Chem. Eur. J. 2011, 17, 3672–3678. [Google Scholar] [CrossRef]
- Mengesha, E.T.; Zehnacker-Rentien, A.; Sepioł, J.; Kijak, M.; Waluk, J. Spectroscopic Study of Jet-Cooled Deuterated Porphycenes: Unusual Isotopic Effects on Proton Tunneling. J. Phys. Chem. B 2015, 119, 2193–2203. [Google Scholar] [CrossRef]
- Peukert, S.; Kijak, M.; Ostapko, J.; Sepioł, J.; Le Bris, C.; Zehnacker-Rentien, A.; Gil, M.; Waluk, J. Supersonic jet spectroscopy of parent hemiporphycene: Structural assignment and vibrational analysis for S0 and S1 electronic states. J. Chem. Phys. 2018, 149, 134307. [Google Scholar] [CrossRef]
- Bondybey, V.E.; Haddon, R.C.; English, J.H. Fluorescence and phosphorescence of 9-hydroxyphenalenone in solid neon and its hydrogen tunneling potential function. J. Chem. Phys. 1984, 80, 5432–5437. [Google Scholar] [CrossRef]
- Kunze, K.L.; De la Vega, J.R. Intramolecular proton exchange in 9-hydroxyphenalen-1-one and methyl-9-hydroxyphenalen-1-one. J. Am. Chem. Soc. 1984, 106, 6528–6533. [Google Scholar] [CrossRef]
- Nishi, K.; Sekiya, H.; Mochida, T.; Sugawara, T.; Nishimura, Y. Coupling between the internal rotation of the methyl group and proton/deuteron transfer in jet-cooled 5-methyl-9-hydroxyphenalenone(OH) and 5-methyl-9-hydroxyphenalenone(OD): Tunneling rate dependence of coupling potential. J. Chem. Phys. 2000, 112, 5002–5011. [Google Scholar] [CrossRef]
- Mori, H.; Sekiya, H.; Miyoshi, E.; Mogi, K.; Sakai, Y. Effects of intermolecular interaction on proton tunneling: Theoretical study on two-dimensional potential energy surfaces for 9-hydroxyphenalenone-CO2/H2O complexes. J. Chem. Phys. 2003, 119, 4159–4165. [Google Scholar] [CrossRef]
- Breitmaier, E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; 1. Reprint Ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Wallach, O. Zur Kenntniss der Terpene und der ätherischen Oele. Ueber neue Verbindungen der Campherreihe und ein neues Terpen. Justus Liebigs Ann. Chem. 1891, 263, 129–156. [Google Scholar] [CrossRef]
- Bhatia, S.; McGinty, D.; Letizia, C.; Api, A. Fragrance material review on fenchyl alcohol. Food Chem. Toxicol. 2008, 46, S157–S159. [Google Scholar] [CrossRef]
- Pate, B.H.; Evangelisti, L.; Caminati, W.; Xu, Y.; Thomas, J.; Patterson, D.; Perez, C.; Schnell, M. Quantitative Chiral Analysis by Molecular Rotational Spectroscopy. In Chiral Analysis; Elsevier: Amsterdam, The Netherlands, 2018; pp. 679–729. [Google Scholar] [CrossRef]
- Neeman, E.M.; Huet, T.R. Identification of the maze in the conformational landscape of fenchol. Phys. Chem. Chem. Phys. 2018, 20, 24708–24715. [Google Scholar] [CrossRef]
- Medel, R.; Suhm, M.A. Predicting OH stretching fundamental wavenumbers of alcohols for conformational assignment: Different correction patterns for density functional and wave-function-based methods. Phys. Chem. Chem. Phys. 2021, 23, 5629–5643. [Google Scholar] [CrossRef]
- Forsting, T.; Suhm, M. Curry-Jet SETUP. Available online: https://figshare.com/articles/dataset/Curry-Jet_SETUP/6395840/1 (accessed on 18 December 2021). [CrossRef]
- Suhm, M.A.; Kollipost, F. Femtisecond single-mole infrared spectroscopy of molecular clusters. Phys. Chem. Chem. Phys. 2013, 15, 10702. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Technical Report; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A At. Mol. Opt. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Lüttschwager, N.O.B. Raman Spectroscopy of Conformational Rearrangements at Low Temperatures Folding and Stretching of Alkanes in Supersonic Jets; Springer International Publishing: Cham, Switzerland, 2014. [Google Scholar]
- Gawrilow, M.; Suhm, M.A. 2-Methoxyethanol: Harmonic tricks, anharmonic challenges and chirality-sensitive chain aggregation. Phys. Chem. Chem. Phys. 2020, 22, 15303–15311. [Google Scholar] [CrossRef]
- Oswald, S. Weak Hydrogen Bonds to Molecular Nitrogen and Oxygen as Experimental Benchmarks for Quantum Chemistry. Ph.D. Thesis, Georg-August-Universität Göttingen, Göttingen, Germany, 2019. [Google Scholar]
- Pearson, J.; Sastry, K.; Herbst, E.; De Lucia, F.C. The Millimeter- and Submillimeter-Wave Spectrum of Gauche-Ethyl Alcohol. J. Mol. Spectrosc. 1996, 175, 246–261. [Google Scholar] [CrossRef]
- Chung-Phillips, A. Torsional energy levels and wave functions. J. Comput. Chem. 1992, 13, 874–882. [Google Scholar] [CrossRef]
- Dieke, G.; Crosswhite, H. The ultraviolet bands of OH Fundamental data. J. Quant. Spectrosc. Radiat. Transf. 1962, 2, 97–199. [Google Scholar] [CrossRef]
- Coxon, J.A. The A2Σ+–X2Πi system of OD. J. Mol. Spectrosc. 1975, 58, 1–28. [Google Scholar] [CrossRef]
- Medel, R.; Stelbrink, C.; Suhm, M.A. Vibrational Signatures of Chirality Recognition Between α-Pinene and Alcohols for Theory Benchmarking. Angew. Chem. Int. Ed. 2019, 58, 8177–8181. [Google Scholar] [CrossRef]
- Zielke, P. Ramanstreuung am Überschallstrahl: Wasserstoffbrückendynamik aus neuer Perspektive. Ph.D. Thesis, Georg-August-Universität Göttingen, Göttingen, Germany, 2007. [Google Scholar]
- Wassermann, T.N.; Suhm, M.A. Ethanol Monomers and Dimers Revisited: A Raman Study of Conformational Preferences and Argon Nanocoating Effects. J. Phys. Chem. A 2010, 114, 8223–8233. [Google Scholar] [CrossRef]
- Wassermann, T.N.; Suhm, M.A.; Roubin, P.; Coussan, S. Isomerization around C–C and C–O bonds in 1-propanol: Collisional relaxation in supersonic jets and selective IR photo-isomerization in cryogenic matrices. J. Mol. Struct. 2012, 1025, 20–32. [Google Scholar] [CrossRef]
- Kollipost, F.; Papendorf, K.; Lee, Y.F.; Lee, Y.P.; Suhm, M.A. Alcohol dimers—How much diagonal OH anharmonicity? Phys. Chem. Chem. Phys. 2014, 16, 15948–15956. [Google Scholar] [CrossRef]
- Castillo-Chará, J.; McIntosh, A.L.; Wang, Z.; Lucchese, R.R.; Bevan, J.W. Near-infrared spectra and rovibrational dynamics on a four-dimensional ab initio potential energy surface of (HBr)2. J. Chem. Phys. 2004, 120, 10426–10441. [Google Scholar] [CrossRef]
- Avila, G.; Fernández, J.; Tejeda, G.; Montero, S. The Raman spectra and cross-sections of H2O, D2O, and HDO in the OH/OD stretching regions. J. Mol. Spectrosc. 2004, 228, 38–65. [Google Scholar] [CrossRef]
- Otto, K.E.; Xue, Z.; Zielke, P.; Suhm, M.A. The Raman spectrum of isolated water clusters. Phys. Chem. Chem. Phys. 2014, 16, 9849. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.W.; Zielke, P.; Suhm, M.A. Hydrogen-bonded OH stretching modes of methanol clusters: A combined IR and Raman isotopomer study. J. Chem. Phys. 2007, 126, 194307. [Google Scholar] [CrossRef] [PubMed]
- Schiel, D.; Richter, W. Effect of the internal rotation of the OH group on the OH stretching vibrational Raman profile of ethanol in the gas phase. J. Chem. Phys. 1983, 78, 6559–6562. [Google Scholar] [CrossRef]
- Schuder, M.D.; Nesbitt, D.J. High resolution near infrared spectroscopy of HCl–DCl and DCl–HCl: Relative binding energies, isomer interconversion rates, and mode specific vibrational predissociation. J. Chem. Phys. 1994, 100, 7250–7267. [Google Scholar] [CrossRef]
- Subramanian, R.; Novick, S.E.; Bohn, R.K. Torsional analysis of 2-butynol. J. Mol. Spectrosc. 2003, 222, 57–62. [Google Scholar] [CrossRef]
- Hirota, E. Internal rotation in propargyl alcohol from microwave spectrum. J. Mol. Spectrosc. 1968, 26, 335–350. [Google Scholar] [CrossRef]
- Pearson, J.; Drouin, B. The ground state torsion–rotation spectrum of propargyl alcohol (HCCCH2OH). J. Mol. Spectrosc. 2005, 234, 149–156. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. Microwave Spectroscopic and Atoms in Molecules Theoretical Investigations on the Ar···Propargyl Alcohol Complex: Ar···H-O, Ar···π, and Ar···C Interactions. ChemPhysChem 2013, 14, 754–763. [Google Scholar] [CrossRef]
- Ruoff, R.S.; Klots, T.D.; Emilsson, T.; Gutowsky, H.S. Relaxation of conformers and isomers in seeded supersonic jets of inert gases. J. Chem. Phys. 1990, 93, 3142–3150. [Google Scholar] [CrossRef]
- Herzberg, G. Molecular Spectra and Molecular Structure. Volume 1: Spectra of Diatomic Molecules, 2nd ed.; 23. Print Ed.; van Nostrand: New York, NY, USA, 1987. [Google Scholar]
- Noether, H.D. The Ratio Rule. J. Chem. Phys. 1943, 11, 97–98. [Google Scholar] [CrossRef]
- Heger, M. Diagonal and Off-Diagonal Anharmonicity in Hydrogen-Bonded Systems. Ph.D. Thesis, Georg-August-Universität Göttingen, Göttingen, Germany, 2016. [Google Scholar]
- Medel, R. Simple models for the quick estimation of ground state hydrogen tunneling splittings in alcohols and other compounds. Phys. Chem. Chem. Phys. 2021, 23, 17591–17605. [Google Scholar] [CrossRef] [PubMed]
- Hirota, E.; Kawashima, Y. Internal Rotation of the Hydroxyl Group in Isopropanol and the Chirality of the Gauche Form: Fourier Transform Microwave Spectroscopy of (CH3)2CHOD. J. Mol. Spectrosc. 2001, 207, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Li, W.; Spada, L.; Evangelisti, L.; Lesarri, A.; Caminati, W. Internal dynamics of cyclohexanol and the cyclohexanol–water adduct. Phys. Chem. Chem. Phys. 2019, 21, 3676–3682. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.N.; Norbury, D.; Sheridan, J. Microwave spectrum, dipole moment and internal rotation potential function of gauche-cyclopropanol. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1365. [Google Scholar] [CrossRef]
- Sibert, E.L.; Castillo-Chará, J. Theoretical studies of the potential surface and vibrational spectroscopy of CH3OH and its deuterated analogs. J. Chem. Phys. 2005, 122, 194306. [Google Scholar] [CrossRef] [PubMed]
- Moruzzi, G.; Xu, L.; Lees, R.; Winnewisser, B.; Winnewisser, M. Investigation of the Ground Vibrational State of CD3OH by a New "Ritz" Program for Direct Energy Level Fitting. J. Mol. Spectrosc. 1994, 167, 156–175. [Google Scholar] [CrossRef]
- Mukhopadhyay, I.; Duan, Y.; Takagi, K. High resolution spectroscopy of CH3OD involving the ground and first excited torsional states in the vibrational ground state. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 1998, 54, 1325–1335. [Google Scholar] [CrossRef]
- Xu, L.H.; Müller, H.; van der Tak, F.; Thorwirth, S. The millimeter-wave spectrum of perdeuterated methanol, CD3OD. J. Mol. Spectrosc. 2004, 228, 220–229. [Google Scholar] [CrossRef]
- Margulès, L.; McGuire, B.A.; Senent, M.L.; Motiyenko, R.A.; Remijan, A.; Guillemin, J.C. Submillimeter spectra of 2-hydroxyacetonitrile (glycolonitrile; HOCH 2 CN) and its searches in GBT PRIMOS observations of Sgr B2(N). Astron. Astrophys. 2017, 601, A50. [Google Scholar] [CrossRef]
- Cazzoli, G.; Lister, D.G.; Mirri, A.M. Rotational isomerism and barriers to internal rotation in hydroxyacetonitrile from microwave spectroscopy. J. Chem. Soc. Faraday Trans. 2 1973, 69, 569. [Google Scholar] [CrossRef]
- Motiyenko, R.A.; Margulès, L.; Senent, M.L.; Guillemin, J.C. Internal Rotation of OH Group in 4-Hydroxy-2-butynenitrile Studied by Millimeter-Wave Spectroscopy. J. Phys. Chem. A 2018, 122, 3163–3169. [Google Scholar] [CrossRef]
- Melandri, S.; Favero, P.G.; Caminati, W. Detection of the syn conformer of allyl alcohol by free jet microwave spectroscopy. Chem. Phys. Lett. 1994, 223, 541–545. [Google Scholar] [CrossRef]
- Motiyenko, R.A.; Margulès, L.; Despois, D.; Guillemin, J.C. Laboratory spectroscopy of methoxymethanol in the millimeter-wave range. Phys. Chem. Chem. Phys. 2018, 20, 5509–5516. [Google Scholar] [CrossRef] [PubMed]
- Suenram, R.; Lovas, F.; Pickett, H. Fluoromethanol: Synthesis, microwave spectrum, and dipole moment. J. Mol. Spectrosc. 1986, 119, 446–455. [Google Scholar] [CrossRef]
- Xu, L.H.; Fraser, G.T.; Lovas, F.J.; Suenram, R.D.; Gillies, C.W.; Warner, H.E.; Gillies, J.Z. The microwave spectrum and OH internal rotation dynamics of gauche-2,2,2-trifluoroethanol. J. Chem. Phys. 1995, 103, 9541–9548. [Google Scholar] [CrossRef]
- Cohen, E.; Drouin, B.; Valenzuela, E.; Woods, R.; Caminati, W.; Maris, A.; Melandri, S. The rotational spectrum of tertiary-butyl alcohol. J. Mol. Spectrosc. 2010, 260, 77–83. [Google Scholar] [CrossRef]
- Li, W.; Spada, L.; Evangelisti, L.; Caminati, W. Conformational Equilibrium and Potential Energy Functions of the O–H Internal Rotation in the Axial and Equatorial Species of 1-Methylcyclohexanol. J. Phys. Chem. A 2016, 120, 4338–4342. [Google Scholar] [CrossRef]
- Leonov, A.; Marstokk, K.M.; de Meijere, A.; Møllendal, H. Microwave Spectrum, Conformational Equilibrium, Intramolecular Hydrogen Bonding, Tunneling, and Quantum Chemical Calculations for 1-Ethenylcyclopropan-1-ol. J. Phys. Chem. A 2000, 104, 4421–4428. [Google Scholar] [CrossRef]
- Giuliano, B.M.; Caminati, W. Isotopomeric Conformational Change in Anisole-Water. Angew. Chem. Int. Ed. 2005, 44, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kye, Y.S.; Harbison, G.S. Isotopomeric Polymorphism. J. Am. Chem. Soc. 2004, 126, 8392–8393. [Google Scholar] [CrossRef] [PubMed]
- Dübal, H.R.; Crim, F.F. Vibrational overtone predissociation spectroscopy of hydrogen peroxide. J. Chem. Phys. 1985, 83, 3863–3872. [Google Scholar] [CrossRef]
- Matthews, J.; Martínez-Avilés, M.; Francisco, J.S.; Sinha, A. Probing OH stretching overtones of CH3OOH through action spectroscopy: Influence of dipole moment dependence on HOOC torsion. J. Chem. Phys. 2008, 129, 074316. [Google Scholar] [CrossRef]
- Hansen, A.S.; Huchmala, R.M.; Vogt, E.; Boyer, M.A.; Bhagde, T.; Vansco, M.F.; Jensen, C.V.; Kjærsgaard, A.; Kjaergaard, H.G.; McCoy, A.B.; et al. Coupling of torsion and OH-stretching in tert-butyl hydroperoxide. I. The cold and warm first OH-stretching overtone spectrum. J. Chem. Phys. 2021, 154, 164306. [Google Scholar] [CrossRef] [PubMed]
- Vogt, E.; Huchmala, R.M.; Jensen, C.V.; Boyer, M.A.; Wallberg, J.; Hansen, A.S.; Kjærsgaard, A.; Lester, M.I.; McCoy, A.B.; Kjaergaard, H.G. Coupling of torsion and OH-stretching in tert-butyl hydroperoxide. II. The OH-stretching fundamental and overtone spectra. J. Chem. Phys. 2021, 154, 164307. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. Rotational spectra of propargyl alcohol dimer: A dimer bound with three different types of hydrogen bonds. J. Chem. Phys. 2014, 141, 164311. [Google Scholar] [CrossRef]
- Saini, J.; Viswanathan, K.S. Discerning Near-Isoergic Isomers. A Matrix Isolation Infrared and ab Initio Study of the Propargyl Alcohol Dimers. J. Phys. Chem. A 2017, 121, 1448–1459. [Google Scholar] [CrossRef]
- Nyquist, R. Vibrational assignments of H-CC-CH2OH and its deuterium analogs. Spectrochim. Acta Part A Mol. Spectrosc. 1971, 27, 2513–2523. [Google Scholar] [CrossRef]
- Hudspeth, E.; McWhorter, D.A.; Pate, B.H. Intramolecular vibrational energy redistribution in the acetylenic C–H and hydroxyl stretches of propynol. J. Chem. Phys. 1998, 109, 4316–4326. [Google Scholar] [CrossRef]
- Sundararajan, K.; Gopi, R.; Ramanathan, N. Conformations of propargyl alcohol and its interaction with acetylene: A matrix isolation infrared and DFT computations. J. Mol. Struct. 2016, 1121, 26–34. [Google Scholar] [CrossRef]
- Altnöder, J.; Bouchet, A.; Lee, J.J.; Otto, K.E.; Suhm, M.A.; Zehnacker-Rentien, A. Chirality-dependent balance between hydrogen bonding and London dispersion in isolated (±)-1-indanol clusters. Phys. Chem. Chem. Phys. 2013, 15, 10167. [Google Scholar] [CrossRef] [PubMed]
- Altnöder, J.; Oswald, S.; Suhm, M.A. Phenyl- vs Cyclohexyl-Substitution in Methanol: Implications for the OH Conformation and for Dispersion-Affected Aggregation from Vibrational Spectra in Supersonic Jets. J. Phys. Chem. A 2014, 118, 3266–3279. [Google Scholar] [CrossRef] [PubMed]
- Neeman, E.M.; Huet, T.R. Conformational changes in hydroxyl functional groups upon hydration: The case study of endo fenchol. Phys. Chem. Chem. Phys. 2021, 23, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Christen, D.; Coudert, L.; Larsson, J.; Cremer, D. The Rotational–Torsional Spectrum of the g′Gg Conformer of Ethylene Glycol: Elucidation of an Unusual Tunneling Path. J. Mol. Spectrosc. 2001, 205, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.; Hearn, I.; Howard, B.J. Chiral recognition in a single molecule: A study of homo and heterochiral butan-2,3-diol by Fourier transform microwave spectroscopy. Mol. Phys. 2007, 105, 825–839. [Google Scholar] [CrossRef]
- Olschewski, M.; Lindner, J.; Vöhringer, P. A Hydrogen-Bond Flip-Flop through a Bjerrum-Type Defect. Angew. Chem. Int. Ed. 2013, 52, 2602–2605. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, L.; Gou, Q.; Spada, L.; Feng, G.; Caminati, W. Conformational analysis of 1,4-butanediol: A microwave spectroscopy study. Chem. Phys. Lett. 2013, 556, 55–58. [Google Scholar] [CrossRef]
- Medel, R. Schwingungsspektroskopische Untersuchungen zur Chiralitätserkennung und Torsionsdynamik bei Alkoholen. Ph.D. Thesis, Georg-August-Universität Göttingen, Göttingen, Germany, 2020. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | ||||
|---|---|---|---|---|
| MP2/6-311++G(d,p) | 0.04 (−0.04) | −8 | 2.71 (1.61) | −3 |
| PBE0-D3(BJ)/may-cc-pVTZ | 0.28 (0.14) | −9 | 0.37 (1.43) | −24 |
| B3LYP-D3(BJ)/may-cc-pVTZ | 0.33 (0.12) | −8 | 0.81 (1.52) | −17 |
| B2PLYP-D3(BJ)/may-cc-pVTZ | 0.22 (0.07) | −8 | 1.51 (1.54) | −12 |
| Computational Level | Model | / cm | / cm |
|---|---|---|---|
| B3LYP | Eckart Barrier [93] | 16.2 | 3.6 |
| B3LYP | Barrier Height [93] | 11.1 | 5.3 |
| B3LYP | Restricted Barrier Height [93] | 15.4 | 5.1 |
| B3LYP | Effective Barrier Height [93] | 17.8 | 7.3 |
| B3LYP | torsion 1D code symmetrization | 15.4 | 3.4 |
| DLPNO-CCSD(T)//B3LYP | torsion 1D code symmetrization | 15.9 | 3.5 |
| DLPNO-CCSD(T)//B3LYP | torsion 1D code localization | 14.4 | 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medel, R.; Springborn, J.R.; Crittenden, D.L.; Suhm, M.A. Hydrogen Delocalization in an Asymmetric Biomolecule: The Curious Case of Alpha-Fenchol. Molecules 2022, 27, 101. https://doi.org/10.3390/molecules27010101
Medel R, Springborn JR, Crittenden DL, Suhm MA. Hydrogen Delocalization in an Asymmetric Biomolecule: The Curious Case of Alpha-Fenchol. Molecules. 2022; 27(1):101. https://doi.org/10.3390/molecules27010101
Chicago/Turabian StyleMedel, Robert, Johann R. Springborn, Deborah L. Crittenden, and Martin A. Suhm. 2022. "Hydrogen Delocalization in an Asymmetric Biomolecule: The Curious Case of Alpha-Fenchol" Molecules 27, no. 1: 101. https://doi.org/10.3390/molecules27010101
APA StyleMedel, R., Springborn, J. R., Crittenden, D. L., & Suhm, M. A. (2022). Hydrogen Delocalization in an Asymmetric Biomolecule: The Curious Case of Alpha-Fenchol. Molecules, 27(1), 101. https://doi.org/10.3390/molecules27010101