Abstract
Propellanes are polycyclic compounds in which tricyclic systems share one carbon–carbon single bond. Propellane frameworks that consist of larger sized rings are found in a variety of natural products. As an approach to the stereoselective synthesis of the propellane framework, one of the efficient methods is forming several rings in a single operation. Lapidilectine B (1) is composed of a propellane framework and was synthesized through the oxidative cyclization of trisubstituted alkenes. When the alkene with an ester moiety was treated with N-iodosuccinimide (NIS), iodocyclization proceeded to give the cyclic carbamate. On the other hand, when PhI(OAc)2 was allowed to react in the carboxyl form, a furoindolin-2-one structure corresponding to the A-B-C ring of lapidilectine B (1) was produced. Furthermore, when Pd(OAc)2 catalyst was used for cyclization under oxidative conditions, the product yield was improved.
1. Introduction
In 1992, lapidilectine B (1) (Figure 1) was isolated from the leaves of Kopsia lapidilecta Sleesen in Malaysia [1]. Biological activity of 1 shows a reverse multidrug resistance in vincristine-resistant KB cells [2].
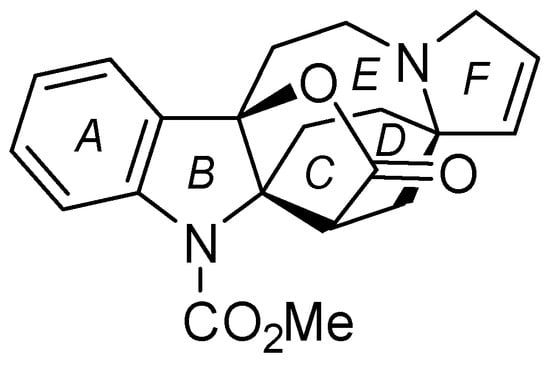
Figure 1.
Lapidilectine B (1).
The structural feature of 1 is possessing a unique propellane structure composed of indoline, γ-lactone, and azocane. In addition, the cyclohexane ring is connected to two pyrrolidine rings via two spirocenters. The first total synthesis of 1 was achieved by Pearson in 2001, where the Smalley cyclization was used to construct the framework of the B and C rings [3,4]. The optically active form of 1 was synthesized by Nishida in 2016 via desymmetrization of the spiro center using enantioselective deprotonation with a chiral lithium amide [5]. In 2018, Ma utilized manganese (III) mediated oxidative cyclization of 2-alkylindole to form the B–C–D rings of 1 in one operation [6]. We focused on the anti-configuration between oxygen and nitrogen atoms in the B–C–E propellane moiety, and postulated that the simultaneous functionalization of the alkene is a key reaction for ring construction in 1.
As a methodology for double functionalization with heteroatoms on C-C double bond, Baran indicated the oxidative coupling reaction of o-iodoaniline and tryptamine using NIS to form the 3a substituted pyrroindoline motif in the synthesis of psychotrimine (Scheme 1A) [7,8]. The electrophilic N-haloaniline and the nucleophilic carbamate of the tryptamine side chain were introduced into the 2,3-positions of the indole. One of promising methods for introducing of two heteroatoms onto an alkene with a transition metal catalyst is Sharpless’s asymmetric aminohydroxylation [9]. In 2007, Muñiz reported the palladium-catalyzed aminoalkoxylation of internal alkene bearing aniline and phenol moieties with PhI(OAc)2 (Scheme 1B) [10,11]. Interestingly in this method, a furoindoline skeleton is assembled by closing two rings of a disubstituted alkene at once. To construct the 5-5 bicyclo B-C ring system in lapidilectine B (1), the Z trisubstituted alkene 2 was selected as the precursor for the ring closing reaction (Scheme 1C). Here, we report the efficient synthesis of furoindolin-2-one 3 by the successive cyclization of trisubstituted alkene 2 with the goal of developing a method for synthesizing the polycyclic skeleton of 1.
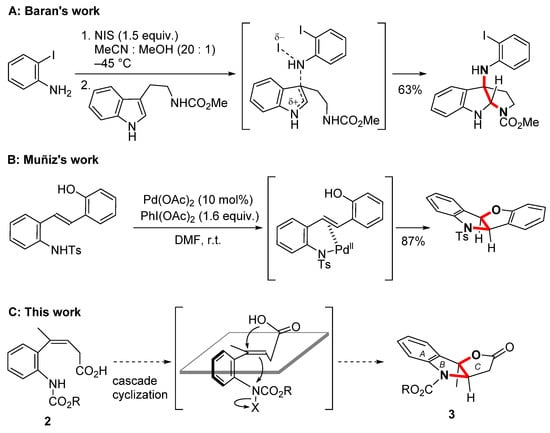
Scheme 1.
Oxidative cyclization of alkenes.
2. Results and Discussion
We synthesized trisubstituted alkenes 2 and 6 bearing an alkoxycarbonyl group on the aniline nitrogen (Scheme 2). Aniline, its hydrochloride, and 4-oxopentanoic acid were condensed under heating at 200 °C to obtain lactam 4 according to the literature [12]. Thereafter, lactam 4 was transformed to imides 5a–e by treating n-butyllithium with alkyl chloroformate or triethylamine with Boc2O, after which imides 5 were hydrolyzed with NaOH in THF-water to prepare carboxylic acids 2a–e. Boc form 5c was solvolyzed with sodium methoxide in MeOH to obtain methyl ester 6.
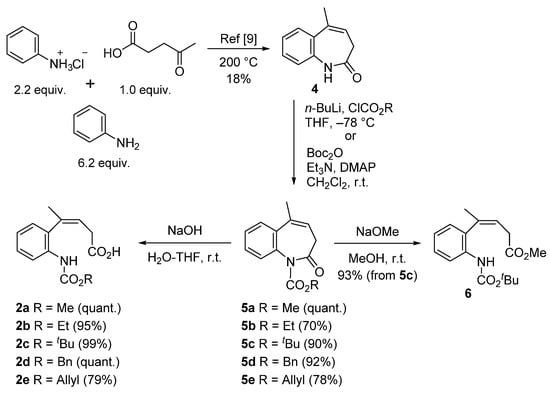
Scheme 2.
Syntheses of trisubstituted alkenes.
First, we investigated a key cyclization using compounds 2c and 6 bearing a Boc group (Scheme 3). The treatment of carboxylic acid 2c with NIS in a solution of MeCN and MeOH resulted in a complex mixture. On the other hand, when methyl ester 6 was treated under the same conditions as above, cyclic carbamate 7 was obtained in 89% yield as a single diastereomer. Iodocyclization triggered by the activation of the alkene with NIS, accompanied by the elimination of isobutylene from the Boc group, gave 7.
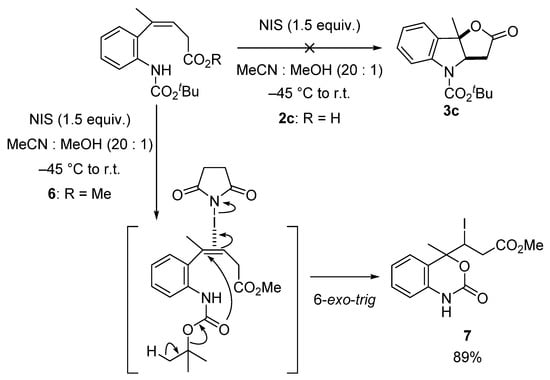
Scheme 3.
Iodocyclization of alkene 6.
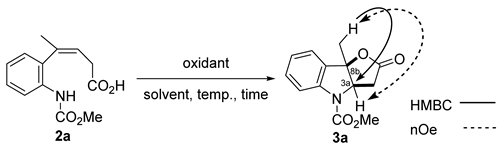
Trisubstituted alkene 2a possessing a carboxylic acid moiety was then subjected to halogenating and oxidizing agents (Table 1). The reaction yielded many products when NIS was added to 2a (entry 1), whereas the reaction did not proceed with NCS (entry 2). When 2a was treated with CAN in MeCN solution, the trace amount of the desired cyclized product 3a was produced (entry 3). When the reaction was carried out with PhI(OAc)2 under refluxing MeCN, product 3a was obtained in 43% yield, along with many decomposition products (entry 4). Other hypervalent iodine reagents, PhI(OCOCF3)2, PhI(OH)OTs, and PhIO were tested, product 3a was slightly detected on TLC, however substrate 2a was mainly decomposed (entries 5–7). The fused ring structure of product 3a was confirmed by the correlation between the methyl proton at position 8b and the 3a carbon in the HMBC experiment, and between the methyl proton at position 8b and the 3a proton in the nOe experiment.

Table 1.
Oxidative cyclization of alkene 2a.
A plausible reaction mechanism is shown in Scheme 4. In conjunction with the oxidative activation of the aniline nitrogen atom in 2a by PhI(OAc)2, the nitrogen atom undergoes nucleophilic attack by the π-electron of the alkene, after which the carboxylic acid cyclized to give furoindolin-2-one 3a.
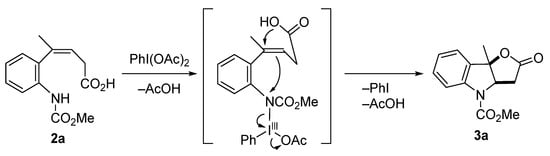
Scheme 4.
Plausible reaction mechanism.
The conditions for palladium(II) mediated oxidative difunctionalization of alkenes reported by Muñiz were applied to trisubstituted alkene 2a (Table 2). When 1 equivalent of the palladium reagent with 1.5 equivalents of PhI(OAc)2 treated with 2a in DMF at room temperature, product 3a was obtained in 19% yield with PdCl2, in 29% yield with Pd(OCOCF3)2, and in 58% yield with Pd(OAc)2 (entries 1–3). Using Pd(OAc)2 in MeCN, 3a was obtained in 50% yield (entry 4). Based on the above results, a catalytic amount of Pd(OAc)2 was examined using DMF or MeCN. First, when 0.2 equivalent of Pd(OAc)2 was reacted at room temperature for 24 h, the yield of 3a decreased to 14% in DMF and was maintained at 51% in MeCN (entries 5 and 6). When the reaction was performed under refluxing MeCN, the reaction time was shortened and the yield of 3a increased to 78% (entry 7). Further reduction the amount of Pd(OAc)2 to 0.1 equivalent decreased the product yield to 47% (entry 8). With the optimum conditions in hand, the substituent effect on nitrogen atom was examined. The ethoxycarbonyl compound 2b gave the corresponding product 3b in 56% yield (entry 9). Even with 2c possessing a bulky Boc group, product 3c was obtained in 38% yield (entry 10). Even when the Cbz and Alloc substituents attached to nitrogen, products 3d and 3e were obtained in 33% and 29% yields, respectively (entries 11 and 12).
Table 2.
Palladium catalyzed cyclization of alkenes 2.
A reaction mechanism for the cyclization was proposed based on the report by Muñiz. [10,13,14,15,16] (Scheme 5). First, Pd(OAc)2 reacts with the nitrogen atom of substrate 2a and coordinates with the alkene to form intermediate 8. Subsequently, anti-oxypalladation occurs on the alkene to generate the six-membered palladacycle intermediate 9. Furthermore, palladium(II) is oxidized to palladium(IV) by PhI(OAc)2 to form intermediate 10, followed by the reductive elimination of palladium(IV) in 10 to produce 3a and palladium(II) species.
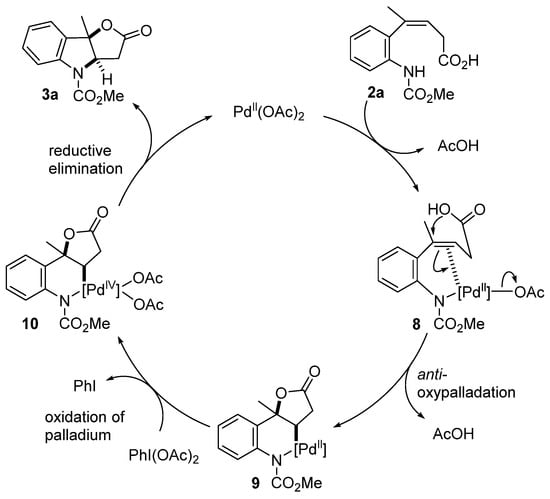
Scheme 5.
Plausible reaction mechanism.
3. Materials and Methods
3.1. General Information
All melting points were measured on a Yanagimoto micro melting point apparatus. IR spectra were recorded on a JASCO FT/IR-4100 spectrometer and absorbance bands are reported in wavenumber (cm−1). 1H NMR spectra were recorded on JEOL JNM-AL 300 and 400 (300 and 400 MHz) spectrometer or JEOL JNM-ECS 400 (400 MHz) spectrometer. Chemical shifts are reported relative to internal standard (tetramethylsilane at δH 0.00, CDCl3 at δH 7.26). Data are presented as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant and integration. 13C NMR spectra were recorded on JEOL JNM-ECA 400 (100 MHz) spectrometer. Chemical shifts are reported relative to internal standard (CDCl3 at δ 77.00). Mass spectra were recorded on a JEOL JMS 700 instrument with a direct inlet system. Column chromatography was carried out on Kanto silica gel 60 N (40–50 mesh). Analytical thin layer chromatography (TLC) was carried out on Merck Kieselgel 60 F254 plates with visualization by ultraviolet, anisaldehyde stain solution or phosphomolybdic acid stain solution. All non-aqueous reactions were carried out in flame-dried glassware under argon atmosphere unless otherwise noted. Reagents were purchased from TCI, nacalai tesque, Kanto chemical, FUJIFILM Wako chemicals or Sigma-Aldrich. Reagents and solvents were used without purification. Substrate 4 was synthesized by a reported method [12], and spectra data agreed with the literature values.
3.2. Preparation and Characterization of Novel Compounds
3.2.1. General Procedure for the Synthesis of Alkyl 5-Methyl-2-oxo-2,3-dihydro-1H-benzo[b]azepine-1-carboxylate (5a,b,d,e)
A solution of 4 in THF was cooled to −78 °C, n-BuLi (1.6 M in n-hexane) was added, and the mixture was stirred for 30 min. Alkyl chloroformate was added to the mixture, and the mixture was stirred for 20 min at same temperature. After confirming the disappearance of substrate 4 by TLC, saturated the NH4Cl aqueous solution was added. The reaction mixture was extracted with CH2Cl2. The organic layer was washed with a saturated aqueous NaCl solution, dried with anhydrous MgSO4, filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 5.
Methyl 5-Methyl-2-oxo-2,3-dihydro-1H-benzo[b]azepine-1-carboxylate (5a)
4 (0.10 g, 0.58 mmol), THF (2.3 mL), n-BuLi (0.73 mL, 1.2 mmol), methyl chloroformate (90 μL, 1.2 mmol), eluent (AcOEt/n-hexane = 1/2), 5a (0.14 g, quant.). Pale yellow oil. IR (KBr): 3547, 2954, 1779, 1224 cm−1. 1H-NMR (400 MHz, CDCl3): δ 2.22 (3H, s), 2.69 (1H, brs), 3.04 (1H, brs), 3.79 (3H s), 5.90 (1H, tq, J = 7.1, 1.4 Hz), 7.24–7.27 (1H, m), 7.30–7.37 (2H, m), 7.47–7.49 (1H, m). 13C-NMR (100 MHz, CDCl3): δ 20.8, 37.4, 54.1, 121.6, 126.5, 127.0, 127.2, 127.6, 135.5, 135.9, 136.4, 153.9, 172.5. MS (EI): m/z (%) 231 (M+, 13), 190 (11), 189 (100), 144 (37). HRMS (EI): m/z calcd for C13H13NO3 [M+] 231.0895, found 231.0892.
Ethyl 5-Methyl-2-oxo-2,3-dihydro-1H-benzo[b]azepine-1-carboxylate (5b)
4 (0.13 g, 0.74 mmol), THF (7.4 mL), n-BuLi (0.69 mL, 1.1 mmol), ethyl chloroformate (0.14 mL, 1.5 mmol), eluent (AcOEt/n-hexane = 1/2), 5b (0.13 g, 70%). Pale yellow oil. IR (KBr): 3381, 2981, 1777, 1219 cm−1. 1H-NMR (300 MHz, CDCl3): δ 1.25 (3H, t, J = 7.1 Hz), 2.21 (3H s), 2.69 (1H, brs), 3.02 (1H, brs), 4.25 (2H, brq, J = 6.2 Hz), 5.90 (1H, tq, J = 7.0, 1.5 Hz), 7.21–7.34 (3H, m), 7.46–7.49 (1H, m). 13C-NMR (100 MHz, CDCl3): δ 14.4, 25.0, 34.3, 61.2, 115.2, 119.5, 121.4, 123.3, 128.0, 128.2, 134.4, 137.9, 153.9, 177.6. MS (EI): m/z (%) 245 (M+, 16), 204 (12), 203 (100), 173 (10), 172 (17), 158 (14), 144 (55), 143 (12), 131 (19), 130 (27). HRMS (EI): m/z calcd for C14H15NO3 [M+] 245.1052, found 245.1050.
tert-Butyl 5-Methyl-2-oxo-2,3-dihydro-1H-benzo[b]azepine-1-carboxylate (5c)
A solution of 4 (0.31 g, 1.8 mmol) in CH2Cl2 (3.0 mL) was added Boc2O (0.62 mL, 2.7 mmol), DMAP (44 mg, 0.36 mmol) and Et3N (0.75 mL, 5.4 mmol) and stirred at rt for 24 h. After confirming the consumption of substrate 4 by TLC, saturated aqueous solution of NaHCO3 was added to the reaction mixture. After extraction with CH2Cl2, the organic layer was washed with a saturated aqueous NaCl solution, dried with anhydrous MgSO4, filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (AcOEt/n-hexane = 1/2) to obtain 5c (0.44 g, 90%) as pale yellow oil. IR (KBr): 3374, 2979, 2253, 1774, 1240 cm−1. 1H-NMR (400 MHz, CDCl3): δ 1.44 (9H, s), 2.21 (3H, s), 2.66 (1H, brs), 2.98 (1H, brs), 5.90 (1H, tq, J = 6.8, 1.4 Hz), 7.22–7.31 (3H, m), 7.45–7.47 (1H, m). 13C-NMR (100 MHz, CDCl3): δ 20.9, 27.6 (3C), 37.2, 83.6, 121.8, 126.4, 126.5, 126.6, 127.3, 135.1, 136.1 136.3, 151.9, 171.7. MS (EI): m/z (%) 273 (M+, 7), 174 (12), 173 (100), 172 (14), 158 (18), 144 (23), 131 (34), 130 (17), 57 (33). HRMS (EI): m/z calcd for C16H19NO3 [M+] 273.1365, found 273.1370.
Benzyl 5-Methyl-2-oxo-2,3-dihydro-1H-benzo[b]azepine-1-carboxylate (5d)
4 (0.19 g, 1.1 mmol), THF (11 mL), n-BuLi (1.0 mL, 1.6 mmol), benzyl chloroformate (0.84 mL, 2.2 mmol), eluent (AcOEt/n-hexane = 1/2), 5d (0.31 g, 92%). Pale yellow oil. IR (KBr): 3535, 2949, 1779, 1218 cm−1. 1H-NMR (400 MHz, CDCl3): δ 2.20 (3H, s), 2.69 (1H, brs), 3.04 (1H, brs), 5.16 (1H, brs), 5.25 (1H, brs), 5.89 (1H, tq, J = 6.9, 1.4 Hz), 7.15 (1H, dd, J = 8.0, 1.3 Hz), 7.14–7.37 (7H, m), 7.46 (1H, dd, J = 7.8, 1.6 Hz). 13C-NMR (100 MHz, CDCl3): δ 20.8, 37.4, 68.6, 121.5, 126.5, 126.9, 127.0, 127.3, 127.4, 128.0, 128.2, 128.4 (2C), 134.9, 135.5, 135.8, 136.4, 153.1, 172.4. MS (EI): m/z (%) 307 (M+, 14), 266 (11), 265 (59), 221 (48), 172 (24), 157 (18), 144 (15), 91 (100). HRMS (EI): m/z calcd for C19H17NO3 [M+] 307.1208, found 307.1206.
Allyl 5-Methyl-2-oxo-2,3-dihydro-1H-benzo[b]azepine-1-carboxylate (5e)
4 (0.10 g, 0.58 mmol), THF (5.8 mL), n-BuLi (0.54 mL, 0.87 mmol), allyl chloroformate (0.12 mL, 1.2 mmol), eluent (AcOEt/n-hexane = 1/2), 5e (0.12 g, 78%). Pale yellow oil. IR (KBr): 3370, 2979, 1773, 1240 cm−1. 1H-NMR (300 MHz, CDCl3): δ 2.21 (3H, s), 2.70 (1H, brs), 3.03 (1H, brs), 4.67 (2H, brs), 5.18–5.29 (2H, m), 5.80–5.93 (2H, m), 7.25–7.37 (3H, m), 7.45–7.49 (1H, m). 13C-NMR (100 MHz, CDCl3): δ 25.1, 34.6, 65.9, 118.1, 119.8, 121.7, 123.6, 128.1, 128.2, 132.4, 134.3, 137.7, 153.6, 162.1, 177.8. MS (EI): m/z (%) 257 (M+, 16), 216 (14), 215 (100), 184 (11), 172 (19), 171 (10), 170 (21), 156 (12), 144 (35), 143 (12), 130 (19). HRMS (EI): m/z calcd for C15H15NO3 [M+] 257.1052, found 257.1052.
3.2.2. General Procedure for the Synthesis of (Z)-4-{2-[(Alkoxycarbonyl)amino]phenyl}pent-3-enoic acids 2
A solution of 5 in THF and H2O was added NaOH and stirred at room temparature for 2 h. After confirming the consumption of substrate 5 by TLC, the reaction mixture was acidified with 10% HCl and extracted with AcOEt. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered. The filtrate was concentrated under reduced pressure to obtain 2.
(Z)-4-{2-[(Methoxycarbonyl)amino]phenyl}pent-3-enoic Acid (2a)
5a (0.11 g, 0.49 mmol), THF (4.9 mL), H2O (4.9 mL), NaOH (58 mg, 2.5 mmol), 2a (0.15 g, quant.). Yellow oil. IR (KBr): 3311, 2959, 1710, 1523, 1219 cm−1. 1H-NMR (500 MHz, CDCl3): δ 2.00 (3H, s), 2.85 (2H, d, J = 7.3 Hz), 3.73 (3H, s), 5.78 (1H, td, J = 7.3, 1.2 Hz), 6.98–7.00 (2H, m), 7.06 (1H, t, J = 7.3 Hz), 7.28 (1H, t, J = 8.8 Hz), 8.02 (1H, brs). 13C-NMR (125 MHz, CDCl3): δ 25.0, 34.2, 52.2, 119.6, 121.4, 123.5, 128.0, 128.3, 130.1, 134.4, 138.1, 154.2, 177.2. MS (FAB): m/z (%) 250 (M+H+, 64), 204 (11). HRMS (FAB): m/z calcd for C13H16NO4 [M+H+] 250.1079, found 250.1078.
(Z)-4-{2-[(Ethoxycarbonyl)amino]phenyl}pent-3-enoic Acid (2b)
5b (0.23 g, 0.92 mmol), THF (9.2 mL), H2O (9.2 mL), NaOH (0.18 g, 4.6 mmol), 2b (0.23 g, 95%). Pale yellow oil. IR (KBr): 3396, 2979, 1731, 1215 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.27 (3H, t, J = 7.3 Hz), 1.99 (3H s), 2.83 (2H, d, J = 6.4 Hz), 4.18 (2H, q, J = 7.0 Hz), 5.77 (1H, brs), 6.98–7.06 (2H, m), 7.27 (1H, t, J = 7.0 Hz), 8.02 (1H, brs). 13C-NMR (125 MHz, CDCl3): δ 14.4, 25.1, 34.5, 61.3, 119.6, 121.6, 123.4, 128.1, 128.2, 130.2, 134.4, 137.8, 154.0, 177.8. MS (EI): m/z (%) 263 (M+, 15), 204 (22), 203 (100), 144 (69), 132 (11), 131 (21), 130 (31). HRMS (EI): m/z calcd for C14H17NO4 [M+] 263.1158, found 263.1154.
(Z)-4-{2-[(tert-Butoxycarbonyl)amino]phenyl}pent-3-enoic Acid (2c)
5c (0.27 g, 0.97 mmol), THF (9.7 mL), H2O (9.7 mL), NaOH (0.20 g, 4.9 mmol), 2c (0.28 g, 99%). Yellow solids. mp: 126–128 °C, IR (KBr): 3410, 2977, 1730, 1164 cm−1. 1H-NMR (300 MHz, CDCl3): δ 1.49 (9H, s), 2.01 (3H, s), 2.86 (2H, d, J = 6.2 Hz), 5.79 (1H, td, J = 7.3, 1.4 Hz), 6.71 (1H, s), 6.96–7.05 (2H, m), 7.23–7.29 (1H, m), 8.01 (1H, d, J = 8.0 Hz). 13C-NMR (100 MHz, CDCl3): δ 25.0, 28.2 (3C), 34.3, 80.5, 119.6, 121.3, 123.0, 128.0, 128.2, 129.8, 134.8, 138.0, 153.0, 177.4. MS (FAB): m/z (%) 292 (M+H+, 42), 236 (68), 192 (45), 146 (23). HRMS (FAB): m/z calcd for C16H22NO4 [M+H+] 292.1549, found 292.1544.
(Z)-4-{2-[(Benzyloxycarbonyl)amino]phenyl}pent-3-enoic Acid (2d)
5d (0.12 g, 0.37 mmol), THF (3.7 mL), H2O (3.7 mL), NaOH (74 mg, 1.9 mmol), 2d (0.13 g, quant.). Pale yellow oil. IR (KBr): 3401, 2965, 1736, 1216 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.98 (3H, s), 2.81 (2H, d, J = 7.0 Hz), 5.16 (2H, brd, J = 9.5 Hz), 5.74 (1H, qt, J = 1.5, 8.0 Hz), 6.98 (1H, d, J = 7.0 Hz), 7.05 (1H, t, J = 7.3 Hz), 7.25–7.39 (6H, m), 8.06 (1H, brs). 13C-NMR (125 MHz, CDCl3): δ 25.1, 34.3, 66.9, 119.7, 121.4, 123.5, 127.0, 128.1, 128.2, 128.3 (2C), 128.5 (2C), 130.1, 134.3, 136.1, 137.9, 153.5, 177.6. MS (EI): m/z (%) 325 (M+, 10), 265 (12), 190 (40), 175 (10), 144 (54), 130 (14), 91 (100). HRMS (EI): m/z calcd for C19H19NO4 [M+] 325.1314, found 325.1312.
(Z)-4-{2-[(Allyloxycarbonyl)amino]phenyl}pent-3-enoic Acid (2e)
5e (0.18 g, 0.65 mmol), THF (6.5 mL), H2O (6.5 mL), NaOH (0.13 g, 3.3 mmol), 2e (0.14 g, 79%). Pale yellow oil. IR (KBr): 3393, 2940, 1732, 1213 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.98 (3H, s), 2.82 (2H, brs), 4.63 (2H, d, J = 4.5 Hz), 5.21 (1H, d, J = 10.5 Hz), 5.32 (1H, d, J = 17.0 Hz), 5.76 (1H, brs), 5.89–5.98 (1H, m), 6.96–7.08 (2H, m), 7.25–7.28 (1H, m), 8.01 (1H, brs). 13C-NMR (125 MHz, CDCl3): δ 25.1, 34.7, 65.9, 118.1, 119.7, 121.8, 123.6, 128.1, 128.2, 130.4, 132.5, 134.3, 137.6, 153.6, 177.8. MS (EI): m/z (%) 275 (M+, 21), 216 (27), 215 (100), 190 (21), 172 (15), 170 (11), 145 (11), 144 (86), 131 (21), 130 (40). HRMS (EI): m/z calcd for C15H17NO4 [M+] 275.1158, found 275.1159.
Methyl (Z)-4-{2-[(tert-Butoxycarbonyl)amino]phenyl}pent-3-enoate (6)
A solution of 5c (0.27 g, 1.0 mmol) in MeOH (10 mL) was added NaOMe (54 mg, 5.0 mmol) and stirred at rt for 1.5 h. After confirming the consumption of substrate 5c by TLC, the reaction mixture was acidified with 10% HCl and extracted with AcOEt, the organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and the filtrate was concentrated under reduced pressure to obtain 6 (0.28 g, 93%). Colorless oil. IR (KBr): 3399, 2978, 1731, 1516, 1160 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.51 (9H, s), 2.01 (3H, s), 2.82 (2H, d, J = 7.5 Hz), 3.65 (3H, s), 5.81 (1H, t, J = 7.5 Hz), 6.72 (1H, brs), 6.96–7.04 (2H, m), 7.25 (1H, t, J = 8.0 Hz), 8.03 (1H, d, J = 8.0 Hz). 13C-NMR (125 MHz, CDCl3): δ 25.0, 28.3 (3C), 34.4, 51.9, 80.3, 119.5, 121.9, 123.0, 128.0, 128.1, 129.9, 134.8, 137.5, 153.0, 172.2. MS (FAB): m/z (%) 306 (M+H+, 45), 250 (69), 206 (100), 146 (29), 131 (27). HRMS (FAB): m/z calcd for C17H24NO4 [M+H+] 306.1705, found 306.1712.
3.2.3. Methyl 3-Iodo-3-(4-methyl-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-4-yl)propanoate (7)
To a solution of 6 (30 mg, 0.10 mmol) in a mixture of MeCN/MeOH (1.2 mL, 20/1) was added a solution of NIS (3.4 mg, 0.15 mmol) in MeCN (0.30 mL) at −45 °C. The reaction mixture was stirred and allowed to room temperature for 5 h. After confirming the consumption of substrate 6 by TLC, the reaction was quenched with aqueous solution of Na2S2O3. After extraction with AcOEt, the organic layer was washed with brine, dried with anhydrous MgSO4, filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (AcOEt/n-hexane = 1/2) to obtain 7 (33 mg, 89%) as a pale yellow oil. IR (KBr): 3619, 3019, 1730, 1046 cm−1. 1H-NMR (400 MHz, CDCl3): δ 1.87 (3H, s), 3.11 (1H, dd, J = 16.5, 10.2 Hz), 3.20 (1H, dd, J = 16.6, 3.9 Hz), 3.70 (3H, s), 4.79 (1H, dd, J = 10.2, 3.6 Hz), 6.91 (1H d, J = 7.8 Hz), 7.08 (1H, t, J = 7.8 Hz), 7.17 (1H, d, J = 7.3 Hz), 7.30 (1H, t, J = 7.8 Hz), 9.21 (1H, s). 13C-NMR (100 MHz, CDCl3): δ 24.5, 33.8, 40.9, 52.3, 85.5, 115.1, 122.2, 123.6, 124.4, 129.9, 133.9, 151.0, 170.7. MS (EI): m/z (%) 375 (M+, 13), 162 (100), 144 (27), 130 (12). HRMS (EI): m/z calcd for C13H14NO4I [M+] 374.9968, found 374.9960.
3.2.4. General Procedure for the Oxidative Cyclization of 2
To a solution of 2 in MeCN was added Pd(OAc)2 and PhI(OAc)2. The reaction mixture was stirred under reflux for 6 h. After confirming the consumption of substrate 2 by TLC, the reaction was quenched with aqueous solution of Na2S2O3. After extraction with AcOEt, the organic layer was washed with brine, dried with anhydrous MgSO4, filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to obtain 3.
Methyl (3aR*,8bR*)-8b-Methyl-2-oxo-2,3,3a,8b-tetrahydro-4H-furo[3,2-b]indole-4-carboxylate (3a)
2a (78 mg, 0.31 mmol), MeCN (3.1 mL), Pd(OAc)2 (14 mg, 60 μmol), PhI(OAc)2 (0.15 g, 0.47 mmol), eluent (AcOEt/n-hexane = 1/2), 3a (59 mg, 78%). Yellow solid. mp: 160–162 °C. IR (KBr): 3021, 1776, 1716 cm−1. 1H-NMR (400 MHz, CDCl3): δ 1.82 (3H, s), 2.96 (1H, d, J = 19.5 Hz), 3.23 (1H, dd, J = 19.5, 8.2 Hz), 3.89 (3H s), 4.60 (1H, d, J = 6.3 Hz), 7.11 (1H, t, J = 7.3 Hz), 7.36 (1H, d, J = 7.8 Hz), 7.40 (1H, d, J = 7.8 Hz), 7.91 (1H, brs). 13C-NMR (100 MHz, CDCl3): δ 24.4, 36.3, 53.1, 64.6, 89.4, 115.2, 123.7, 124.1, 129.8, 131.1, 141.8, 152.6, 174.1. MS (EI): m/z (%) 247 (M+, 83), 202 (60), 188 (100), 158 (35), 146 (17), 144 (83), 143 (21). HRMS (EI): m/z calcd for C13H13NO4 [M+] 247.0845, found 247.0847.
Ethyl (3aR*,8bR*)-8b-Methyl-2-oxo-2,3,3a,8b-tetrahydro-4H-furo[3,2-b]indole-4-carboxylate (3b)
2b (0.23 g, 0.87 mmol), Pd(OAc)2 (40 mg, 0.18 mmol), PhI(OAc)2 (0.42 g, 1.3 mmol), MeCN (8.7 mL), eluent (AcOEt/n-hexane = 1/2), 3b (0.10 g, 56%). Yellow solid. mp: 116–118 °C. IR (KBr): 3532, 2980, 1778, 1713, 1225 cm−1. 1H-NMR (400 MHz, CDCl3): δ 1.38 (3H, brs), 1.83 (3H, s), 2.98 (1H, d, J = 19.0 Hz), 3.23 (1H, dd, J = 19.2, 8.2 Hz), 4.35 (2H, brq, J = 6.6 Hz), 4.60 (1H, dd, J = 8.2, 2.0 Hz), 7.10 (1H, t, J = 7.3 Hz), 7.36 (1H, d, J = 7.7 Hz), 7.41 (1H, d, J = 8.2 Hz), 7.93 (1H, brs). 13C-NMR (100 MHz, CDCl3): δ 14.5, 24.4, 36.5, 62.2, 64.6, 89.4, 115.2, 123.6, 124.1, 130.0, 131.1, 141.7, 152.2, 174.3. MS (EI): m/z (%) 261 (M+, 71), 188 (42), 146 (23), 144 (100), 130 (30). HRMS (EI): m/z calcd for C14H15NO4 [M+] 261.1001, found 261.0999.
tert-Butyl (3aR*,8bR*)-8b-Methyl-2-oxo-2,3,3a,8b-tetrahydro-4H-furo[3,2-b]indole-4-carboxylate (3c)
2c (0.16 g, 0.55 mmol), Pd(OAc)2 (25 mg, 0.11 mmol), PhI(OAc)2 (0.27 g, 0.83 mmol), MeCN (5.5 mL), eluent (AcOEt/n-hexane = 1/2), 3c (60 mg, 38%). Yellow solid. mp: 172–174 °C. IR (KBr): 3534, 2976, 1778, 1711, 1381, 1166 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.58 (9H, s), 1.82 (3H, s), 2.99 (1H, brs), 3.20 (1H, dd, J = 19.5, 8.0 Hz), 4.54 (1H, brs), 7.08 (1H, t, J = 7.5 Hz), 7.35 (1H, t, J = 8.0 Hz), 7.39 (1H, d, J = 7.5 Hz), 7.92 (1H, brs). 13C-NMR (100 MHz, CDCl3): δ 24.4, 28.3 (3C), 36.7, 64.6, 82.1, 89.5, 115.1, 123.3, 124.1, 129.6, 131.0, 142.3, 152.1, 174.5. MS (EI): m/z (%) 289 (M+, 27), 233 (100), 189 (41), 188 (23), 144 (58), 57 (68). HRMS (EI): m/z calcd for C16H19NO4 [M+] 289.1314, found 289.1311.
Benzyl (3aR*,8bR*)-8b-Methyl-2-oxo-2,3,3a,8b-tetrahydro-4H-furo[3,2-b]indole-4-carboxylate (3d)
2d (0.28 g, 0.85 mmol), Pd(OAc)2 (38 mg, 0.17 mmol), PhI(OAc)2 (0.41 g, 1.3 mmol), MeCN (8.5 mL)), eluent (AcOEt/n-hexane = 1/2), 3d (90 mg, 33%). Yellow solid. mp: 140–142 °C. IR (KBr): 3532, 2974, 1777, 1719, 1402, 1226 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.81 (3H, s), 2.93 (1H, brs), 3.17 (1H, brs), 4.60 (1H, brs), 5.27 (1H, brs), 5.31 (1H, brs), 7.10 (1H, brs), 7.37-7.41 (7H, m), 7.96 (1H, brs). 13C-NMR (100 MHz, CDCl3): δ 24.5, 36.6, 64.6, 67.9, 89.6, 115.3, 123.8, 124.1, 128.3 (2C), 128.6, 128.8 (2C), 129.8, 131.1, 135.4, 141.7, 151.8, 174.1. MS (EI): m/z (%) 323 (M+, 30), 279 (16), 144 (18), 91 (100). HRMS (EI): m/z calcd for C19H17NO4 [M+] 323.1158, found 323.1155.
Allyl (3aR*,8bR*)-8b-Methyl-2-oxo-2,3,3a,8b-tetrahydro-4H-furo[3,2-b]indole-4-carboxylate (3e)
2e (0.14 g, 0.52 mmol), Pd(OAc)2 (22 mg, 0.10 mmol), PhI(OAc)2 (0.25 g, 0.78 mmol), MeCN (5.2 mL), eluent (AcOEt/n-hexane = 1/2), 3e (41 mg, 29%). Yellow solid. mp: 70–72 °C. IR (KBr): 3533, 2933, 1778, 1720, 1397, 1226 cm−1. 1H-NMR (300 MHz, CDCl3): δ 1.83 (3H, s), 2.98 (1H, d, J = 18.0 Hz), 3.24 (1H, dd, J = 18.0, 8.1 Hz), 4.62 (1H dd, J = 8.1, 2.7 Hz), 4.77 (2H, brs), 5.32 (1H, d, J = 10.5 Hz), 5.39 (1H, d, J = 17.1 Hz), 5.95–6.05 (1H, m), 7.11 (1H, t, J = 7.5 Hz), 7.28–7.43 (2H, m), 7.90 (1H, brs). 13C-NMR (125 MHz, CDCl3, major): δ 24.5, 36.6, 64.6, 66.7, 89.5, 115.3, 119.1, 123.8, 124.1, 129.8, 131.2, 131.8, 141.9, 151.7, 174.2. MS (EI): m/z (%) 273 (M+, 100), 188 (22), 146 (56), 144 (85), 41 (20). HRMS (EI): m/z calcd for C15H15NO4 [M+] 273.1001, found 273.1000.
4. Conclusions
We investigated the construction of the furoindolin-2-one structure corresponding to the A to C ring system of lapidilectine B (1). The furoindolin-2-one structure was constructed directly by double functionalization of the internal alkene with carboxyl and carbamoyl groups under oxidative conditions using PhI(OAc)2. Furthermore, we succeeded in obtaining the desired product in higher yield with a palladium(II) catalyst under oxidative conditions. We are currently synthesizing more advanced substrates for the synthesis of lapidilectine B (1).
Author Contributions
Conceptualization, K.H.; methodology, K.H. and K.M.; formal analysis, K.H., K.M., T.A. and M.I.; investigation, K.M. and T.A.; writing—original draft preparation, K.H. and K.M.; writing—review and editing, K.H., M.I. and S.S.; supervision, S.S.; project administration, K.H. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not available.
Informed Consent Statement
Not available.
Data Availability Statement
The data presented in this study are available in this article.
Acknowledgments
We are grateful to Koseki, T. and Yamada, S. at the Analytical Center of Meiji Pharmaceutical University for performing mass spectrometry and NMR measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Awang, K.; Sévenet, T.; Hamid, A.; Hadi, A.; David, B.; Païs, M. Lapidilectine A and Lapidilectine B, two new alkaloids from Kopsia lapidilecta. Tetrahedron Lett. 1992, 33, 2493–2496. [Google Scholar] [CrossRef]
- Yap, W.-S.; Gan, C.-Y.; Low, Y.-Y.; Choo, Y.-M.; Etoh, T.; Hayashi, M.; Komiyama, K.; Kam, T.-S. Grandilodines A–C, biologically active indole alkaloids from Kopsia grandifolia. J. Nat. Prod. 2011, 74, 1309–1312. [Google Scholar] [CrossRef]
- Pearson, W.H.; Mi, Y.; Lee, I.Y.; Stoy, P. Total synthesis of the Kopsia lapidilecta alkaloid (±)-Lapidilectine B. J. Am. Chem. Soc. 2001, 123, 6724–6725. [Google Scholar] [CrossRef]
- Pearson, W.H.; Lee, I.Y.; Mi, Y.; Stoy, P. Total synthesis of the Kopsia lapidilecta alkaloid (±)-Lapidilectine B. J. Org. Chem. 2004, 69, 9109–9122. [Google Scholar] [CrossRef]
- Nakajima, M.; Arai, S.; Nishida, A. Total syntheses of (+)-Grandilodine C and (+)-Lapidilectine B and determination of their absolute stereochemistry. Angew. Chem. Int. Ed. 2016, 55, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, M.; Geng, Q.; Ma, D. Total synthesis of Lapidilectine B enabled by manganese(III)-mediated oxidative cyclization of indoles. Chem. Eur. J. 2018, 24, 6547–6550. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S. Total synthesis of (±)-Psychotrimine. J. Am. Chem. Soc. 2008, 130, 10886–10887. [Google Scholar] [CrossRef]
- Newhouse, T.; Lewis, C.A.; Eastman, K.J.; Baran, P.S. Scalable total syntheses of N-linked tryptamine dimers by direct indole-aniline coupling: Psychotrimine and Kapakahines B and F. J. Am. Chem. Soc. 2010, 132, 7119–7137. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Chang, H.-T.; Sharpless, K.B. Catalytic asymmetric aminohydroxylation (AA) of olefins. Angew. Chem. Int. Ed. 1996, 35, 451–454. [Google Scholar] [CrossRef]
- Muñiz, K. Advancing palladium-catalyzed C-N bond formation: Bisindoline construction from successive amide transfer to internal alkenes. J. Am. Chem. Soc. 2007, 129, 14542–14543. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, S.H.; Chang, S. Intramolecular oxidative diamination and aminohydroxylation of olefins under metal-free conditions. Org. Lett. 2012, 14, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Candeloro, V.; Bowie, J.H. 1H-1-Benzazepines. The reactions of laevulinic acid and β-benzoylpropionic acid with aniline and methoxyanilines. Aust. J. Chem. 1978, 31, 2031–2037. [Google Scholar] [CrossRef]
- Muñiz, K. High-oxidation-state palladium catalysis: New reactivity for organic synthesis. Angew. Chem. Int. Ed. 2009, 48, 9412–9423. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jiang, H. Palladium-catalyzed oxidation of unsaturated hydrocarbons using molecular oxygen. Acc. Chem. Res. 2012, 45, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Kočovský, P.; Bäckvall, J.-E. The syn/anti-dichotomy in the palladium-catalyzed addition of nucleophiles to alkenes. Chem. Euro. J. 2015, 21, 36–56. [Google Scholar] [CrossRef] [Green Version]
- Yin, G.; Mu, X.; Liu, G. Palladium(II)-catalyzed oxidative difunctionalization of alkenes: Bond forming at a high-valent palladium center. Acc. Chem. Res. 2016, 49, 2413–2423. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).