
Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
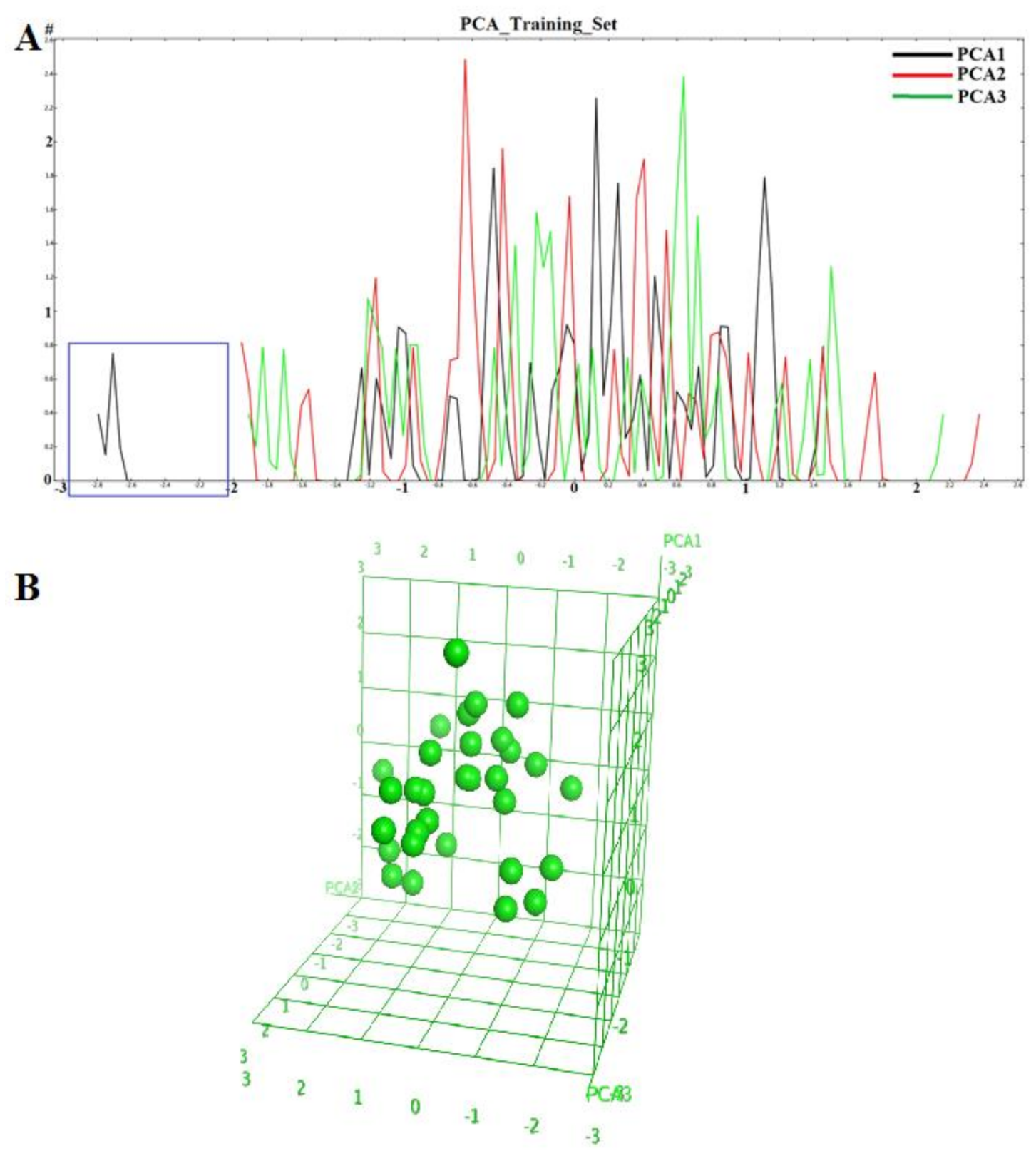
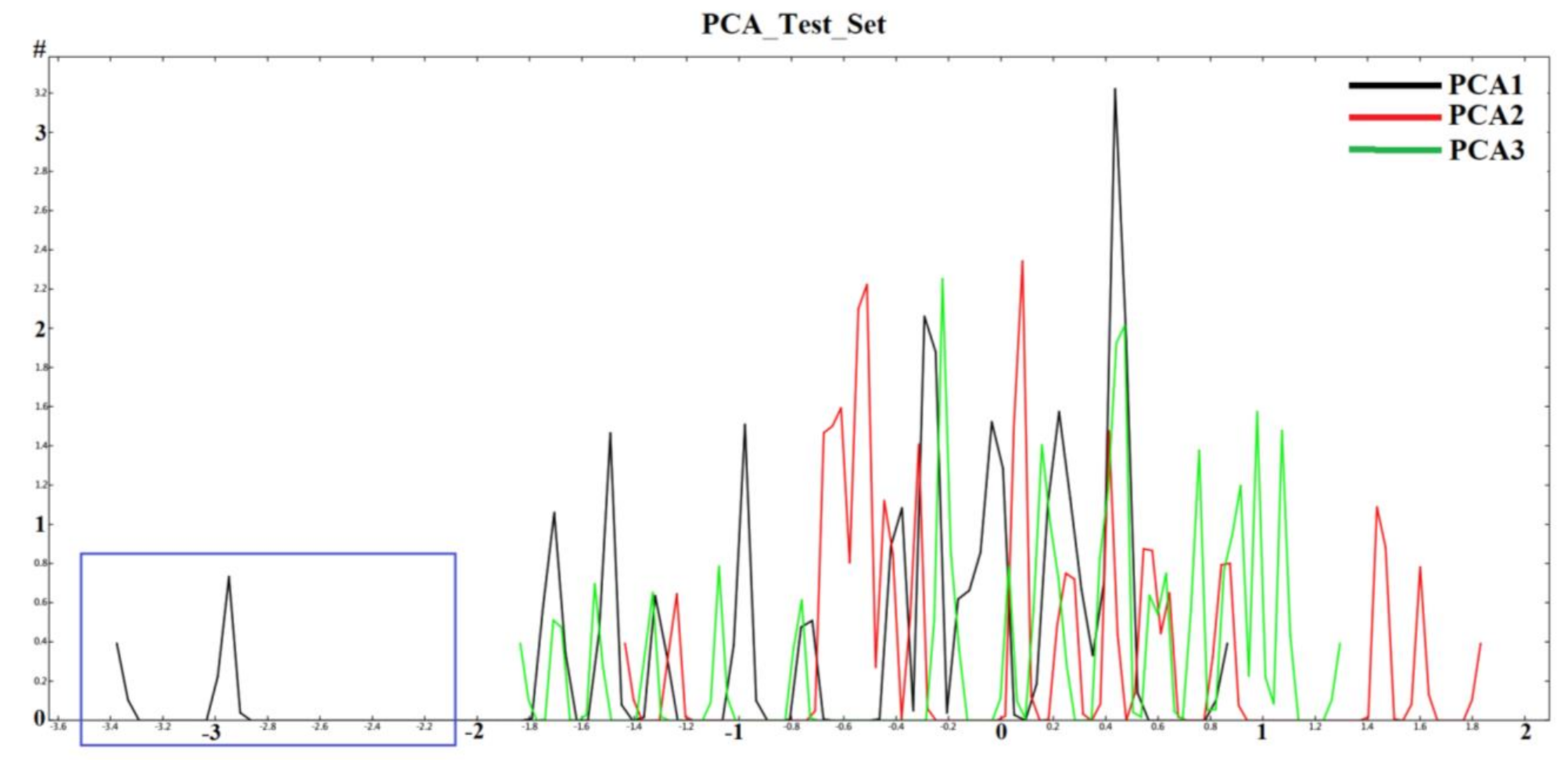
2.1. Diversity Analysis of Dataset(s)
2.2. Molecular Docking
2.3. Analysis of BFE
2.4. Derived QSAR Models and Their Validation
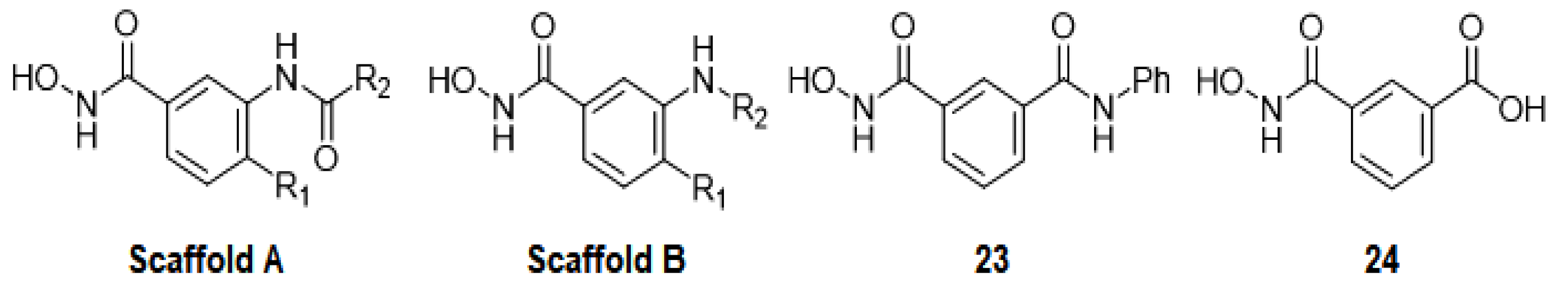
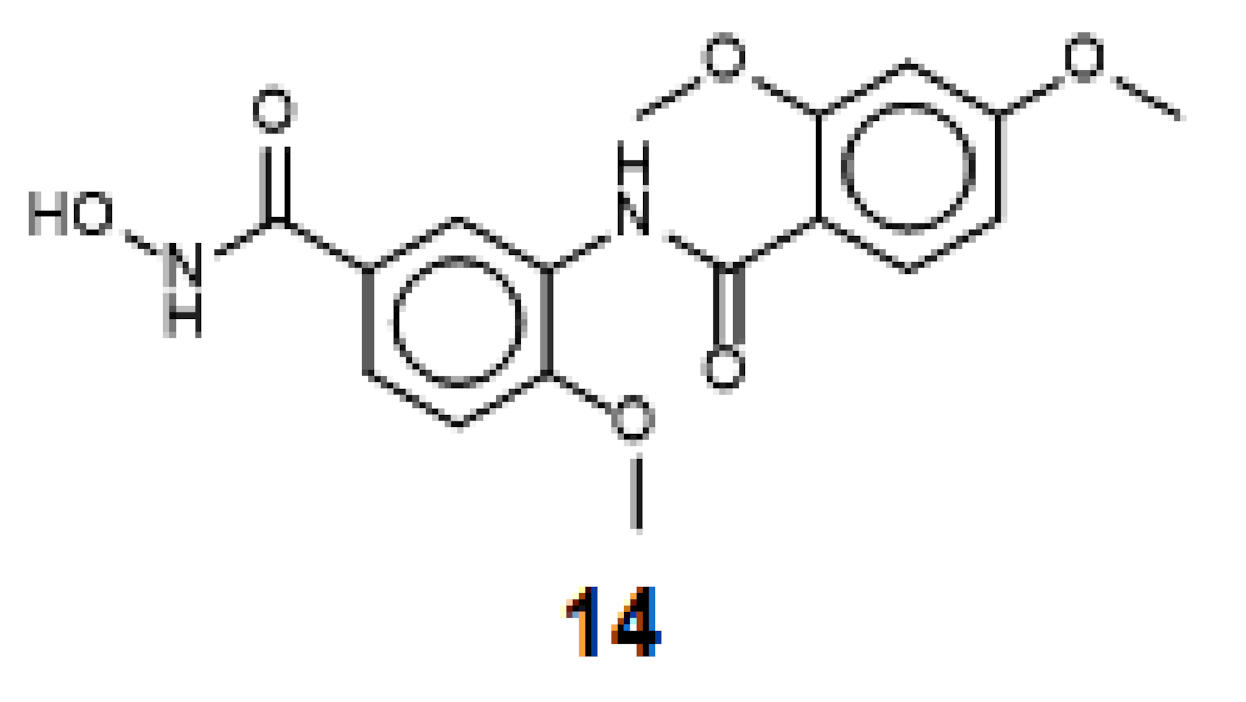
2.5. Evaluation of Novel Designed smHDAC8 Inhibitors
3. Materials and Methods
3.1. Dataset Source, Preparation and Analysis
3.1.1. Dataset
3.1.2. Calculation of Molecular Descriptors and Dataset Diversity Analysis
3.2. Molecular Docking
3.2.1. Ligand Preparation
3.2.2. Protein Preparation
3.2.3. Grid Generation and Docking
3.3. Binding Free Energy (BFE) Calculations
3.3.1. Ligand and Ligand–Protein Complex Preparation
3.3.2. MM-PB/GBSA and QM/MM Based BFE Prediction
3.4. Quantitative Structure–Activity Relationship (QSAR) Model Development and Selection
3.5. Test Set Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- McManus, D.P.; Dunne, D.W.; Sacko, M.; Utzinger, J.; Vennervald, B.J.; Zhou, X.N. 1236 Schistosomiasis. Nat. Rev. Dis. Primers 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, R.S.; Esmat, G.; El-Baz, T. Human schistosomiasis: Clinical perspective. J. Adv. Res. 2013, 4, 433–444. [Google Scholar] [CrossRef]
- World Health Organization. Schistosomiasis Facts Sheet. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 18 November 2020).
- King, C.H.; Sutherland, L.J.; Bertsch, D. Systematic review and meta-analysis of the impact of chemical-based mollusciciding for control of Schistosoma mansoni and S. haematobium transmission. PLoS Negl. Trop. Dis. 2015, 9, e0004290. [Google Scholar] [CrossRef]
- van der Werf, M.J.; Bosompem, K.M.; de Vlas, S.J. Schistosomiasis control in Ghana: Case management and means for diagnosis and treatment within the health system. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 146–152. [Google Scholar] [CrossRef]
- Gray, D.J.; Ross, A.G.; Li, Y.S.; McManus, D.P. Diagnosis and management of schistosomiasis. BMJ 2011, 342, d2651. [Google Scholar] [CrossRef] [PubMed]
- Olveda, D.U.; Olveda, R.M.; Lam, A.K.; Chau, T.N.; Li, Y.; Gisparil, A.D. Utility of diagnostic imaging in the diagnosis and management of schistosomiasis. Clin. Microbiol. 2014, 3, 142. [Google Scholar] [CrossRef]
- Ricciardi, A.; Ndao, M. Diagnosis of parasitic infections: What’s going on? J. Biomol. Screen. 2015, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Da Paixão Siqueira, L.; Fontes, D.A.F.; Aguilera, C.S.B.; Timóteo, T.R.R.; Ângelos, M.A.; Silva, L.C.P.B.B.; de Melo, C.G.; Rolim, L.A.; da Silva, R.M.F.; Neto, P.J.R. Schistosomiasis: Drugs used and treatment strategies. Acta Trop. 2017, 176, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L.; Basso, A.; Guidi, A. Schistosomiasis control: Praziquantel forever? Mol. Biochem. Parasitol. 2014, 195, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Hailu, G.S.; Robaa, D.; Forgione, M.; Sippl, W.; Rotili, D.; Mai, A. Lysine deacetylase inhibitors in parasites: Past, present, and future perspectives. J. Med. Chem. 2017, 60, 4780–4804. [Google Scholar] [CrossRef]
- Jeffers, V.; Yang, C.; Huang, S.; Sullivan, W.J. Bromodomains in protozoan parasites: Evolution, function, and opportunities for drug development. Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef] [PubMed]
- Zuma, A.A.; Souza, W.D. Histone deacetylases as targets for antitrypanosomal drugs. Future Sci. OA 2018, 4, FSO325. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef]
- Ruijter, A.J.D.; Gennip, A.H.V.; Caron, H.N.; Kemp, S.; Kuilenburg, A.B.V. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.W.; Gallo, C.M.; Smith, J.S. Diversity in the Sir2 family of protein deacetylases. J. Leukoc. Biol. 2004, 75, 939–950. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166. [Google Scholar]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Haque, A.; Jones, M.K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Tran, T.N.; Fairlie, D.P. Towards histone deacetylase inhibitors as new antimalarial drugs. Curr. Pharm. Des. 2012, 18, 3467–3479. [Google Scholar] [CrossRef]
- Ingram, A.K.; Horn, D. Histone deacetylases in Trypanosoma brucei: Two are essential and another is required for normal cell cycle progression. Mol. Microbiol. 2002, 45, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ouaissi, M.; Ouaissi, A. Histone deacetylase enzymes as potential drug targets in cancer and parasitic diseases. J. Biomed. Biotechnol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.J.; Dubois-Abdesselem, F.; Lancelot, J.; Andrade, L.; Oliveira, G. Targeting schistosome histone modifying enzymes for drug development. Curr. Pharm. Des. 2012, 18, 3567–3578. [Google Scholar] [CrossRef]
- Ning, Z.Q.; Li, Z.B.; Newman, M.J.; Shan, S.; Wang, X.H.; Pan, D.S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; De Francesco, R.; Paz, O.G.; Kinzel, O.; Mesiti, G.; Monteagudo, E.; Pescatore, G.; Rowley, M.; Verdirame, M.; et al. A novel series of potent and selective ketone histone deacetylase inhibitors with antitumor activity in vivo. J. Med. Chem. 2008, 51, 2350–2353. [Google Scholar] [CrossRef]
- Lee, J.H.; Mahendran, A.; Yao, Y.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Kim, N.; Ham, W.S.; Breslow, R.; Marks, P.A. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2013, 110, 15704–15709. [Google Scholar] [CrossRef]
- Lee, J.H.; Yao, Y.; Mahendran, A.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Breslow, R.; Marks, P.A. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2015, 112, 12005–12010. [Google Scholar] [CrossRef]
- Ito, T.; Ouchida, M.; Morimoto, Y.; Yoshida, A.; Jitsumori, Y.; Ozaki, T.; Sonobe, H.; Inoue, H.; Shimizu, K. Significant growth suppression of synovial sarcomas by the histone deacetylase inhibitor FK228 in vitro and in vivo. Cancer Lett. 2005, 224, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.B.; Sullivan, E.D.; Chen, Y.; Chan, J.C.; Jennings, P.A.; Fierke, C.A.; Cohen, S.M. Dual-mode HDAC prodrug for covalent modification and subsequent inhibitor release. J. Med. Chem. 2015, 58, 4812–4821. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Yoshida, M.; Horinouchi, S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 87–92. [Google Scholar] [CrossRef]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [CrossRef]
- Day, J.A.; Cohen, S.M. Investigating the selectivity of metalloenzyme inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting metalloenzymes for therapeutic intervention. Chem. Rev. 2018, 119, 1323–1455. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Brosch, G.; Kölle, D.; Scherf, H.; Gerhäuser, C.; Loidl, P. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J. Med. Chem. 1999, 42, 4669–4679. [Google Scholar] [CrossRef]
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef]
- Moradei, O.; Maroun, C.R.; Paquin, I.; Vaisburg, A. Histone deacetylase inhibitors: Latest developments, trends and prospects. Curr. Med. Chem. Anti Cancer Agents 2005, 5, 529–560. [Google Scholar] [CrossRef]
- Paris, M.; Porcelloni, M.; Binaschi, M.; Fattori, D. Histone deacetylase inhibitors: From bench to clinic. J. Med. Chem. 2008, 51, 1505–1529. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [Google Scholar] [CrossRef] [PubMed]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef]
- Marek, M.; Kannan, S.; Hauser, A.T.; Mourao, M.M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef]
- Melesina, J.; Praetorius, L.; Simoben, C.V.; Robaa, D.; Sippl, W. Design of selective histone deacetylase inhibitors: Rethinking classical pharmacophore. Future Med. Chem. 2018, 10, 1537–1540. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A novel class of Schistosoma mansoni histone deacetylase 8 (HDAC8) inhibitors identified by structure-based virtual screening and in vitro testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef]
- Kannan, S.; Melesina, J.; Hauser, A.T.; Chakrabarti, A.; Heimburg, T.; Schmidtkunz, K.; Walter, A.; Marek, M.; Pierce, R.J.; Romier, C.; et al. Discovery of inhibitors of Schistosoma mansoni HDAC8 by combining homology modeling, virtual screening, and in vitro validation. J. Chem. Inf. Model. 2014, 54, 3005–3019. [Google Scholar] [CrossRef]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from Schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A.; Brooks, C.L., III. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Mongan, J.; Simmerling, C.; McCammon, J.A.; Case, D.A.; Onufriev, A. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 2007, 3, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the generalized Born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins: Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Dewar, M.J.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. [Erratum to document cited in CA103 (2): 11627f]. J. Am. Chem. Soc. 1993, 115, 5348. [Google Scholar] [CrossRef]
- Stewart, J.J. Semiempirical molecular orbital methods. Rev. Comput. Chem. 1990, 1, 45–81. [Google Scholar]
- Stewart, J.J. Optimization of parameters for semiempirical methods II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods. III Extension of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Te, Hg, Tl, Pb, and Bi. J. Comput. Chem. 1991, 12, 320–341. [Google Scholar]
- Stewart, J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Stolfa, D.A.; Marek, M.; Lancelot, J.; Hauser, A.T.; Walter, A.; Leproult, E.; Melesina, J.; Rumpf, T.; Wurtz, J.M.; Cavarelli, J.; et al. Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen Schistosoma mansoni. J. Mol. Biol. 2014, 426, 3442–3453. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) MOE. 2013.08; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2019. Available online: https://www.chemcomp.com/Products.htm (accessed on 2 August 2019).
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Carey, R.N.; Wold, S.; Westgard, J.O. Principal component analysis. Alternative to referee methods in method comparison studies. Anal. Chem. 1975, 47, 1824–1829. [Google Scholar] [CrossRef]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef]
- Schrödinger. LigPrep; Release Version 2017-2; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
- Release, S. ConfGen; 2017-2; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Christie, C.; Duarte, J.M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Suite 2017-1 Protein Preparation Wizard; Release 2017-1; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Release 2017-2: Epik; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Glide; Release 2017-2; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- David, L.; Luo, R.; Gilson, M.K. Comparison of generalized Born and Poisson models: Energetics and dynamics of HIV protease. J. Comput. Chem. 2000, 21, 295–309. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Liu, H.Y.; Zou, X. Electrostatics of ligand binding: Parametrization of the generalized born model and comparison with the Poisson− Boltzmann approach. J. Phys. Chem. B 2006, 110, 9304–9313. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M., Jr. Rational design of particle mesh Ewald compatible Lennard-Jones parameters for+ 2 metal cations in explicit solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M., Jr. Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef]
- Najjar, A.; Platzer, C.; Luft, A.; Aßmann, C.A.; Elghazawy, N.H.; Erdmann, F.; Sippl, W.; Schmidt, M. Computer-aided design, synthesis and biological characterization of novel inhibitors for PKMYT1. Eur. J. Med. Chem. 2019, 161, 479–492. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins: Struct. Funct. Bioinform. 2004, 55, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M., Jr. Parameterization of highly charged metal ions using the 12-6-4 LJ-type nonbonded model in explicit water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef]
- Lee, T.S.; Hu, Y.; Sherborne, B.; Guo, Z.; York, D.M. Toward fast and accurate binding affinity prediction with pmemdGTI: An efficient implementation of GPU-accelerated thermodynamic integration. J. Chem. Theory Comput. 2017, 13, 3077–3084. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Sagui, C.; Pedersen, L.G.; Darden, T.A. Towards an accurate representation of electrostatics in classical force fields: Efficient implementation of multipolar interactions in biomolecular simulations. J. Chem. Phys. 2004, 120, 73–87. [Google Scholar] [CrossRef]
- Toukmaji, A.; Sagui, C.; Board, J.; Darden, T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000, 113, 10913–10927. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Wollacott, A.M.; Merz, K.M. Development of a parametrized force field to reproduce semiempirical geometries. J. Chem. Theory Comput. 2006, 2, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Voityuk, A.A.; Bliznyuk, A.A.; Burshtein, K.Y. Revised semiempirical parameters for Br, I, Sn, Hg, and Pb in the MNDO method. J. Struct. Chem. 1987, 28, 9–12. [Google Scholar] [CrossRef]
- Åqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. Des. Sel. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Holst, M.; Saied, F. Multigrid solution of the Poisson—Boltzmann equation. J. Comput. Chem. 1993, 14, 105–113. [Google Scholar] [CrossRef]
- Holst, M.J.; Saied, F. Numerical solution of the nonlinear Poisson–Boltzmann equation: Developing more robust and efficient methods. J. Comput. Chem. 1995, 16, 337–364. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson—Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. FEW: A workflow tool for free energy calculations of ligand binding. J. Comput. Chem. 2013, 34, 965–973. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Lee, M.R.; Duan, Y.; Kollman, P.A. Use of MM-PB/SA in estimating the free energies of proteins: Application to native, intermediates, and unfolded villin headpiece. Proteins Struct. Funct. Bioinform. 2000, 39, 309–316. [Google Scholar] [CrossRef]
- Thalheim, T. cvq2-Package: Calculate the Predictive Squared Correlation Coefficient. 2013. Available online: https://CRAN.R-project.org/package=cvq2 (accessed on 14 February 2020).
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015; Available online: http://www.rstudio.com/ (accessed on 1 February 2020).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Compound Code * | Scaffold | R1 | R2 | smHDAC8 IC50 (nM) | pIC50-smHDAC8 | GLIDE SP_Score |
|---|---|---|---|---|---|---|---|
| TH100 | 13r | A | methoxy | 4-ethoxyphenyl | 305 ± 35 | 6.52 | −7.14 |
| TH101 | 13s | A | methoxy | benzyl | 183 ± 39 | 6.74 | −7.51 |
| TH104 | 13z | A | chloro | 2,4-dichlorophenyl | 191 ± 17 | 6.72 | −7.20 |
| TH31 | 13a | A | hydrogen | phenyl | 468 ± 79 | 6.33 | −8.52 |
| TH33 | 13b | A | methyl | phenyl | 154 ± 0.03 | 6.81 | −7.48 |
| TH39 | 13c | A | methoxy | phenyl | 135 ±0.03 | 6.87 | −7.60 |
| TH60 | 13k | A | methyl | 2-quinolyl | 96 ± 13 | 7.02 | −7.04 |
| TH61 | 13e | A | chloro | phenyl | 67 ± 10 | 7.17 | −7.31 |
| TH65 | 13l | A | methoxy | 4-biphenyl | 293 ±35 | 6.53 | −7.35 |
| TH66 | 13h | A | ethoxy | phenyl | 129 ± 8 | 6.89 | −7.43 |
| TH67 | 13i | A | propoxy | phenyl | 267 ± 49 | 6.57 | −7.51 |
| TH68 | 13n | A | methoxy | 4-chlorophenyl | 146 ± 4 | 6.84 | −7.34 |
| TH69 | 13m | A | methoxy | 4-methoxyphenyl | 106 ± 18 | 6.97 | −7.31 |
| TH74 | 13t | A | chloro | 4-methoxyphenyl | 147 ± 5 | 6.83 | −7.65 |
| TH75 | 13f | A | bromo | phenyl | 150 ± 9 | 6.82 | −7.41 |
| TH76 | 13d | A | fluoro | phenyl | 178 ± 8 | 6.75 | −7.28 |
| TH83 | 13j | A | isopropoxy | phenyl | 220 ± 56 | 6.66 | −8.58 |
| TH85 | 13o | A | methoxy | 2-chlorophenyl | 351 ± 16 | 6.45 | −7.22 |
| TH86 | 13p | A | methoxy | 2,4-dichlorophenyl | 122 ± 19 | 6.92 | −7.98 |
| TH92 | 13za | A | ethoxy | 4-biphenyl | 92 ± 26 | 7.04 | −8.58 |
| TH93 | 13x | A | chloro | 4-chlorophenyl | 235 ± 10 | 6.63 | −7.36 |
| TH94 | 13g | A | trifluoromethyl | phenyl | 140 ± 8 | 6.86 | −7.10 |
| TH95 | 13q | A | methoxy | 3-biphenyl | 290 ± 20 | 6.54 | −7.28 |
| TH96 | 13y | A | chloro | 4-nitropheny | 394 ± 50 | 6.40 | −7.06 |
| TH77 | 13u | A | chloro | 3-benzyloxyphenyl | 378 ± 45 | 6.42 | −9.49 |
| TH78 | 13v | A | chloro | 3-phenoxyphenyl | 396 ± 43 | 6.40 | −9.55 |
| TH81 | 13w | A | chloro | 4-phenoxyphenyl | 620 ± 70 | 6.21 | −8.85 |
| TH58 | 14a | - | - | - | 8210 ± 1300 | 5.09 | −7.51 |
| TH36 | 10c | - | - | - | 1722 ± 910 | 5.76 | −7.41 |
| TH70 | 15a | - | - | - | 268 ± 21 | 6.57 | −7.34 |
| TH71 | 16a | - | - | - | 485 ± 158 | 6.31 | −7.58 |
| TH28 | 10a | B | hydrogen | benzyl | 1040 ± 250 | 5.98 | −7.15 |
| TH32 | 10b | B | hydrogen | cyclohexyl | 3630 ± 620 | 5.44 | −6.80 |
| TH35 | 10e | B | methyl | cyclohexyl | 600 ± 196 | 6.22 | −7.16 |
| Model Number | Method | Frame | Number of Molecules | lm | LOOCV | Leave_3out CV | 3fold CV | Outlier | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r2 | RMSE | q2 | QMSE | q2 | QMSE | q2 | QMSE | |||||
| 1 | 34 | 0.01 | 0.44 | - | - | - | - | - | - | |||
| 6 | AM1/GB1 | Emin2 | 34 | 0.41 | 0.34 | 0.30 | 0.37 | - | - | - | - | |
| 7 | AM1/GB1 | Emin2 | 34 | 0.51 | 0.31 | 0.38 | 0.35 | - | - | - | - | |
| 22 | GBHCT (igb = 1) | Emin2 | 34 | 0.31 | 0.37 | 0.13 | 0.42 | - | - | - | - | |
| 23 | GBHCT (igb = 1) | Emin2 | 34 | 0.51 | 0.31 | 0.32 | 0.36 | - | - | - | - | |
| 33 | GBOBC (igb = 2) | Emin1 | 33 | 0.30 | 0.37 | 0.19 | 0.40 | - | - | - | - | |
| 34 | GBOBC (igb = 2) | Emin1 | 34 | 0.46 | 0.32 | 0.33 | 0.36 | - | - | - | - | |
| 48 | GBOBC (igb = 5) | Emin1 | 34 | 0.31 | 0.36 | 0.20 | 0.40 | - | - | - | - | |
| 49 | GBOBC (igb = 5) | Emin1 | 34 | 0.47 | 0.32 | 0.34 | 0.36 | - | - | - | - | |
| 65 | GBn (igb = 8) | Emin2 | 34 | 0.19 | 0.40 | 0.02 | 0.48 | - | - | - | - | |
| 77 | PB-mbondi | Emin2 | 34 | 0.27 | 0.38 | 0.08 | 0.43 | - | - | - | - | |
| 94 | PB-bondi | MD51-100 | 34 | 0.45 | 0.33 | 0.36 | 0.35 | 0.39 | 0.36 | 0.38 | 0.36 | |
| 95 | PB-bondi | MD51-100 | 34 | 0.61 | 0.27 | 0.53 | 0.30 | 0.55 | 0.30 | 0.54 | 0.31 | |
| 96 | PB-bondi | MD51-100 | 33 | 0.62 | 0.22 | 0.53 | 0.25 | 0.56 | 0.25 | 0.54 | 0.26 | TH58 |
| 97 | PB-bondi | MD51-100 | 31 | 0.73 | 0.19 | 0.66 | 0.22 | 0.70 | 0.21 | 0.69 | 0.22 | TH58, TH70, TH74 |
| 105 | PB-PARSE | Emin2 | 34 | 0.17 | 0.40 | 0.01 | 0.46 | - | - | - | - | |
| 117 | PM3/GB1 | Emin2 | 34 | 0.37 | 0.35 | 0.26 | 0.38 | - | - | - | - | |
| 118 | PM3/GB1 | Emin2 | 34 | 0.52 | 0.31 | 0.41 | 0.34 | - | - | - | - | |
| Compound Number | Compoud Code | Scaffold | R1 | R2 | smHDAC8_IC50 nM- | pIC50_exp | pIC50_pred | Res a (Exp_pIC50-Pred_pIC50) | Res b Avg_Exp_pIC50-Pred_pIC50 | Glide SP_Score |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AT_T4 | A | methoxy | 9H-fluoren-1-yl | 163 ± 17 | 6.79 | 7.01 | −0.22 | −0.44 | −9.20 |
| 2 | SD14 | A | methoxy | 3-methyl-1,2,3,4-tetrahydro-ɣ-carbolin-8-yl | 197 ± 19 | 6.71 | 6.64 | 0.06 | −0.07 | −9.24 |
| 3 | TH112 | A | ethoxy | 2,4-dichlorophenyl | 103 ± 7 | 6.99 | 6.77 | 0.22 | −0.20 | −8.99 |
| 4 | TH117 | A | chloro | 4-biphenyl | 404 ± 90 | 6.39 | 6.42 | −0.03 | 0.15 | −8.69 |
| 5 | TH119 | A | methanethiolyl | 4-biphenyl | 101 ± 7 | 6.99 | 6.68 | 0.31 | −0.11 | −8.47 |
| 6 | TH120 | A | chloro | benzo[b]thien-7-yl | 97 ± 16 | 7.01 | 6.81 | 0.20 | −0.24 | −8.90 |
| 7 | TH125 | A | methoxy | 1H-benzo[d]imidazol-2-yl | 575 ±72 | 6.24 | 6.86 | −0.62 | −0.29 | −9.48 |
| 8 | TH127 | A | methoxy | 3-benzyloxyphenyl | 605 ± 68 | 6.22 | 6.66 | −0.44 | −0.09 | −9.23 |
| 9 | TH128 | A | methyl | 3-benzyloxyphenyl | 447 ± 31 | 6.35 | 6.43 | −0.08 | 0.14 | −8.63 |
| 10 | TH132 | A | methoxy | 2-chloro-4-biphenyl | 101 ± 77 | 7.00 | 6.68 | 0.31 | −0.11 | −8.71 |
| 11 | TH133 | A | chloro | 2-chloro-4-biphenyl | 112 ± 11 | 6.95 | 6.77 | 0.18 | −0.20 | −8.35 |
| 12 | TH134 | A | methoxy | 4-propoxyphenyl | 729 ± 86 | 6.14 | 6.19 | −0.05 | 0.38 | −8.51 |
| 13 | TH135 | A | methoxy | 4-isopropoxyphenyl | 725 ± 52 | 6.14 | 6.86 | −0.72 | −0.29 | −8.76 |
| 14 | TH136 | A | methoxy | 2,4-dimethoxyphenyl | 2078 ± 273 | 5.68 | 6.90 | −1.22 | −0.33 | −9.29 |
| 15 | TH137 | A | chloro | 2,4-dimethoxyphenyl | 220 ± 13 | 6.66 | 6.51 | 0.15 | 0.06 | −9.22 |
| 16 | TH138 | A | methoxy | 2-chloro-4-(4-fluorophenyl)phenyl | 318 ± 19 | 6.50 | 6.37 | 0.13 | 0.20 | −8.99 |
| 17 | TH139 | A | chloro | 2-chloro-4-(4-fluorophenyl)phenyl | 281 ± 37 | 6.55 | 6.70 | −0.14 | −0.13 | −8.66 |
| 18 | TH142 | A | methoxy | quinolin-8-yl | 332 ± 51 | 6.48 | 6.98 | −0.50 | −0.41 | −9.84 |
| 19 | TH143 | A | methoxy | 4-dibenzofuranyl | 271 ± 30 | 6.57 | 6.91 | −0.34 | −0.34 | −9.76 |
| 20 | TH156 | B | methoxy | 4-dibenzofuranyl | 451 ± 90 | 6.35 | 5.98 | 0.37 | 0.59 | −9.42 |
| 21 | TH34 | B | methyl | benzyl | 1260 ± 170 | 5.90 | 6.18 | −0.28 | 0.39 | −9.15 |
| 22 | TH42 | B | methoxy | benzyl | 620 ± 0 | 6.21 | 6.13 | 0.08 | 0.44 | −9.74 |
| 23 | TH97 | - | - | - | 220 ± 67 | 6.66 | 6.77 | −0.11 | −0.26 | −8.13 |
| 24 | TH98 | - | - | - | 1590 ± 190 | 5.80 | 6.49 | −0.69 | 0.08 | −9.59 |
| Notation | Molecular Descriptors |
|---|---|
| a_heavy | Number of heavy atoms |
| b_1rotN | Number of rotatable single bonds. Conjugated single bonds are not included |
| b_single | Number of single bonds (including implicit hydrogens). Aromatic bonds are not considered to be single bonds. |
| lip_acc | The number of O and N atoms. |
| lip_don | The number of OH and NH atoms. |
| mr | Molecular refractivity (including implicit hydrogens). |
| PEOE_VSA_POL | The number of OH and NH atoms. |
| TPSA | Polar surface area (Å2) calculated using group contributions to approximate the polar surface area from connection table information only. |
| h_logD | The octanol/water distribution coefficient at pH 7. |
| PEOE_VSA_FPPOS * | Fractional positive polar van der Waals surface area. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simoben, C.V.; Ghazy, E.; Zeyen, P.; Darwish, S.; Schmidt, M.; Romier, C.; Robaa, D.; Sippl, W. Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors. Molecules 2021, 26, 2584. https://doi.org/10.3390/molecules26092584
Simoben CV, Ghazy E, Zeyen P, Darwish S, Schmidt M, Romier C, Robaa D, Sippl W. Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors. Molecules. 2021; 26(9):2584. https://doi.org/10.3390/molecules26092584
Chicago/Turabian StyleSimoben, Conrad V., Ehab Ghazy, Patrik Zeyen, Salma Darwish, Matthias Schmidt, Christophe Romier, Dina Robaa, and Wolfgang Sippl. 2021. "Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors" Molecules 26, no. 9: 2584. https://doi.org/10.3390/molecules26092584
APA StyleSimoben, C. V., Ghazy, E., Zeyen, P., Darwish, S., Schmidt, M., Romier, C., Robaa, D., & Sippl, W. (2021). Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors. Molecules, 26(9), 2584. https://doi.org/10.3390/molecules26092584