
Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics




Abstract
1. Introduction
2. Results
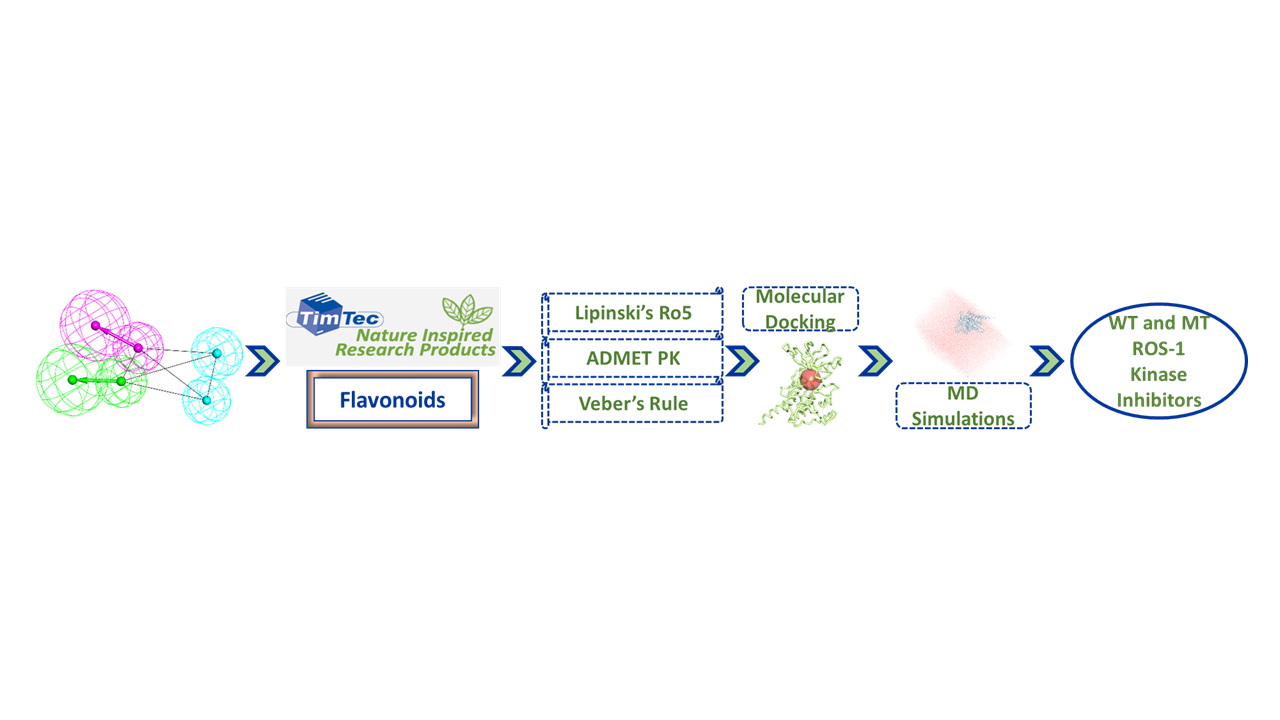
2.1. Receptor-Ligand Pharmacophore Model
2.2. Güner-Henry Validation of the Receptor-Ligand Pharmacophore Model
2.3. Drug-Like Flavonoids Retrieved by Virtual Screening
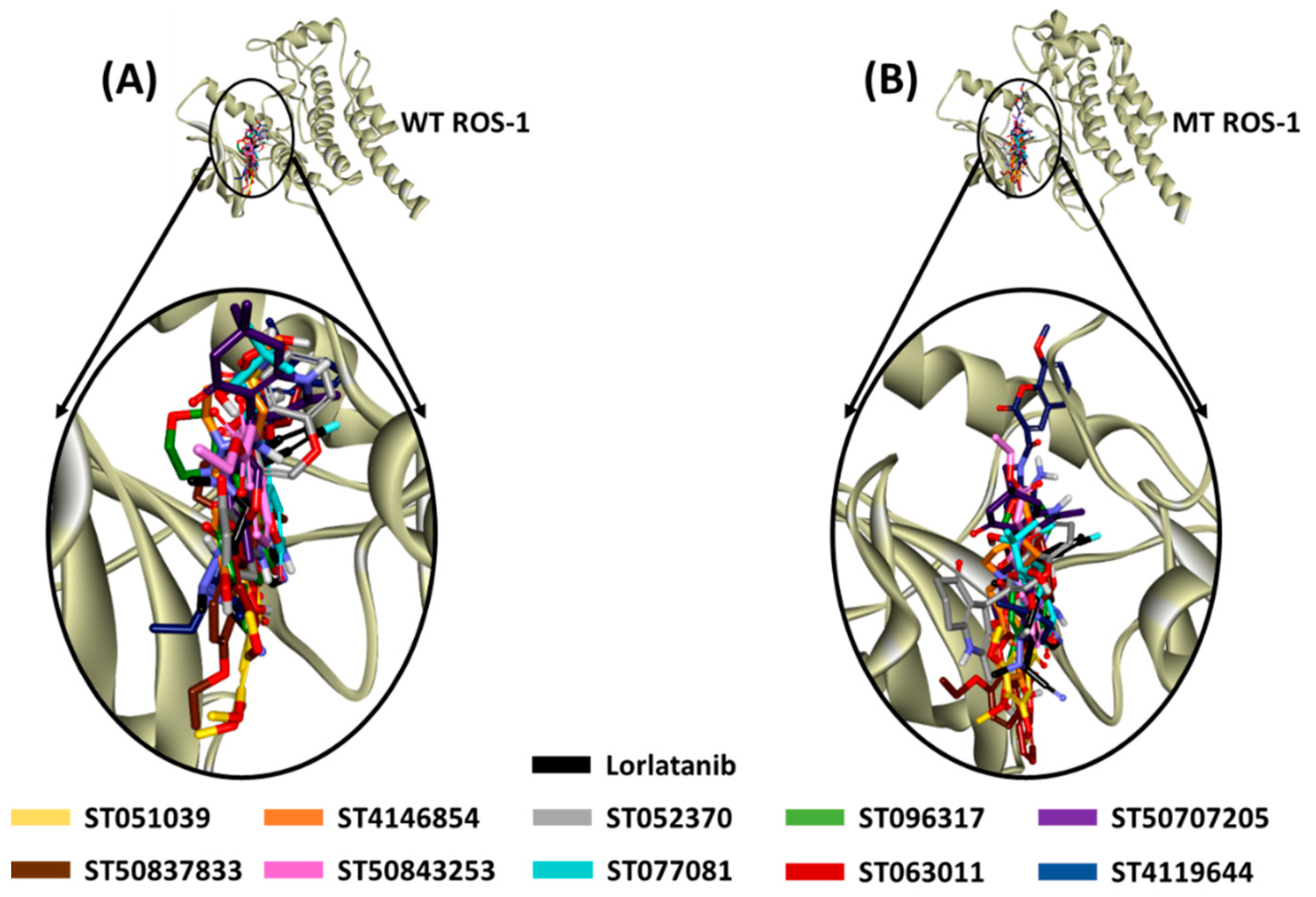
2.4. Molecular Docking of Retrieved Flavonoids with ROS-1 Tyrosine Kinase Domain
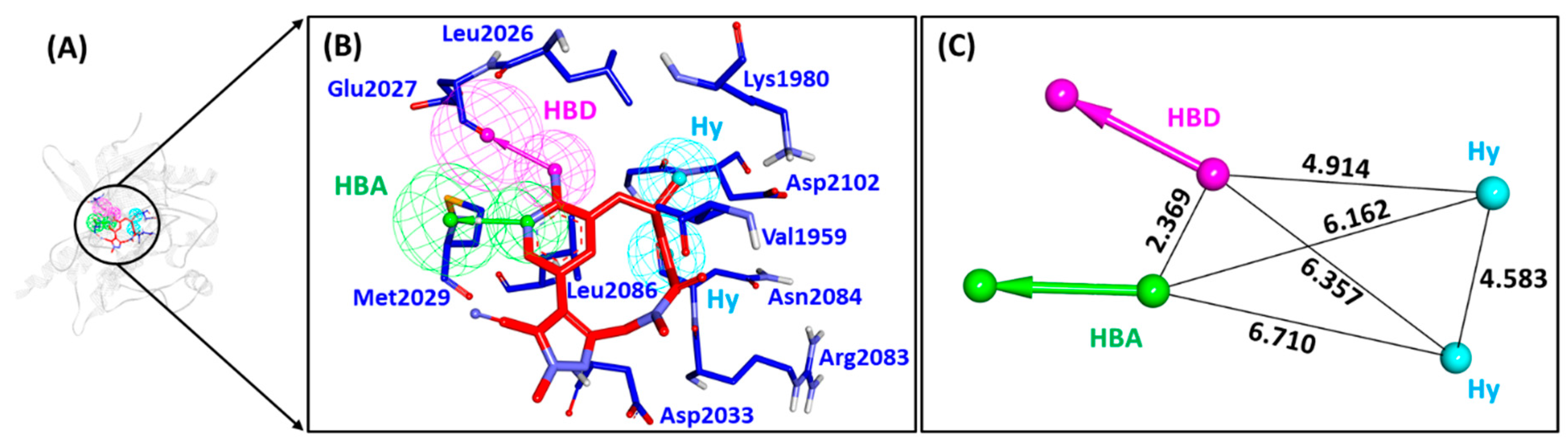
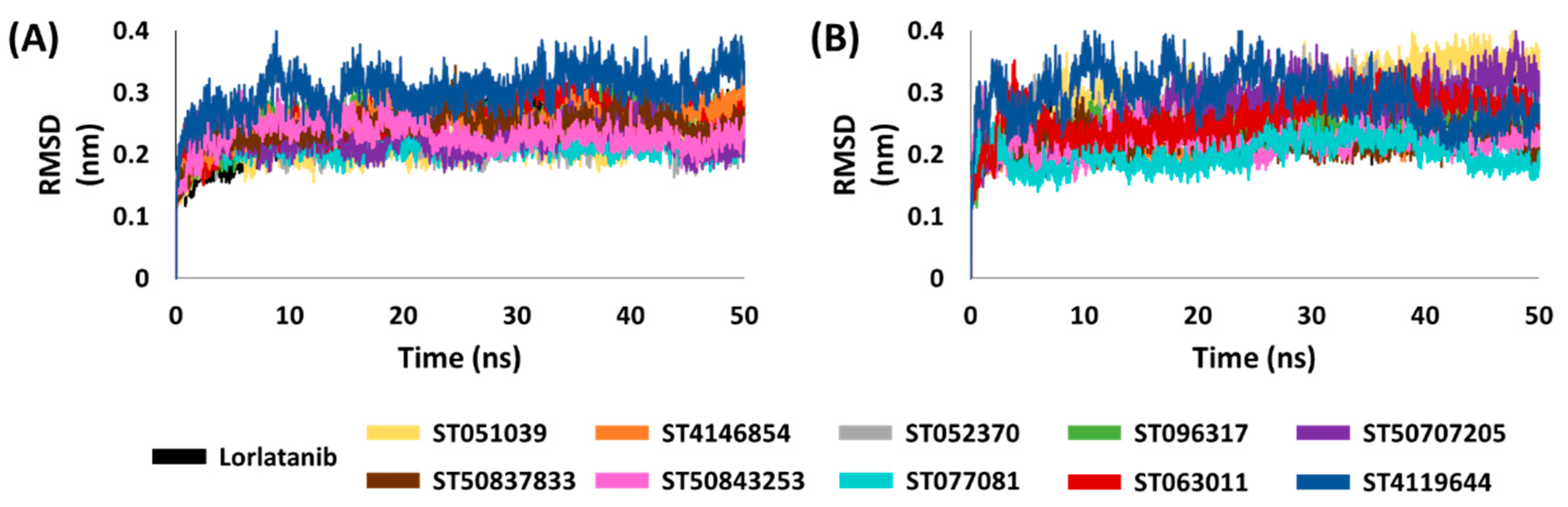
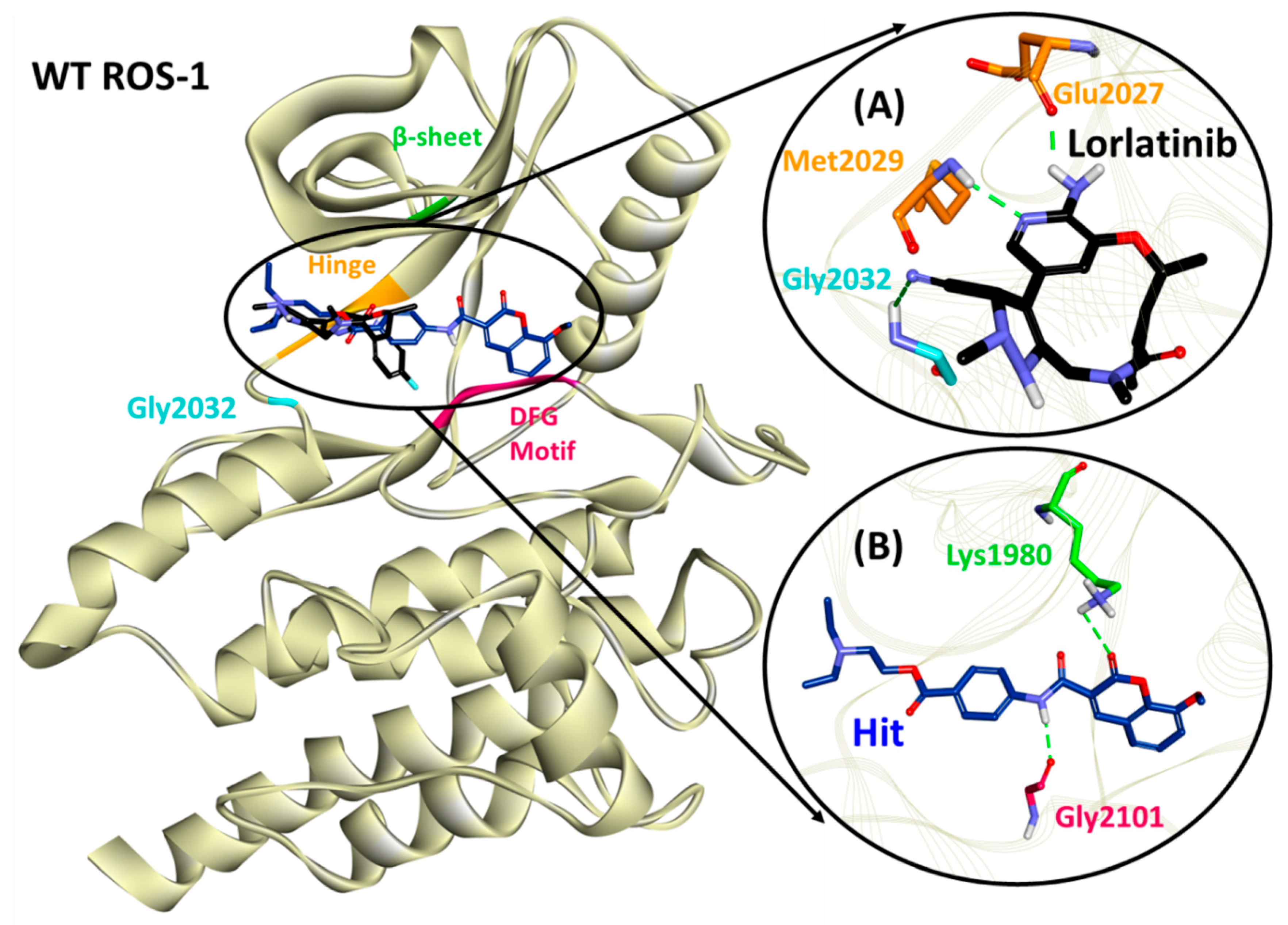
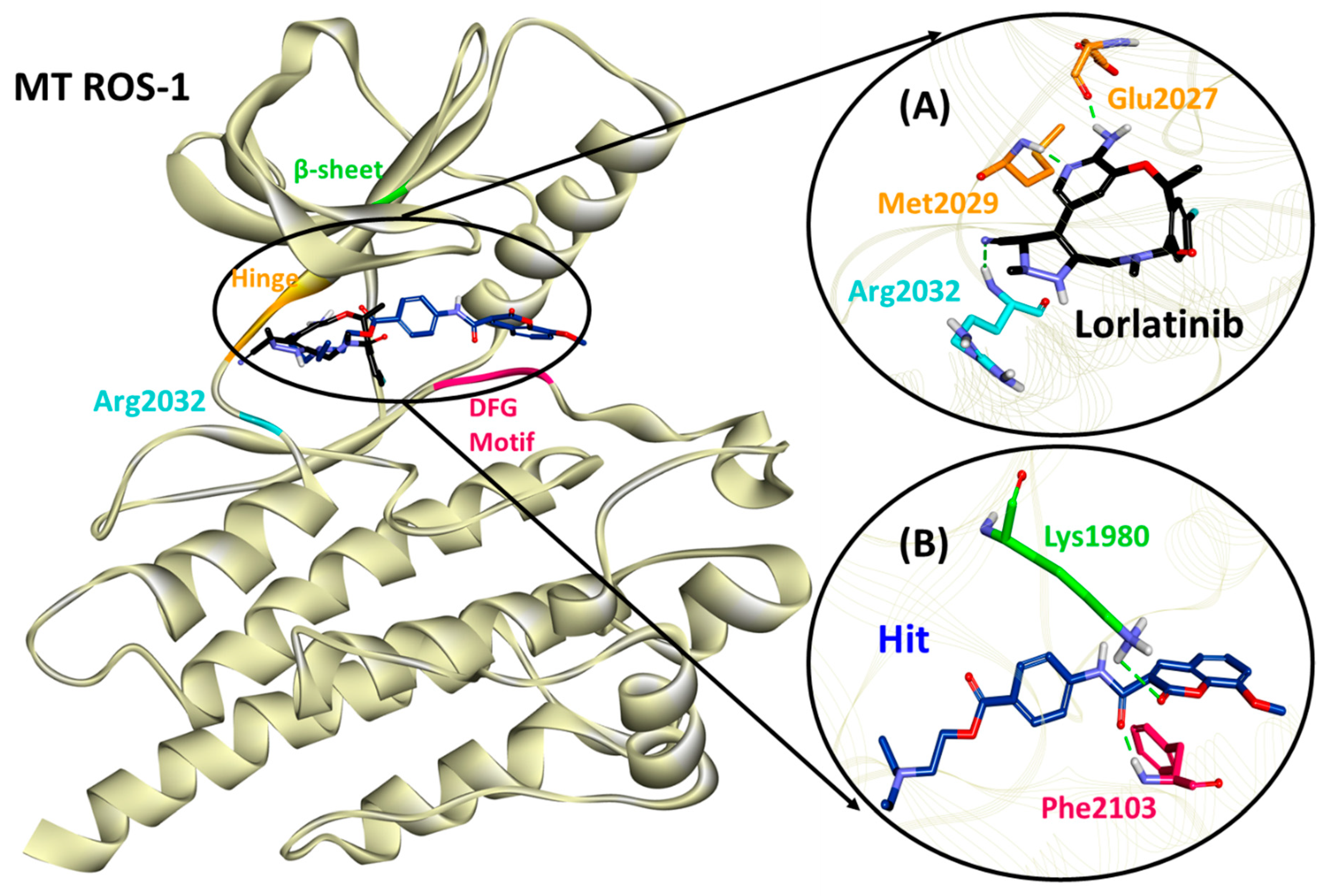
2.5. Binding Mode, Interaction and Free Energy Analysis of Identified Flavonoids by Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Receptor-Ligand Pharmacophore Model Generation
4.2. Validation of Receptor-Ligand Pharmacophore Model
4.3. Virtual Screening of TimTec Flavonoids Database
4.4. Molecular Docking of Drug-Like Compounds with ROS-1 Tyrosine Kinase
4.5. Molecular Dynamics Simulation and Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMET | absorption: distribution, metabolism, excretion, toxicity |
| AKT | protein kinase B |
| ALK | anaplastic lymphoma kinase |
| BFE | binding free energy |
| DFG | Asp-Phe-Gly |
| EGF | epidermal growth factor |
| FDA | food and drug administration |
| GF | goodness of fit |
| GFA | genetic function approximation |
| GH | Güner-Henry |
| GOLD | genetic optimization for ligand docking |
| GROMACS | GROningen MAchine for Chemical Simulations |
| HBD | hydrogen bond donor |
| HBA | hydrogen bond acceptor |
| Hy | hydrophobic |
| IC50 | half maximal inhibitory concentration |
| LINCS | LINear Constraint Solver |
| LUAD | lung adenocarcinoma |
| LUSC | lung squamous cell carcinoma |
| MD | molecular dynamics |
| MT | mutant |
| mTOR | mammalian target of rapamycin |
| MM/PBSA | molecular mechanics/poisson-boltzmann surface area |
| NI | negative ionizable |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| NVT | constant number of particles, volume and temperature |
| NPT | constant number of particles, pressure and temperature |
| NSCLC | non-small cell lung cancer |
| PDB | protein data bank |
| PI | positive ionizable |
| PI3K | phosphoinositide 3-kinase |
| P-loop | phosphate binding loop |
| PME | Particle mesh Ewald |
| RA | ring aromatic |
| RTK | receptor tyrosine kinase |
| ROS-1 | c-ros oncogene 1 |
| Ro5 | rule of five |
| RMSD | root mean square deviation |
| VMD | visual molecular dynamics |
| WT | wild-type |
References
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo. Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Rajurkar, S.; Mambetsariev, I.; Pharaon, R.; Leach, B.; Tan, T.; Kulkarni, P.; Salgia, R. Non-Small Cell Lung Cancer from Genomics to Therapeutics: A Framework for Community Practice Integration to Arrive at Personalized Therapy Strategies. J. Clin. Med. 2020, 9, 1870. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Büttner, R.; Al-Dayel, F.; Dietel, M.; Elmberger, G.; Kerr, K.; López-Ríos, F.; Marchetti, A.; Öz, B.; Pauwels, P.; et al. Testing for ROS1 in non-small cell lung cancer: A review with recommendations. Virchows Arch. 2016, 469, 489–503. [Google Scholar] [CrossRef]
- Camp, E.; Anderson, P.J.; Zannettino, A.C.W.; Gronthos, S. Tyrosine kinase receptor c-ros-oncogene 1 mediates TWIST-1 regulation of human mesenchymal stem cell lineage commitment. Bone 2017, 94, 98–107. [Google Scholar] [CrossRef]
- Zhu, V.W.; Upadhyay, D.; Schrock, A.B.; Gowen, K.; Ali, S.M.; Ou, S.H.I. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016, 97, 48–50. [Google Scholar] [CrossRef]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [CrossRef]
- Shan, L.; Lian, F.; Guo, L.; Qiu, T.; Ling, Y.; Ying, J.; Lin, D. Detection of ROS1 gene rearrangement in lung adenocarcinoma: Comparison of IHC, FISH and real-time RT-PCR. PLoS ONE 2015, 10, e0120422. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Blumenthal, G.M.; Luo, L.; He, K.; Fran, I.; Lemery, S.; Pazdur, R. Benefit-Risk Summary of Crizotinib for the Treatment of Patients With ROS1 Alteration-Positive, Metastatic Non-Small Cell Lung Cancer. Oncologist 2016, 21, 974–980. [Google Scholar] [CrossRef]
- Patil, T.; Simons, E.; Mushtaq, R.; Pacheco, J.M.; Doebele, R.C.; Bowles, D.W. Targeted therapies for ROS1-rearranged non-small cell lung cancer. Drugs Today. 2019, 55, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Bai, Z.; Hsieh, S.C.; Kelly, S.E.; Chen, L.; Skidmore, B.; Yousef, S.; Zheng, C.; Stewart, D.J.; Wells, G.A. ALK inhibitors for non-small cell lung cancer: A systematic review and network meta-analysis. PLoS ONE 2020, 15, e0229179. [Google Scholar] [CrossRef]
- Sgambato, A.; Casaluce, F.; Maione, P.; Gridelli, C. Targeted therapies in non-small cell lung cancer: A focus on ALK/ROS1 tyrosine kinase inhibitors. Expert Rev. Anticancer Ther. 2018, 18, 71–80. [Google Scholar] [CrossRef]
- Davare, M.A.; Saborowski, A.; Eide, C.A.; Tognon, C.; Smith, R.L.; Elferich, J.; Agarwal, A.; Tyner, J.W.; Shinde, U.P.; Lowe, S.W.; et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 19519–19524. [Google Scholar] [CrossRef]
- Davare, M.A.; Vellore, N.A.; Wagner, J.P.; Eide, C.A.; Goodman, J.R.; Drilon, A.; Deininger, M.W.; O’Hare, T.; Druker, B.J. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E5381–E5390. [Google Scholar] [CrossRef]
- Ye, M.; Zhang, X.; Li, N.; Zhang, Y.; Jing, P.; Chang, N.; Wu, J.; Ren, X.; Zhang, J. ALK and ROS1 as targeted therapy paradigms and clinical implications to overcome crizotinib resistance. Oncotarget 2016, 7, 12289–12304. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Engstrom, L.D.; West, M.; Appleman, V.; Wong, K.A.; McTigue, M.; Deng, Y.L.; Liu, W.; Brooun, A.; et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 3493–3498. [Google Scholar] [CrossRef] [PubMed]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Somwar, R.; Wagner, J.P.; Vellore, N.A.; Eide, C.A.; Zabriskie, M.S.; Arcila, M.E.; Hechtman, J.F.; Wang, L.; Smith, R.S.; et al. A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin. Cancer Res. 2016, 22, 2351–2358. [Google Scholar] [CrossRef]
- D’Angelo, A.; Sobhani, N.; Chapman, R.; Bagby, S.; Bortoletti, C.; Traversini, M.; Ferrari, K.; Voltolini, L.; Darlow, J.; Roviello, G. Focus on ros1-positive non-small cell lung cancer (Nsclc): Crizotinib, resistance mechanisms and the newer generation of targeted therapies. Cancers 2020, 12, 3293. [Google Scholar] [CrossRef]
- Zanoaga, O.; Braicu, C.; Jurj, A.; Rusu, A.; Buiga, R.; Berindan-Neagoe, I. Progress in research on the role of flavonoids in lung cancer. Int. J. Mol. Sci. 2019, 20, 4291. [Google Scholar] [CrossRef]
- Cui, Y.; Morgenstern, H.; Greenland, S.; Tashkin, D.P.; Mao, J.T.; Cai, L.; Cozen, W.; Mack, T.M.; Lu, Q.Y.; Zhang, Z.F. Dietary flavonoid intake and lung cancer-A population-based case-control study. Cancer 2008, 112, 2241–2248. [Google Scholar] [CrossRef]
- Le Marchand, L.; Murphy, S.P.; Hankin, J.H.; Wilkens, L.R.; Kolonel, L.N. Intake of Flavonoids and Lung Cancer. JNCI J. Natl. Cancer Inst. 2000, 92, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, B.; Yu, Z.; He, Q.; Hu, Z.; Zhou, S.; Chen, M.; Zhu, L. Trifolium Flavonoids Overcome Gefitinib Resistance of Non-Small-Cell Lung Cancer Cell by Suppressing ERK and STAT3 Signaling Pathways. Biomed Res. Int. 2020, 1–8. [Google Scholar] [CrossRef]
- Han, L.; Fang, S.; Li, G.; Wang, M.; Yu, R. Total flavonoids suppress lung cancer growth via the COX–2–mediated Wnt/β–catenin signaling pathway. Oncol. Lett. 2020, 19, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Park, K.I.; Park, H.S.; Kim, M.K.; Hong, G.E.; Nagappan, A.; Lee, H.J.; Yumnam, S.; Lee, W.S.; Won, C.K.; Shin, S.C.; et al. Flavonoids identified from Korean Citrus aurantium L. inhibit Non-Small Cell Lung Cancer growth in vivo and in vitro. J. Funct. Foods 2014, 7, 287–297. [Google Scholar] [CrossRef]
- Chen, H.; Yang, J.; Hao, J.; Lv, Y.; Chen, L.; Lin, Q.; Yuan, J.; Yang, X. A Novel Flavonoid Kushenol Z from Sophora flavescens Mediates mTOR Pathway by Inhibiting Phosphodiesterase and Akt Activity to Induce Apoptosis in Non-Small-Cell Lung Cancer Cells. Molecules 2019, 24, 4425. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Maheshwari, U.; Muthukrishnan, A.; Muthuswamy, R.; Anand, K.; Ravindran, B.; Dhanaraj, P.; Balamuralikrishnan, B.; Chang, S.W.; Chung, W.J. Myricetin: Versatile plant based flavonoid for cancer treatment by inducing cell cycle arrest and ROS–reliant mitochondria-facilitated apoptosis in A549 lung cancer cells and in silico prediction. Mol. Cell. Biochem. 2020, 476, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.W. Reversal of multidrug resistance in cancer by multi-functional flavonoids. Front. Oncol. 2019, 9, 487. [Google Scholar] [CrossRef]
- Pathak, D.; Chadha, N.; Silakari, O. Identification of non-resistant ROS-1 inhibitors using structure based pharmacophore analysis. J. Mol. Graph. Model. 2016, 70, 85–93. [Google Scholar] [CrossRef]
- Rampogu, S.; Park, C.; Ravinder, D.; Son, M.; Baek, A.; Zeb, A.; Bavi, R.; Kumar, R.; Lee, G.; Parate, S.; et al. Pharmacotherapeutics and molecular mechanism of phytochemicals in alleviating hormone-responsive breast cancer. Oxid. Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, T.; Long, L.; Li, Z.; Wan, S.; Wang, G.; Yu, Y.; Hou, J.; Wu, X.; Zhang, J. Design, synthesis, biological evaluation and molecular modeling of novel 2-amino-4-(1-phenylethoxy) pyridine derivatives as potential ROS1 inhibitors. Eur. J. Med. Chem. 2018, 143, 182–199. [Google Scholar] [CrossRef]
- Tian, Y.; Yu, Y.; Shen, Y.; Wan, H.; Chang, S.; Zhang, T.; Wan, S.; Zhang, J. Molecular Simulation Studies on the Binding Selectivity of Type-I Inhibitors in the Complexes with ROS1 versus ALK. J. Chem. Inf. Model. 2017, 57, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.; Awad, M.D.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.; Brooun, A.; Friboulet, L.; Huang, D.; Matthew, D.; et al. Acquired Resistance to Crizotinib from a Mutation in CD74–ROS1. N. Engl. J. Med. 2013, 368, 1–9. [Google Scholar] [CrossRef]
- Basit, S.; Ashraf, Z.; Lee, K.; Latif, M. First macrocyclic 3rd-generation ALK inhibitor for treatment of ALK/ROS1 cancer: Clinical and designing strategy update of lorlatinib. Eur. J. Med. Chem. 2017, 134, 348–356. [Google Scholar] [CrossRef]
- Pathak, D.; Choudhary, S.; Singh, P.K.; Singh, M.; Chadha, N.; Silakari, O. Pharmacophore-based designing of putative ROS-1 targeting agents for NSCLC. Mol. Divers. 2020. [Google Scholar] [CrossRef]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in cancer treatment: From preclinical studies to clinical practice. Front. Pharmacol. 2020, 10, 1614. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in cancer and apoptosis. Cancers 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, B.; Xu, D.; Li, Z.; Liu, H.; Huang, Z.; Huang, K.; Lin, X.; Yao, H. Molecular mechanism and pharmacokinetics of flavonoids in the treatment of resistant EGF receptor-mutated non-small-cell lung cancer: A narrative review. Br. J. Pharmacol. 2021, 6, 1388–1406. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, B.; Sun, J.; Lu, L.; Liu, L.; Qiu, J.; Li, Q.; Yan, C.; Jiang, S.; Mohammadtursun, N.; et al. Scutellaria flavonoids effectively inhibit the malignant phenotypes of non-small cell lung cancer in an id1-dependent manner. Int. J. Biol. Sci. 2019, 15, 1500–1513. [Google Scholar] [CrossRef]
- Yan, W.; Wu, T.H.Y.; Leung, S.S.Y.; To, K.K.W. Flavonoids potentiated anticancer activity of cisplatin in non-small cell lung cancer cells in vitro by inhibiting histone deacetylases. Life Sci. 2020, 258, 118211. [Google Scholar] [CrossRef]
- Fouzder, C.; Mukhuty, A.; Kundu, R. Kaempferol inhibits Nrf2 signalling pathway via downregulation of Nrf2 mRNA and induces apoptosis in NSCLC cells. Arch. Biochem. Biophys. 2021, 697, 108700. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Lee, G.; Rampogu, S.; Hong, J.C.; Lee, K.W. Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals 2021, 14, 282. [Google Scholar] [CrossRef]
- Weng, C.-W.; Li, J.-H.; Tsai, J.-Y.; Lin, S.-H.; Chang, G.-C.; Liu, C.-C.; Chen, J.J. Pharmacophore-based virtual screening for the identification of the novel Src inhibitor SJG-136 against lung cancer cell growth and motility. Am. J. Cancer Res. 2020, 10, 1668–1690. [Google Scholar]
- Kumar, A.; Rai, S.; Rathi, E.; Agarwal, P.; Kini, S.G. Pharmacophore-guided fragment-based design of novel mammalian target of rapamycin inhibitors: Extra precision docking, fingerprint-based 2D and atom-based 3D-QSAR modelling. J. Biomol. Struct. Dyn. 2020, 39, 1155–1173. [Google Scholar] [CrossRef] [PubMed]
- Vanajothi, R.; Vedagiri, H.; Al-Ansari, M.M.; Al-Humaid, L.A.; Kumpati, P. Pharmacophore based virtual screening, molecular docking and molecular dynamic simulation studies for finding ROS1 kinase inhibitors as potential drug molecules. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Rampogu, S.; Parate, S.; Parameswaran, S.; Park, C.; Baek, A.; Son, M.; Park, Y.; Park, S.J.; Lee, K.W. Natural compounds as potential Hsp90 inhibitors for breast cancer-Pharmacophore guided molecular modelling studies. Comput. Biol. Chem. 2019, 83, 1–12. [Google Scholar] [CrossRef]
- Luo, L.X.; Fan, X.X.; Li, Y.; Peng, X.; Ji, Y.C.; Hsiao, W.W.L.; Liu, L.; Leung, E.L.H.; Yao, X.J. Identification of mitoxantrone as a new inhibitor of ROS1 fusion protein in non-small cell lung cancer cells. Medchemcomm 2017, 8, 621–624. [Google Scholar] [CrossRef]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Meslamani, J.; Li, J.; Sutter, J.; Stevens, A.; Bertrand, H.O.; Rognan, D. Protein-ligand-based pharmacophores: Generation and utility assessment in computational ligand profiling. J. Chem. Inf. Model. 2012, 52, 943–955. [Google Scholar] [CrossRef]
- Sutter, J.; Li, J.; J. Maynard, A.; Goupil, A.; Luu, T.; Nadassy, K. New Features that Improve the Pharmacophore Tools from Accelrys. Curr. Comput. Aided-Drug Des. 2011, 7, 173–180. [Google Scholar] [CrossRef]
- Khedkar, S.; Malde, A.; Coutinho, E.; Srivastava, S. Pharmacophore Modeling in Drug Discovery and Development: An Overview. Med. Chem. (Los Angeles) 2007, 3, 187–197. [Google Scholar] [CrossRef]
- Lin, S.-K. Pharmacophore Perception, Development and Use in Drug Design. Edited by Osman, F. Güner. Molecules 2000, 5, 987–989. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Parameswaran, S.; Bavi, R.; Baek, A.; Son, M.; Rampogu, S.; Park, C.; Lee, G.; Zeb, A.; Parate, S.; et al. Investigation of novel chemical scaffolds targeting prolyl oligopeptidase for neurological therapeutics. J. Mol. Graph. Model. 2019, 88, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Zhu, X.; Lopes, P.E.M.; MacKerell, A.D. Recent developments and applications of the CHARMM force fields. WIREs Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Models | Number of Features | Feature Set * | Selectivity Score |
|---|---|---|---|
| Pharmacophore_01 | 4 | ADHH | 7.0747 |
| Set No. | Parameters | Values |
|---|---|---|
| 1 | Total number of compounds in the database (D) | 78 |
| 2 | Total number of active compounds in the database (A) | 20 |
| 3 | Total number of hits retrieved by pharmacophore model from the database (Ht) | 25 |
| 4 | Total number of active compounds in the hit list (Ha) | 20 |
| 5 | % Yield of active ((Ha/Ht) × 100) | 80 |
| 6 | % Ratio of actives ((Ha/A) × 100) | 100 |
| 7 | False negatives (A-Ha) | 0 |
| 8 | False positives (Ht-Ha) | 5 |
| 9 | Goodness of fit score (GF) | 0.77 |
| Complex | Hydrogen Bond Interactions (Distance in Å) | Carbon Hydrogen Bond Interactions | Hydrophobic (π) Interactions | Van der Waals Interactions |
|---|---|---|---|---|
| Lorlatinib (with WT ROS-1) | Glu2027 (1.96), Met2029 (2.21), Gly2032 (3.05) | Leu1951, Met2029, Leu2030, Gly2101 | Val1959, Ala1978, Lys1980, Leu2026, Leu2028 | Gly1952, Leu2010, Leu2028, Asp2033, Arg2083, Asp2102, |
| Hit (with WT ROS-1) | Lys1980 (2.81), Gly2101 (2.03) | Met2029 | Leu1951, Met2001, Leu2010, Leu2028, Leu2086, Phe2103 | Ser1953, Glu1961, Glu1997, Leu2000, Phe2004, Ile2009, Leu2026, Glu2030, Gly2032, Asp2102 |
| Lorlatinib (with MT ROS-1) | Glu2027 (1.81), Met2029 (1.95), Arg2032 (2.12) | Leu1951, Met2029 | Val1959, Ala1978, Lys1980, Leu2026, Leu2086 | Gly1952, Leu2010, Leu2028, Glu2030, Gly2031, Asp2033, Asn2084, Asp2102 |
| Hit (with MT ROS-1) | Lys1980 (2.97), Phe2103 (1.85) | Ala2106 | Leu1951, Glu1997, Met2001, Phe2004, Leu2026, Leu2028, Arg2032, Phe2103 | Gly1952, Val1959, Ala1978, Leu2000, Leu2010, Met2029, Phe2075, Leu2086, Gly2101, Asp2102, Gly2104 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics. Molecules 2021, 26, 2114. https://doi.org/10.3390/molecules26082114
Parate S, Kumar V, Hong JC, Lee KW. Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics. Molecules. 2021; 26(8):2114. https://doi.org/10.3390/molecules26082114
Chicago/Turabian StyleParate, Shraddha, Vikas Kumar, Jong Chan Hong, and Keun Woo Lee. 2021. "Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics" Molecules 26, no. 8: 2114. https://doi.org/10.3390/molecules26082114
APA StyleParate, S., Kumar, V., Hong, J. C., & Lee, K. W. (2021). Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics. Molecules, 26(8), 2114. https://doi.org/10.3390/molecules26082114