
Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol—A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling

 , , , and
, , , and 
Abstract
1. Introduction
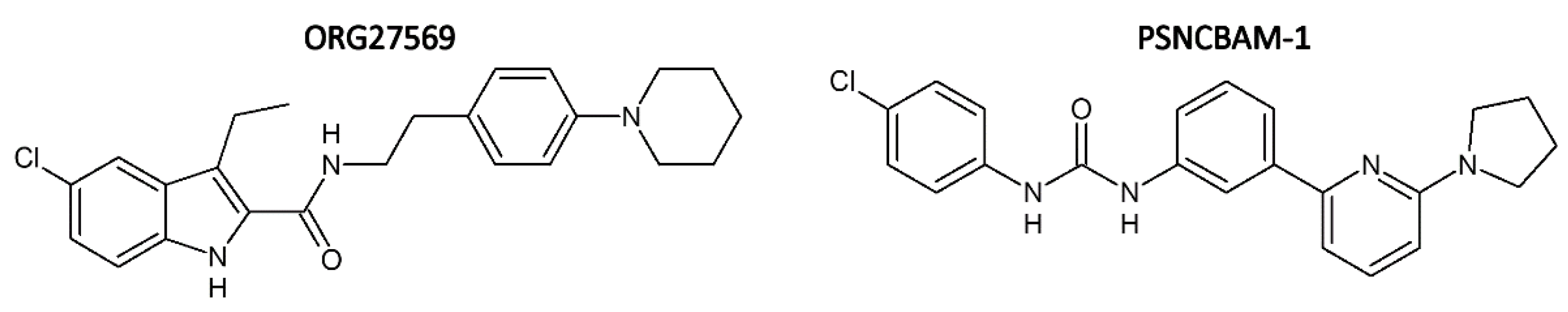
1.1. Advantages of Allosteric over Orthosteric Ligands of CB1R
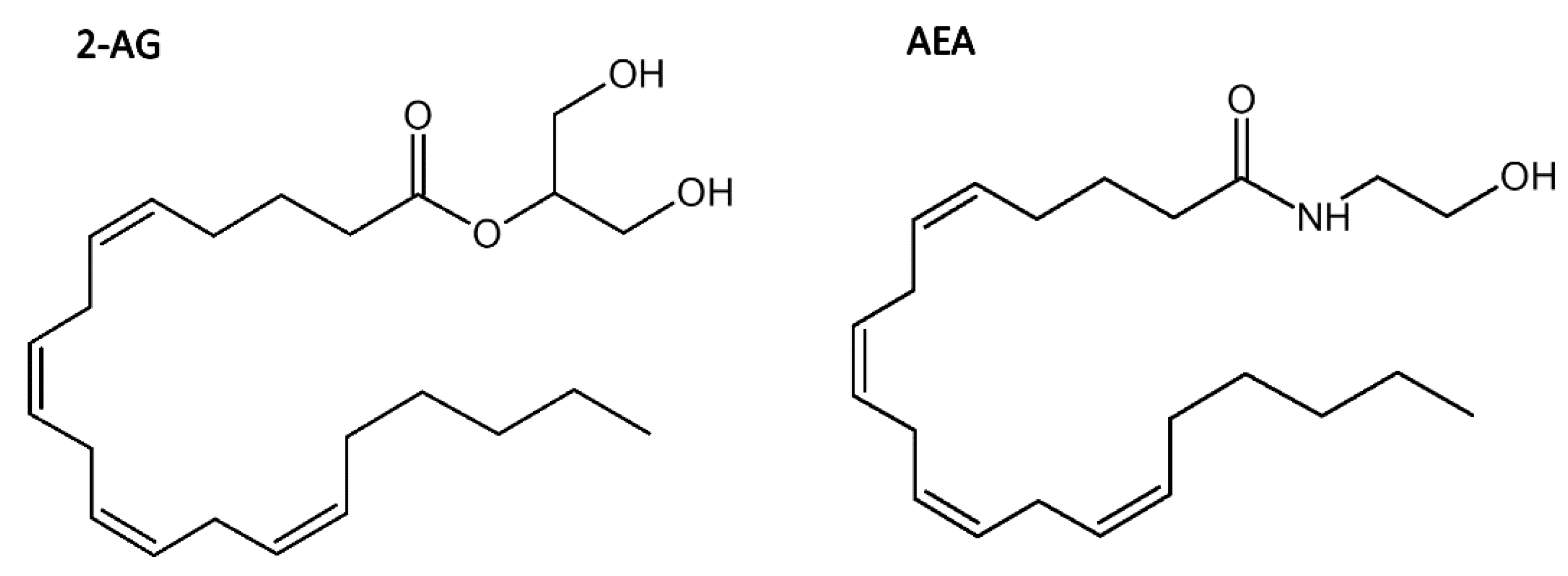
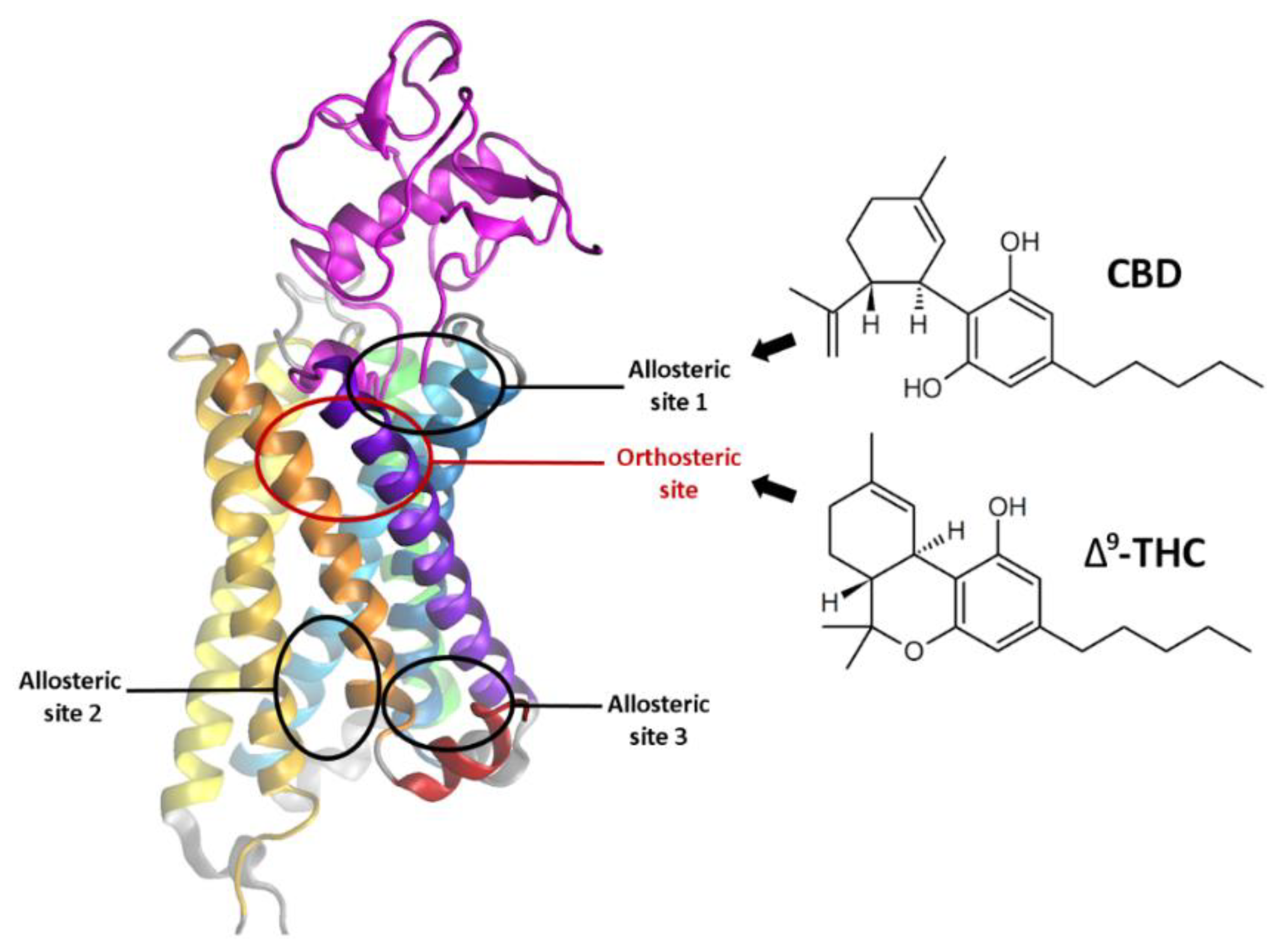
1.2. Cannabidiol—Its Properties and the Binding Site
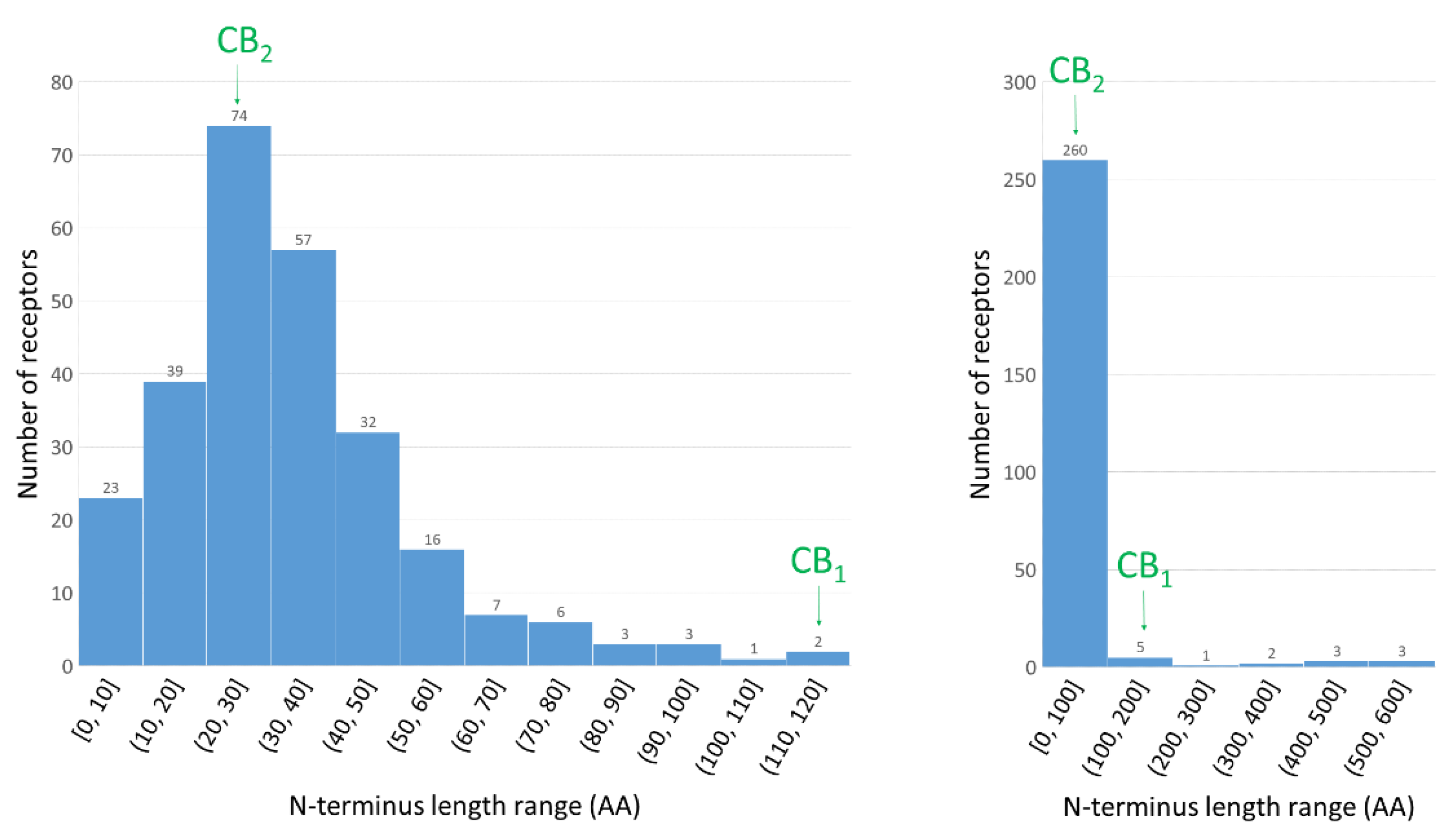
1.3. The Importance of N-Terminus for Allosteric and Orthosteric Binding
2. Results and Discussion
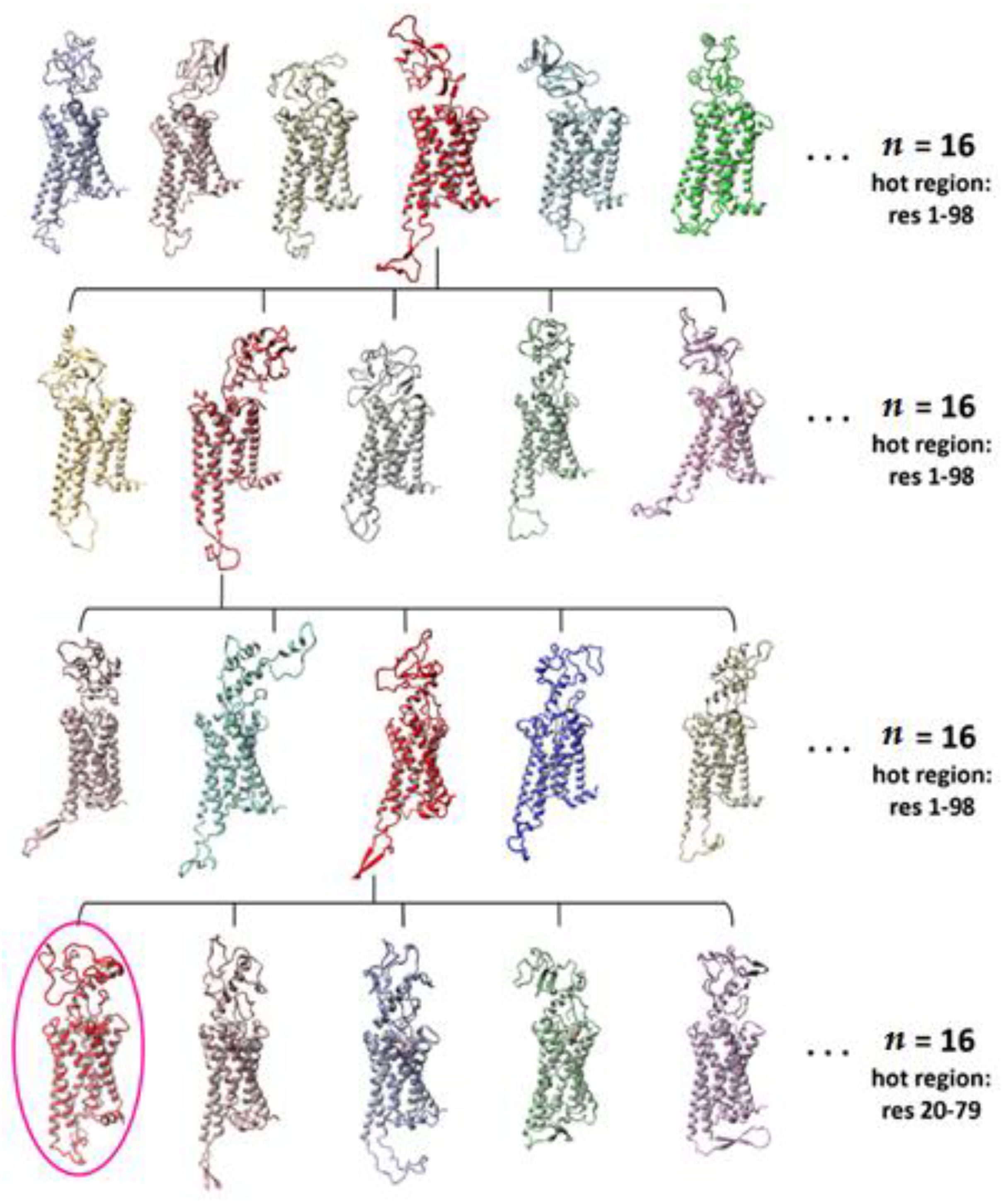
2.1. Construction of the N-Terminus of CB1R
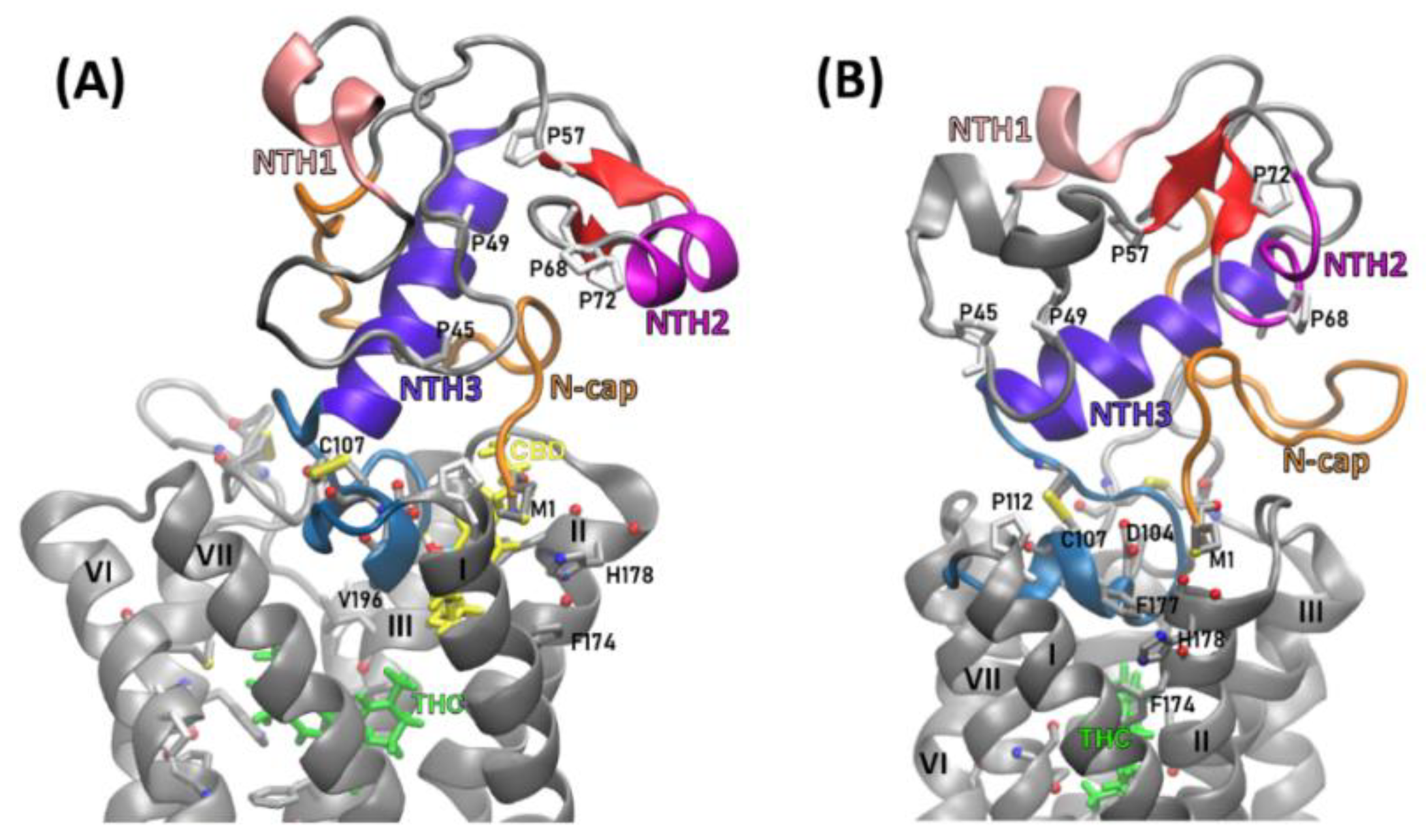
2.2. Structural Features of the CB1R N-Terminal Domain
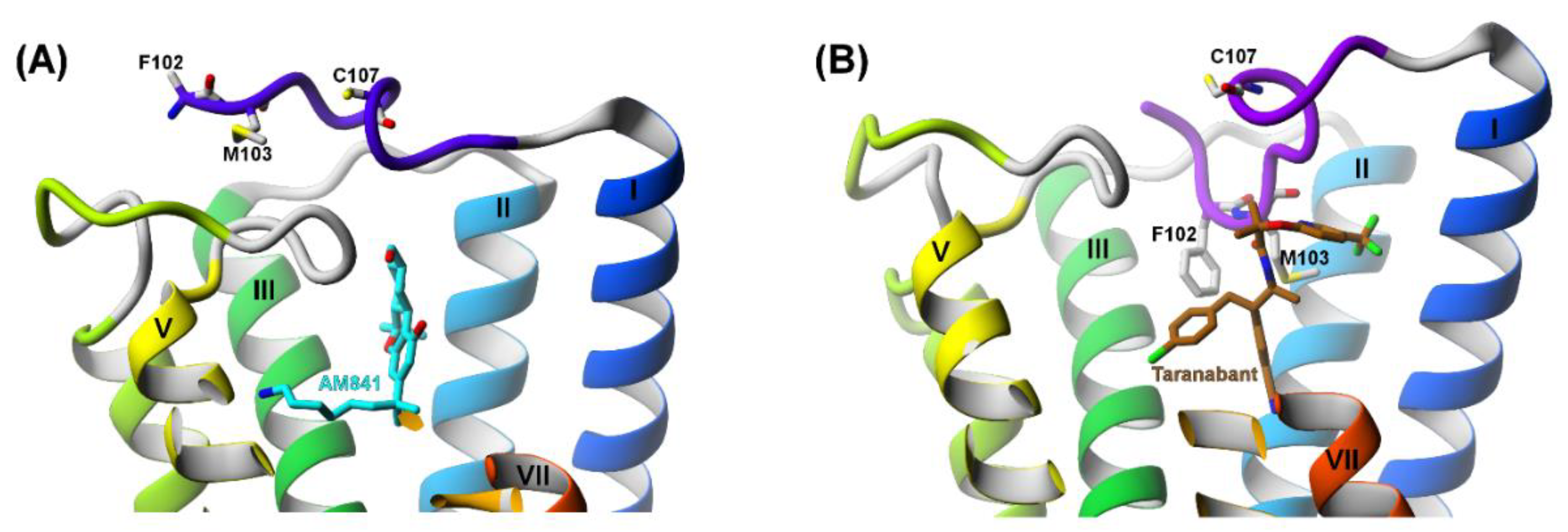
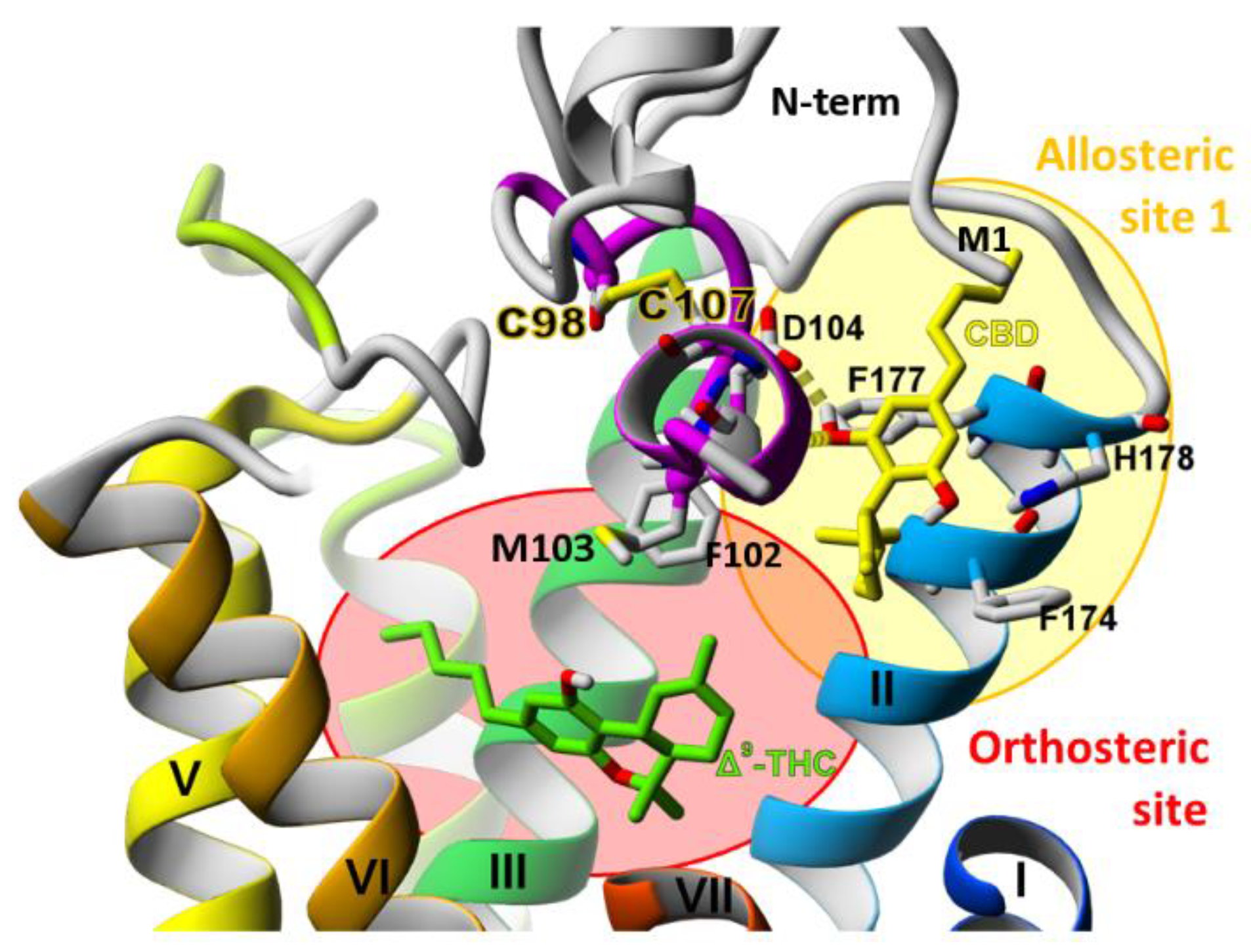
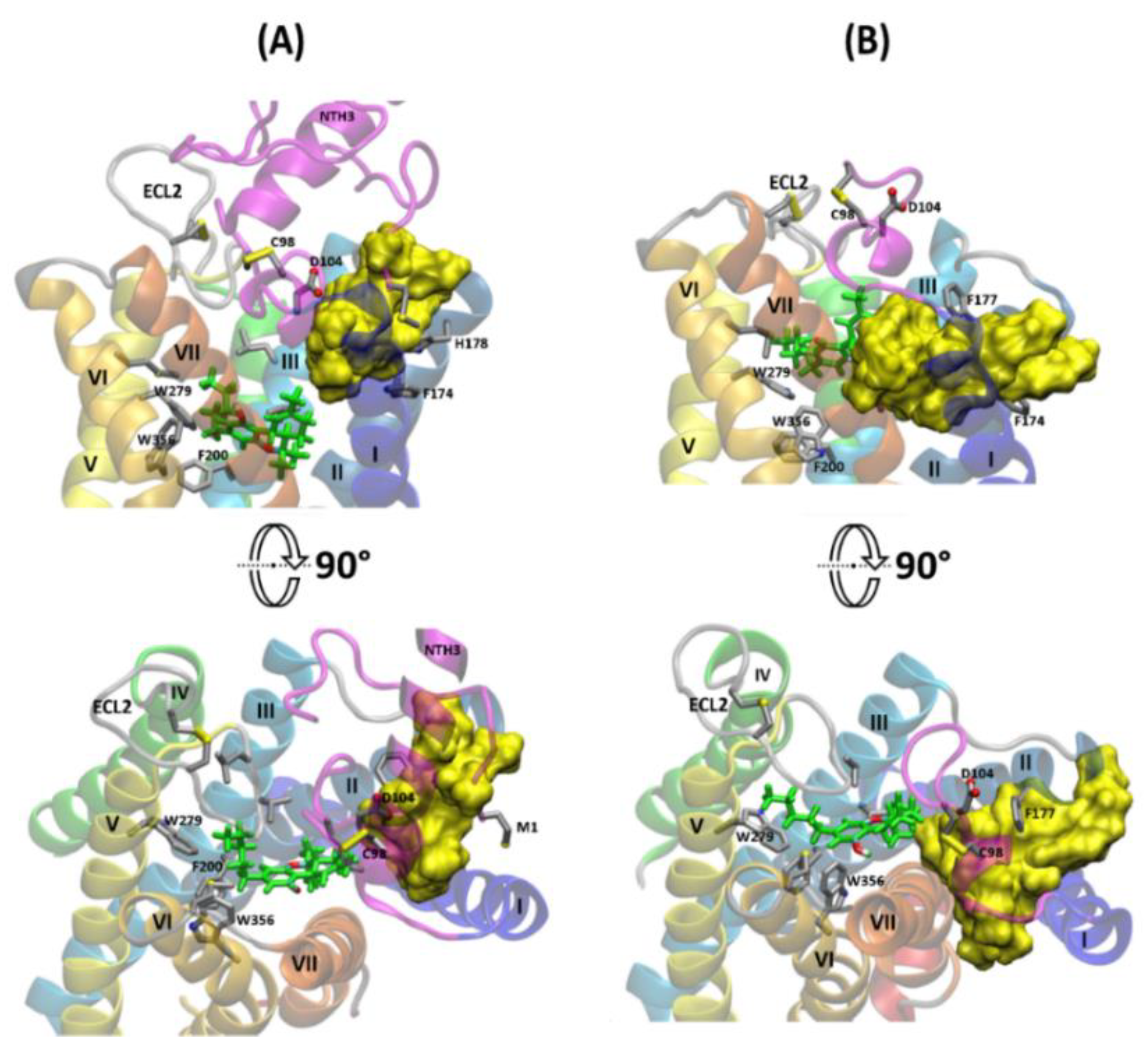
2.3. The Orthosteric and Allosteric Ligand Binding
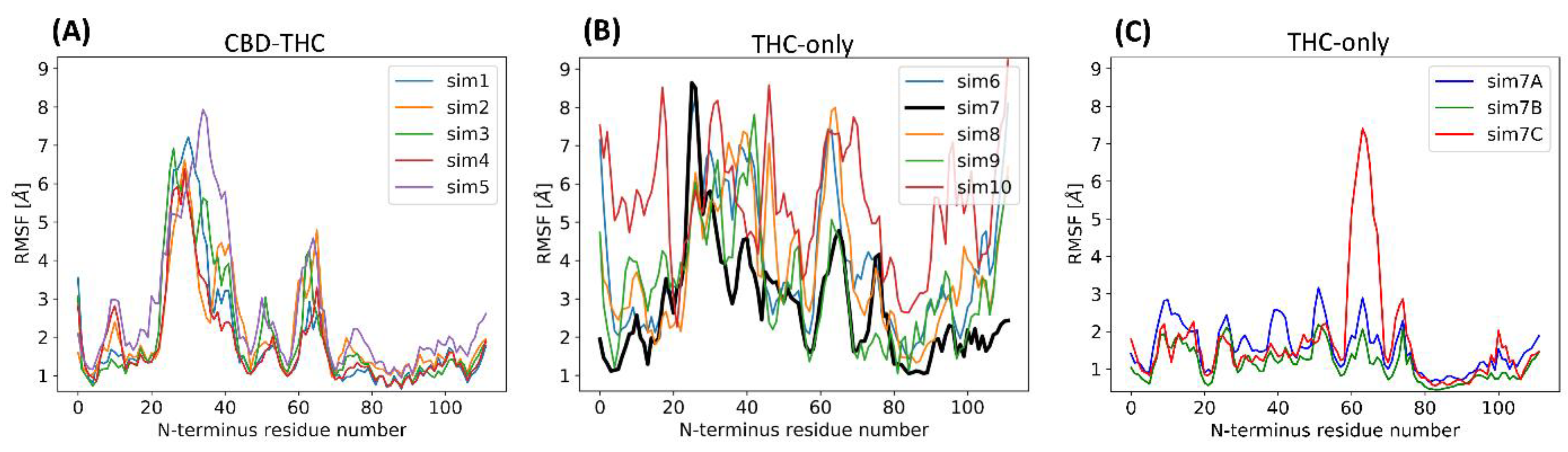
2.4. Stability of the N-Terminal Domain
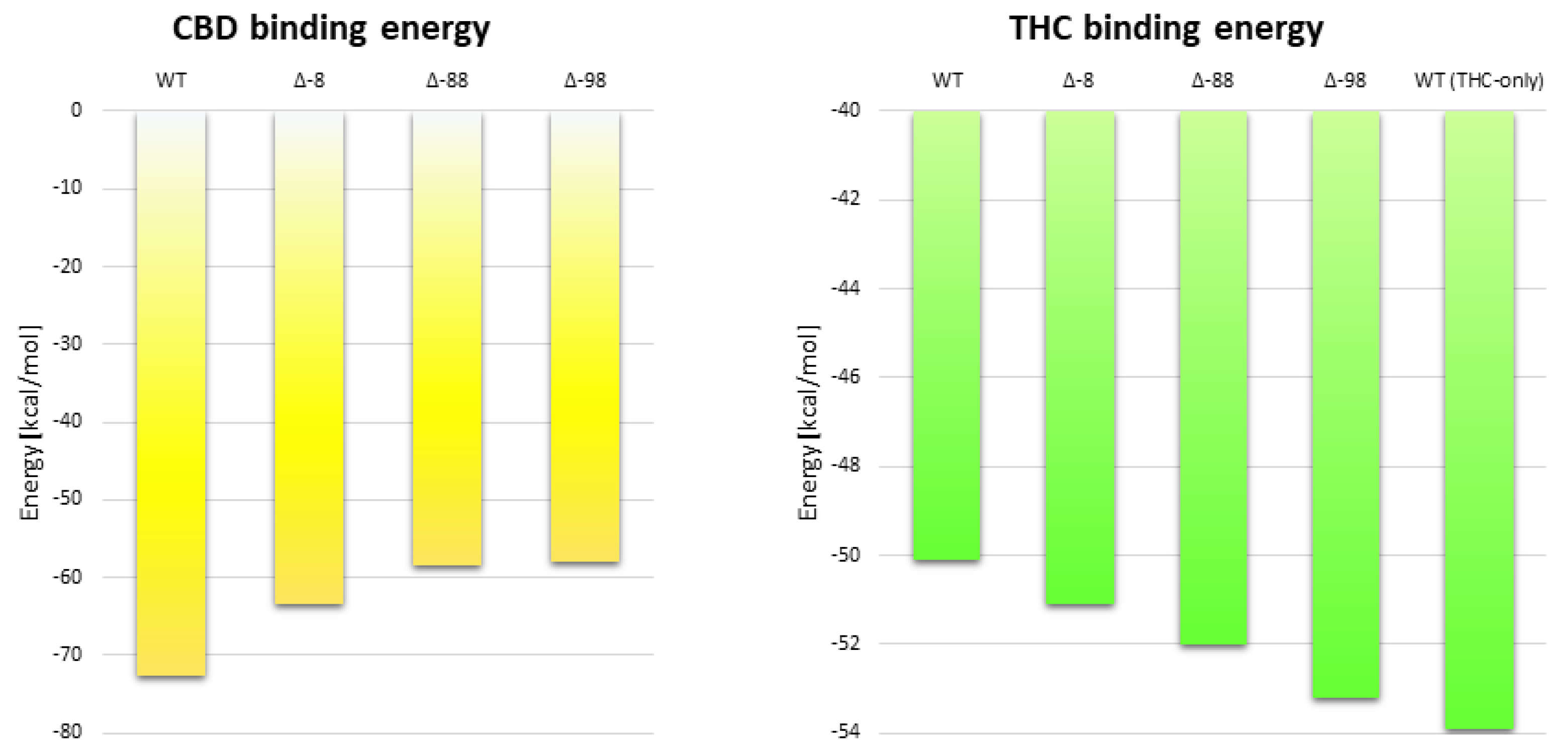
2.5. The Effect of Truncation of the N-Terminus on the Energy of CBD Binding
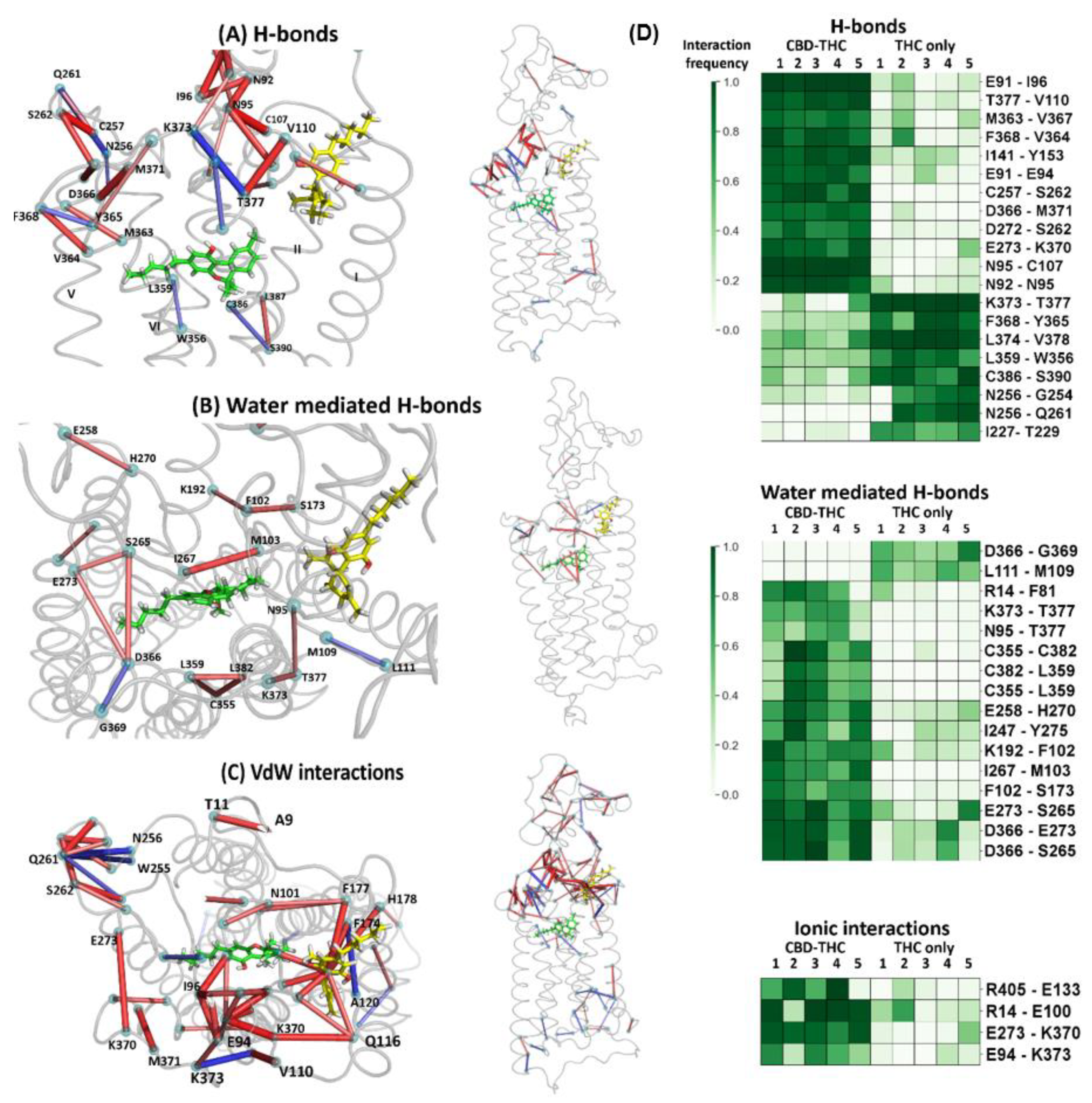
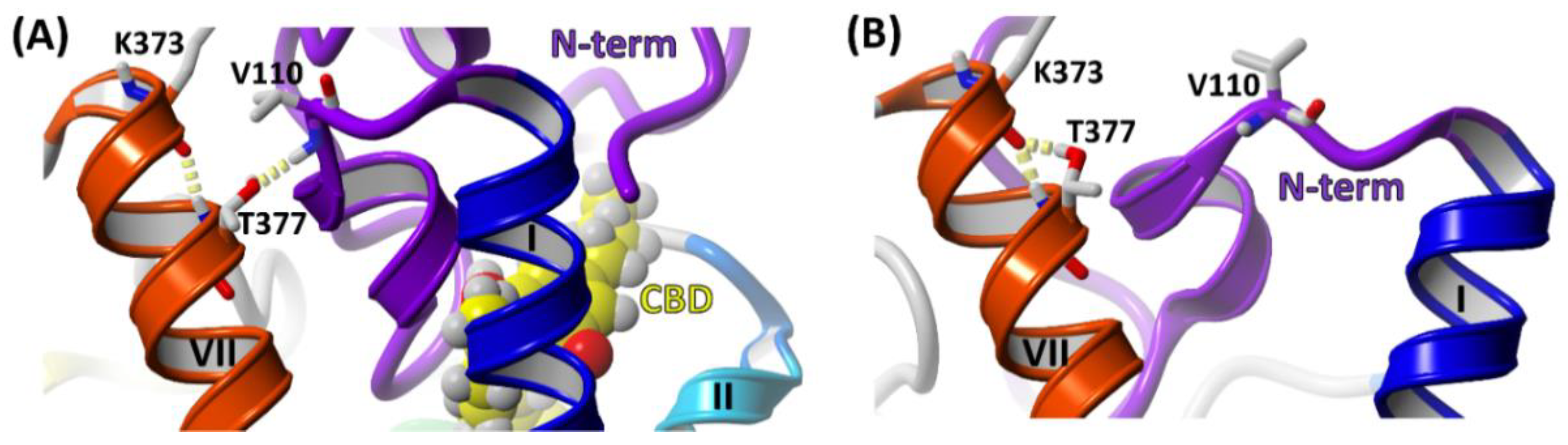
2.6. The Dynamic Contact Networks
3. Methods
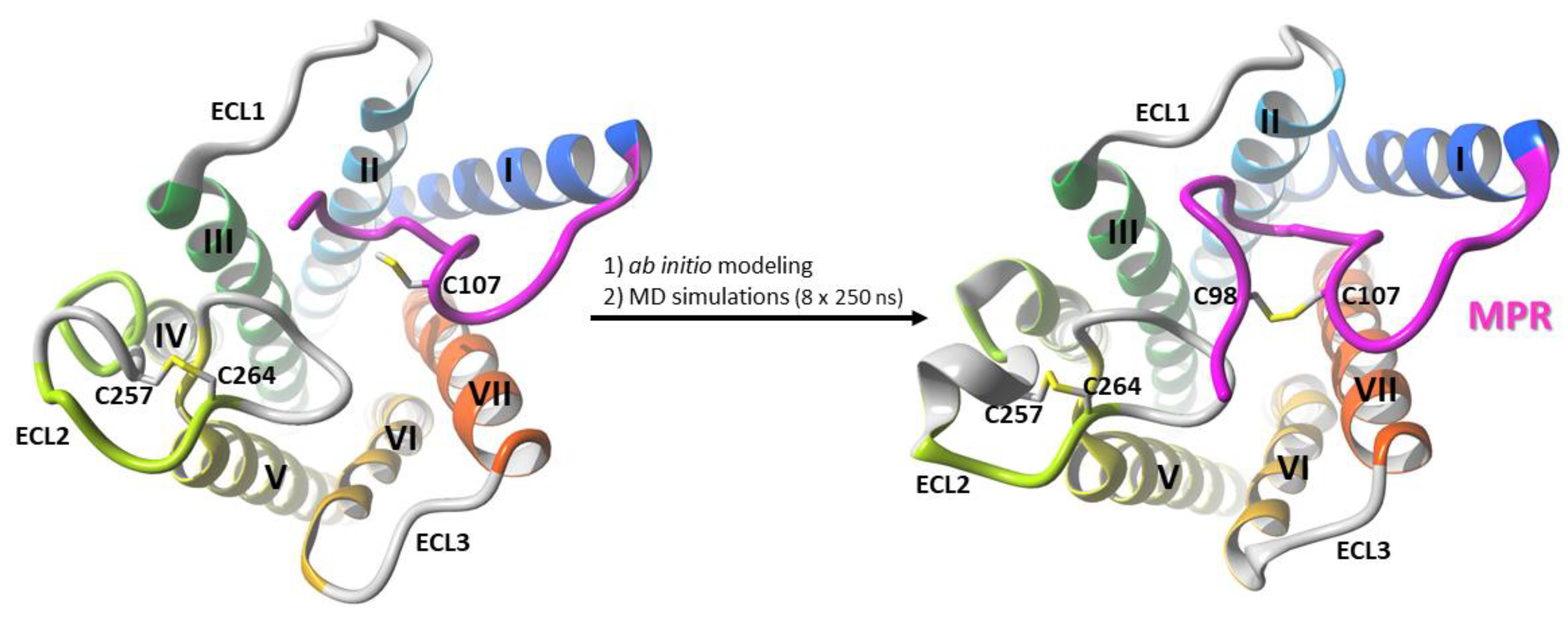
3.1. Modeling of the Membrane-Proximal Region (MPR) of CB1R
3.2. Building and Elucidating the Structure of Whole N-Terminus
3.3. Ligand Docking
3.4. All-Atom MD Simulations of CB1R Receptor with Ligands
3.5. Contact Network Analysis
3.6. Mutational Study of N-Terminus on CBD Binding to the CB1 Receptor
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| 2-AG | 2-arachidonylogycerol |
| AEA | N-arachidonoylethanolamine (anandamide) |
| CB1R | CB1 cannabinoid receptor |
| CBD | cannabidiol |
| COVID-19 | coronavirus disease 19 |
| GPCR | G protein coupled receptor |
| MD | molecular dynamics |
| MPR | membrane-proximal region |
| NAM | negative allosteric modulator |
| PAM | positive allosteric modulator |
| REST2 | replica exchange. molecular dynamics with solute tempering |
| RMSD | root mean square deviation |
| RMSF | root mean square fluctuation |
| THC | (−)-trans-Δ9-tetrahydrocannabinol |
References
- Howlett, A.C.; Breivogel, C.S.; Childers, S.R.; Deadwyler, S.A.; Hampson, R.E.; Porrino, L.J. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology 2004, 47 (Suppl. 1), 345–358. [Google Scholar] [CrossRef]
- Stasiulewicz, A.; Znajdek, K.; Grudzien, M.; Pawinski, T.; Sulkowska, A.J.I. A Guide to Targeting the Endocannabinoid System in Drug Design. Int. J. Mol. Sci. 2020, 21, 2778. [Google Scholar] [CrossRef]
- Alaverdashvili, M.; Laprairie, R.B. The future of type 1 cannabinoid receptor allosteric ligands. Drug Metab. Rev. 2018, 50, 14–25. [Google Scholar] [CrossRef]
- Lazzari, P.; Pau, A.; Tambaro, S.; Asproni, B.; Ruiu, S.; Pinna, G.; Mastinu, A.; Curzu, M.M.; Reali, R.; Bottazzi, M.E.; et al. Synthesis and pharmacological evaluation of novel 4-alkyl-5-thien-2’-yl pyrazole carboxamides. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 254–276. [Google Scholar] [CrossRef]
- Mastinu, A.; Pira, M.; Pani, L.; Pinna, G.A.; Lazzari, P. NESS038C6, a novel selective CB1 antagonist agent with anti-obesity activity and improved molecular profile. Behav. Brain Res. 2012, 234, 192–204. [Google Scholar] [CrossRef]
- Lu, D.; Immadi, S.S.; Wu, Z.; Kendall, D.A. Translational potential of allosteric modulators targeting the cannabinoid CB1 receptor. Acta Pharmacol. Sin. 2019, 40, 324–335. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, J.X.; Thomas, B.F.; Wiley, J.L.; Kenakin, T.P.; Zhang, Y. Allosteric Modulation: An Alternate Approach Targeting the Cannabinoid CB1 Receptor. Med. Res. Rev. 2017, 37, 441–474. [Google Scholar] [CrossRef]
- Price, M.R.; Baillie, G.L.; Thomas, A.; Stevenson, L.A.; Easson, M.; Goodwin, R.; McLean, A.; McIntosh, L.; Goodwin, G.; Walker, G.; et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 2005, 68, 1484–1495. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Bracey, M.H.; Hanson, M.A.; Masuda, K.R.; Stevens, R.C.; Cravatt, B.F. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 2002, 298, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Meini, S.; Bertini, S.; Digiacomo, M.; Macchia, M.; Manera, C. Allosteric modulators targeting cannabinoid cb1 and cb2 receptors: Implications for drug discovery. Future Med. Chem. 2019, 11, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Gentry, P.R.; Sexton, P.M.; Christopoulos, A. Novel Allosteric Modulators of G Protein-coupled Receptors. J. Biol. Chem. 2015, 290, 19478–19488. [Google Scholar] [CrossRef] [PubMed]
- Slivicki, R.A.; Xu, Z.; Kulkarni, P.M.; Pertwee, R.G.; Mackie, K.; Thakur, G.A.; Hohmann, A.G. Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence. Biol. Psychiatry 2018, 84, 722–733. [Google Scholar] [CrossRef]
- Hryhorowicz, S.; Kaczmarek-Rys, M.; Andrzejewska, A.; Staszak, K.; Hryhorowicz, M.; Korcz, A.; Slomski, R. Allosteric Modulation of Cannabinoid Receptor 1-Current Challenges and Future Opportunities. Int. J. Mol. Sci. 2019, 20, 5874. [Google Scholar] [CrossRef]
- Christopoulos, A.; Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef]
- Morales, P.; Jagerovic, N. Novel approaches and current challenges with targeting the endocannabinoid system. Expert. Opin. Drug Discov. 2020, 15, 917–930. [Google Scholar] [CrossRef]
- Mastinu, A.; Ribaudo, G.; Ongaro, A.; Bonini, S.A.; Memo, M.; Gianoncelli, A. Critical Review on the Chemical Aspects of Cannabidiol (CBD) and Harmonization of Computational Bioactivity Data. Curr. Med. Chem. 2021, 28, 213–237. [Google Scholar] [CrossRef]
- Esposito, G.; Pesce, M.; Seguella, L.; Sanseverino, W.; Lu, J.; Corpetti, C.; Sarnelli, G. The potential of cannabidiol in the COVID-19 pandemic. Br. J. Pharmacol. 2020, 177, 4967–4970. [Google Scholar] [CrossRef]
- Costiniuk, C.T.; Jenabian, M.A. Acute inflammation and pathogenesis of SARS-CoV-2 infection: Cannabidiol as a potential anti-inflammatory treatment? Cytokine Growth Factor Rev. 2020, 53, 63–65. [Google Scholar] [CrossRef]
- Salles, E.L.; Khodadadi, H.; Jarrahi, A.; Ahluwalia, M.; Paffaro, V.A., Jr.; Costigliola, V.; Yu, J.C.; Hess, D.C.; Dhandapani, K.M.; Baban, B. Cannabidiol (CBD) modulation of apelin in acute respiratory distress syndrome. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Byrareddy, S.N.; Mohan, M. SARS-CoV2 induced respiratory distress: Can cannabinoids be added to anti-viral therapies to reduce lung inflammation? Brain. Behav. Immun. 2020, 87, 120–121. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Manuzak, J.A.; Gott, T.M.; Kirkwood, J.S.; Coronado, E.; Hensley-McBain, T.; Miller, C.; Cheu, R.K.; Collier, A.C.; Funderburg, N.T.; Martin, J.N.; et al. Heavy Cannabis Use Associated with Reduction in Activated and Inflammatory Immune Cell Frequencies in Antiretroviral Therapy-Treated Human Immunodeficiency Virus-Infected Individuals. Clin. Infect. Dis. 2018, 66, 1872–1882. [Google Scholar] [CrossRef]
- Wang, B.; Kovalchuk, A.; Li, D.; Ilnytskyy, Y.; Kovalchuk, I.; Kovalchuk, O. In Search of Preventative Strategies: Novel Anti-Inflammatory High-CBD Cannabis Sativa Extracts Modulate ACE2 Expression in COVID-19 Gateway Tissues. Preprints 2020. [Google Scholar] [CrossRef]
- Russo, E.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Ross, R.A.; Craib, S.J.; Thomas, A. (-)-Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur. J. Pharmacol. 2002, 456, 99–106. [Google Scholar] [CrossRef]
- Fay, J.F.; Farrens, D.L. The membrane proximal region of the cannabinoid receptor CB1 N-terminus can allosterically modulate ligand affinity. Biochemistry 2013, 52, 8286–8294. [Google Scholar] [CrossRef]
- Baillie, G.L.; Horswill, J.G.; Anavi-Goffer, S.; Reggio, P.H.; Bolognini, D.; Abood, M.E.; McAllister, S.; Strange, P.G.; Stephens, G.J.; Pertwee, R.G.; et al. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol. Pharmacol. 2013, 83, 322–338. [Google Scholar] [CrossRef]
- Sabatucci, A.; Tortolani, D.; Dainese, E.; Maccarrone, M. In silico mapping of allosteric ligand binding sites in type-1 cannabinoid receptor. Biotechnol. Appl. Biochem. 2018, 65, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Fierro, A.; Pessoa-Mahana, C.D. Cannabidiol binding and negative allosteric modulation at the cannabinoid type 1 receptor in the presence of delta-9-tetrahydrocannabinol: An In Silico study. PLoS ONE 2019, 14, e0220025. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Mordalski, S.; Pandy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keseru, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Murphy, J.W.; Kendall, D.A. Integrity of extracellular loop 1 of the human cannabinoid receptor 1 is critical for high-affinity binding of the ligand CP 55,940 but not SR 141716A. Biochem. Pharmacol. 2003, 65, 1623–1631. [Google Scholar] [CrossRef]
- Jakowiecki, J.; Orzel, U.; Chawananon, S.; Miszta, P.; Filipek, S. The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations. Molecules 2020, 25, 1930. [Google Scholar] [CrossRef]
- Jo, S.; Jiang, W. A generic implementation of replica exchange with solute tempering (REST2) algorithm in NAMD for complex biophysical simulations. Comput. Phys. Commun. 2015, 197, 304–311. [Google Scholar] [CrossRef]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed. 2015, 54, 556–559. [Google Scholar] [CrossRef]
- Yuan, S.; Peng, Q.; Palczewski, K.; Vogel, H.; Filipek, S. Mechanistic Studies on the Stereoselectivity of the Serotonin 5-HT1A Receptor. Angew. Chem. Int. Ed. 2016, 55, 8661–8665. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers—Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Mandell, D.J.; Coutsias, E.A.; Kortemme, T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 2009, 6, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wires Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Canutescu, A.A.; Dunbrack, R.L., Jr. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003, 12, 963–972. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman , P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MD Simulation Type | Average Length [μs] | Repeats | REST2 Replicas | Cumulative Length [μs] |
|---|---|---|---|---|
| CB1R with MPR | 0.25 | 8 | 1 | 2 |
| REST2 of CB1R with complete N-terminus | 1.2 | 6 | 16 | 115.2 |
| CB1R with CBD-THC (for contact map) | 1 | 5 | 1 | 5 |
| CB1R with THC-only (for contact map) | 1 | 5 | 1 | 5 |
| CB1R N-terminus stability corroboration (with and without CBD) | 2 × 3 | 3 | 1 | 18 |
| CB1R with truncated N-term (mutation studies) | 3 × 1 | 2 | 1 | 6 |
| Total | 151.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakowiecki, J.; Abel, R.; Orzeł, U.; Pasznik, P.; Preissner, R.; Filipek, S. Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol—A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling. Molecules 2021, 26, 2456. https://doi.org/10.3390/molecules26092456
Jakowiecki J, Abel R, Orzeł U, Pasznik P, Preissner R, Filipek S. Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol—A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling. Molecules. 2021; 26(9):2456. https://doi.org/10.3390/molecules26092456
Chicago/Turabian StyleJakowiecki, Jakub, Renata Abel, Urszula Orzeł, Paweł Pasznik, Robert Preissner, and Sławomir Filipek. 2021. "Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol—A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling" Molecules 26, no. 9: 2456. https://doi.org/10.3390/molecules26092456
APA StyleJakowiecki, J., Abel, R., Orzeł, U., Pasznik, P., Preissner, R., & Filipek, S. (2021). Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol—A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling. Molecules, 26(9), 2456. https://doi.org/10.3390/molecules26092456