
Towards Conformation-Sensitive Inhibition of Gyrase: Implications of Mechanistic Insight for the Identification and Improvement of Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: The Bacterial Type IIA Topoisomerase Gyrase
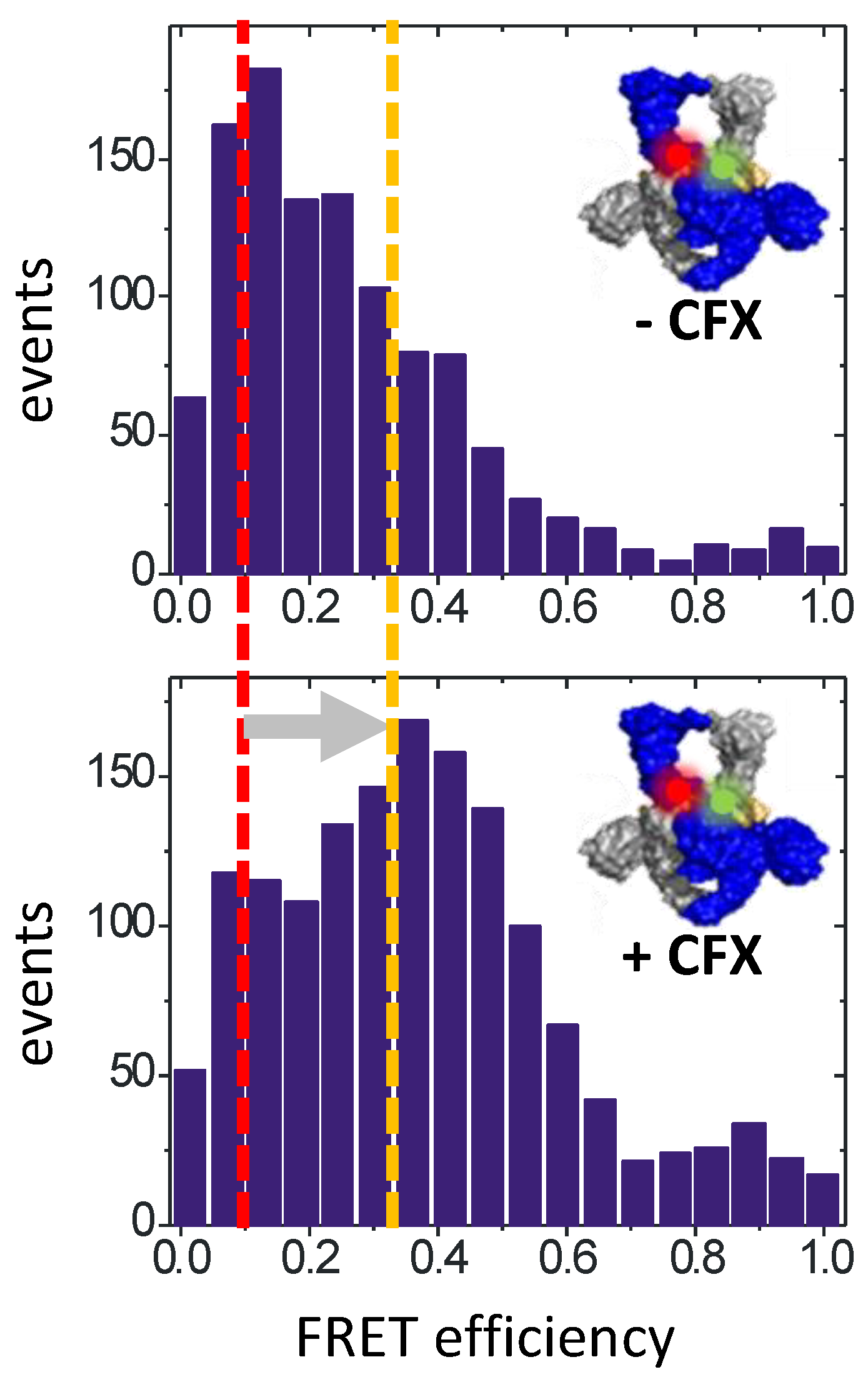
2. Inhibiting Gyrase: Current Strategies
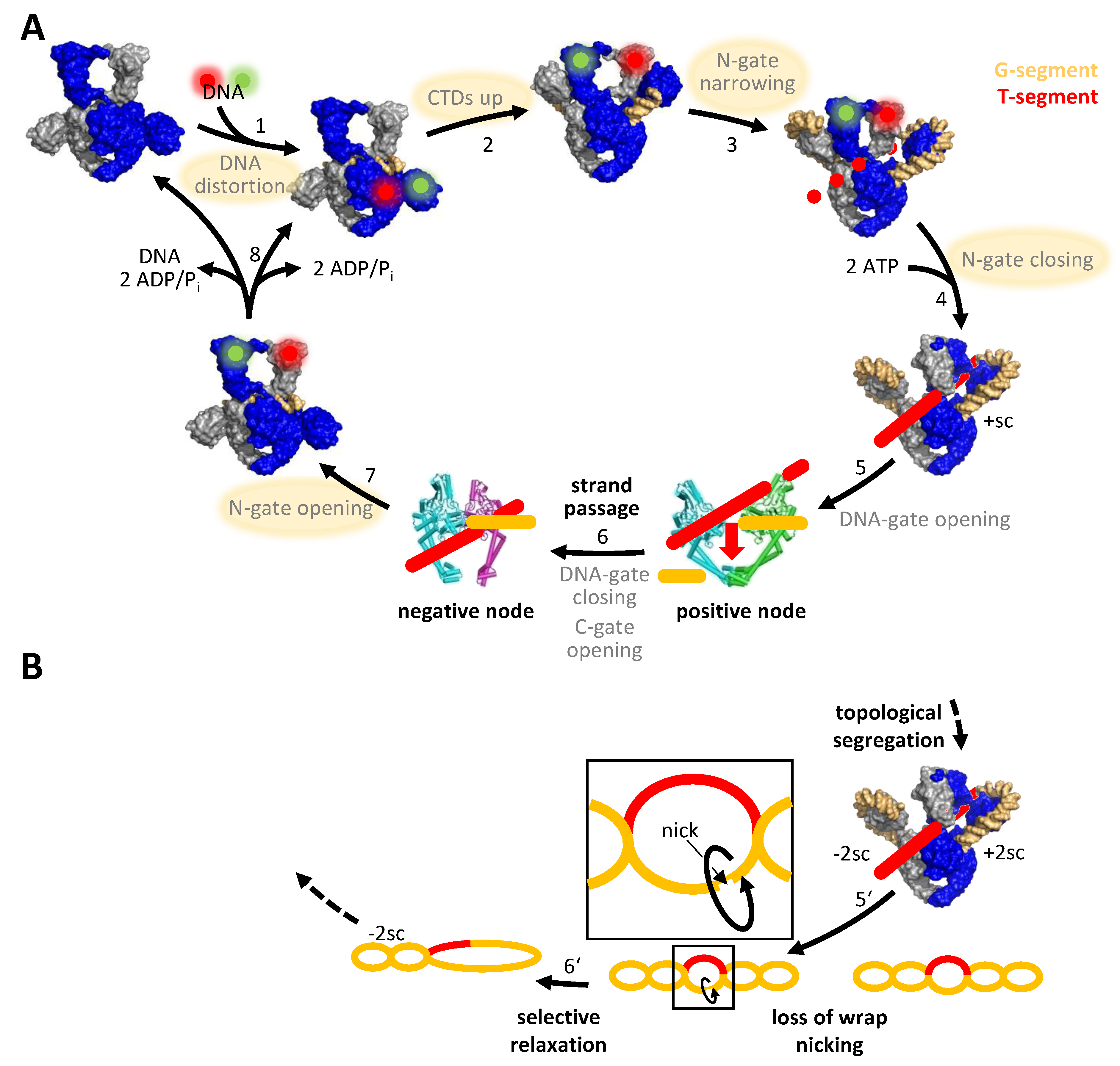
3. DNA Supercoiling Requires a Series of ATP- and DNA-Induced Conformational Changes of Gyrase
4. The Role of Symmetry: An Alternative Mechanism for DNA Supercoiling without Strand Passage
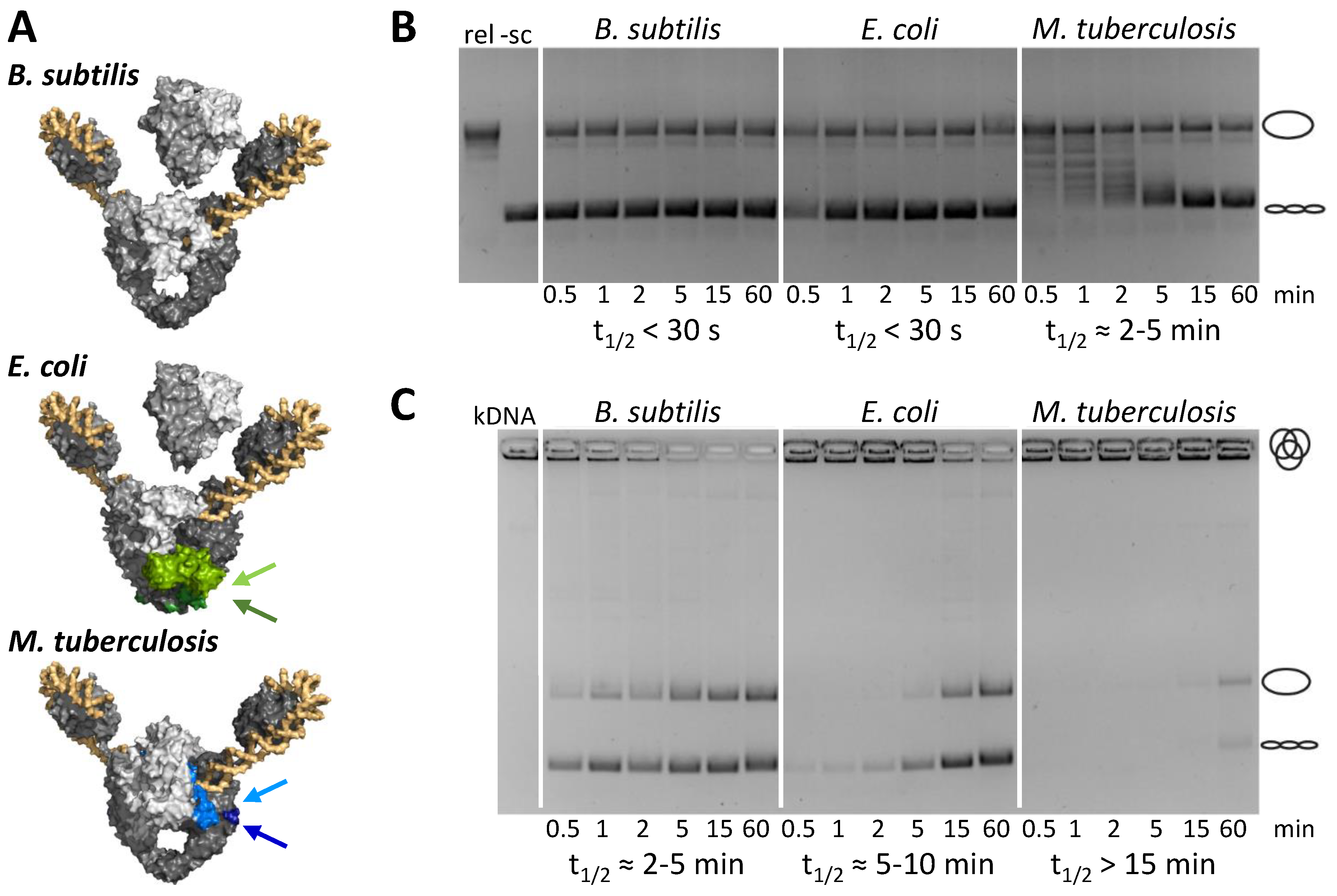
5. Species-Specific Elements Modulate Gyrase Activity
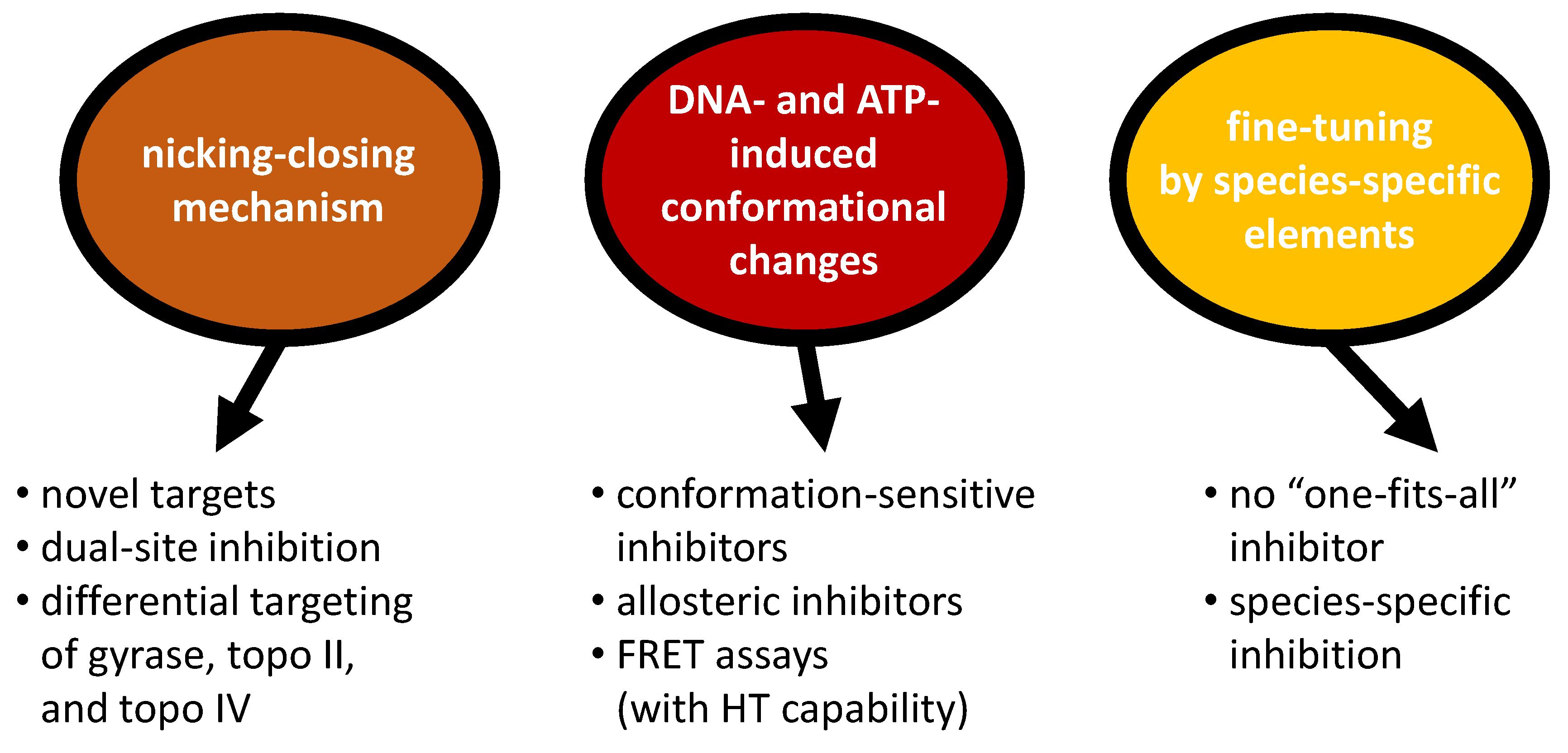
6. Implications for Gyrase Inhibition: Conclusions and Outlook
Funding
Conflicts of Interest
References
- Gellert, M.; Mizuuchi, K.; O’Dea, M.H.; Nash, H.A. DNA gyrase: An enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci. USA 1976, 73, 3872–3876. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, K.; O’Dea, M.H.; Gellert, M. DNA gyrase: Subunit structure and ATPase activity of the purified enzyme. Proc. Natl. Acad. Sci. USA 1978, 75, 5960–5963. [Google Scholar] [CrossRef]
- Mizuuchi, K.; Fisher, L.M.; O’Dea, M.H.; Gellert, M. DNA gyrase action involves the introduction of transient double-strand breaks into DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 1847–1851. [Google Scholar] [CrossRef]
- Fisher, L.M.; Mizuuchi, K.; O’Dea, M.H.; Ohmori, H.; Gellert, M. Site-specific interaction of DNA gyrase with DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 4165–4169. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.; Gellert, M. The DNA dependence of the ATPase activity of DNA gyrase. J. Biol. Chem. 1984, 259, 14472–14480. [Google Scholar] [CrossRef]
- Cozzarelli, N.R. DNA gyrase and the supercoiling of DNA. Science 1980, 207, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Zechiedrich, E.L.; Cozzarelli, N.R. Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes Dev. 1995, 9, 2859–2869. [Google Scholar] [CrossRef]
- Zechiedrich, E.L.; Khodursky, A.B.; Cozzarelli, N.R. Topoisomerase IV, not gyrase, decatenates products of site-specific recombination in Escherichia coli. Genes Dev. 1997, 11, 2580–2592. [Google Scholar] [CrossRef]
- Sissi, C.; Palumbo, M. In front of and behind the replication fork: Bacterial type IIA topoisomerases. Cell. Mol. Life Sci. 2010, 67, 2001–2024. [Google Scholar] [CrossRef]
- Mizuuchi, K.; Mizuuchi, M.; O’Dea, M.H.; Gellert, M. Cloning and simplified purification of Escherichia coli DNA gyrase A and B proteins. J. Biol. Chem. 1984, 259, 9199–9201. [Google Scholar] [CrossRef]
- Morais Cabral, J.H.; Jackson, A.P.; Smith, C.V.; Shikotra, N.; Maxwell, A.; Liddington, R.C. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature 1997, 388, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Reece, R.J.; Maxwell, A. The C-terminal domain of the Escherichia coli DNA gyrase A subunit is a DNA-binding protein. Nucleic Acids Res. 1991, 19, 1399–1405. [Google Scholar] [CrossRef][Green Version]
- Corbett, K.D.; Shultzaberger, R.K.; Berger, J.M. The C-terminal domain of DNA gyrase A adopts a DNA-bending beta-pinwheel fold. Proc. Natl. Acad. Sci. USA 2004, 101, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Wigley, D.B.; Davies, G.J.; Dodson, E.J.; Maxwell, A.; Dodson, G. Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature 1991, 351, 624–629. [Google Scholar] [CrossRef]
- Fu, G.; Wu, J.; Liu, W.; Zhu, D.; Hu, Y.; Deng, J.; Zhang, X.E.; Bi, L.; Wang, D.C. Crystal structure of DNA gyrase B’ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 2009, 37, 5908–5916. [Google Scholar] [CrossRef]
- Papillon, J.; Menetret, J.F.; Batisse, C.; Helye, R.; Schultz, P.; Potier, N.; Lamour, V. Structural insight into negative DNA supercoiling by DNA gyrase, a bacterial type 2A DNA topoisomerase. Nucleic Acids Res. 2013, 41, 7815–7827. [Google Scholar] [CrossRef]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. [Google Scholar] [CrossRef]
- Gubaev, A.; Klostermeier, D. DNA-induced narrowing of the gyrase N-gate coordinates T-segment capture and strand passage. Proc. Natl. Acad. Sci. USA 2011, 108, 14085–14090. [Google Scholar] [CrossRef]
- Hartmann, S.; Gubaev, A.; Klostermeier, D. Binding and Hydrolysis of a Single ATP Is Sufficient for N-Gate Closure and DNA Supercoiling by Gyrase. J. Mol. Biol. 2017, 429, 3717–3729. [Google Scholar] [CrossRef]
- Williams, N.L.; Howells, A.J.; Maxwell, A. Locking the ATP-operated clamp of DNA gyrase: Probing the mechanism of strand passage. J. Mol. Biol. 2001, 306, 969–984. [Google Scholar] [CrossRef]
- Tingey, A.P.; Maxwell, A. Probing the role of the ATP-operated clamp in the strand-passage reaction of DNA gyrase. Nucleic Acids Res. 1996, 24, 4868–4873. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brown, P.O.; Cozzarelli, N.R. A sign inversion mechanism for enzymatic supercoiling of DNA. Science 1979, 206, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Wang, J.C. DNA-DNA gyrase complex: The wrapping of the DNA duplex outside the enzyme. Cell 1978, 15, 979–984. [Google Scholar] [CrossRef]
- Horowitz, D.S.; Wang, J.C. Mapping the active site tyrosine of Escherichia coli DNA gyrase. J. Biol. Chem. 1987, 262, 5339–5344. [Google Scholar] [CrossRef]
- Forterre, P.; Gribaldo, S.; Gadelle, D.; Serre, M.C. Origin and evolution of DNA topoisomerases. Biochimie 2007, 89, 427–446. [Google Scholar] [CrossRef]
- Drlica, K.; Franco, R.J. Inhibitors of DNA topoisomerases. Biochemistry 1988, 27, 2253–2259. [Google Scholar] [CrossRef]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol 2019, 431, 3427–3449. [Google Scholar] [CrossRef]
- Hansen, F.G.; von Meyenburg, K. Characterization of the dnaA, gyrB and other genes in the dnaA region of the Escherichia coli chromosome on specialized transducing phages lambda-tna. Mol. Gen. Genet. 1979, 175, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Gubaev, A.; Klostermeier, D. The mechanism of negative DNA supercoiling: A cascade of DNA-induced conformational changes prepares gyrase for strand passage. DNA Repair 2014, 16, 23–34. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Klostermeier, D. Mapping the Spectrum of Conformational States of the DNA- and C-Gates in Bacillus subtilis Gyrase. J. Mol. Biol. 2013, 425, 2632–2640. [Google Scholar] [CrossRef]
- Lanz, M.A.; Klostermeier, D. The GyrA-box determines the geometry of DNA bound to gyrase and couples DNA binding to the nucleotide cycle. Nucleic Acids Res. 2012, 40, 10893–10903. [Google Scholar] [CrossRef][Green Version]
- Lanz, M.A.; Klostermeier, D. Guiding strand passage: DNA-induced movement of the gyrase C-terminal domains defines an early step in the supercoiling cycle. Nucleic Acids Res. 2011, 39, 9681–9694. [Google Scholar] [CrossRef] [PubMed]
- Gubaev, A.; Hilbert, M.; Klostermeier, D. The DNA gate of Bacillus subtilis gyrase is predominantly in the closed conformation during the DNA supercoiling reaction. Proc. Natl. Acad. Sci. USA 2009, 106, 13278–13283. [Google Scholar] [CrossRef]
- Chan, P.F.; Germe, T.; Bax, B.D.; Huang, J.; Thalji, R.K.; Bacque, E.; Checchia, A.; Chen, D.; Cui, H.; Ding, X.; et al. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc. Natl. Acad. Sci. USA 2017, 114, 4492–4500. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Andrews, B. Allosteric inhibition of the DNA-dependent ATPase activity of Escherichia coli DNA gyrase by a representative of a novel class of inhibitors. Biochem. Pharmacol. 2012, 84, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.A.; Jackson, A.P.; Howells, A.J.; Maxwell, A. The 43-kilodalton N-terminal fragment of the DNA gyrase B protein hydrolyzes ATP and binds coumarin drugs. Biochemistry 1993, 32, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Sugino, A.; Higgins, N.P.; Brown, P.O.; Peebles, C.L.; Cozzarelli, N.R. Energy coupling in DNA gyrase and the mechanism of action of novobiocin. Proc. Natl. Acad. Sci. USA 1978, 75, 4838–4842. [Google Scholar] [CrossRef] [PubMed]
- Sugino, A.; Cozzarelli, N.R. The intrinsic ATPase of DNA gyrase. J. Biol. Chem. 1980, 255, 6299–6306. [Google Scholar] [CrossRef]
- Lewis, R.J.; Singh, O.M.; Smith, C.V.; Skarzynski, T.; Maxwell, A.; Wonacott, A.J.; Wigley, D.B. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996, 15, 1412–1420. [Google Scholar] [CrossRef]
- Lamour, V.; Hoermann, L.; Jeltsch, J.M.; Oudet, P.; Moras, D. Crystallization of the 43 kDa ATPase domain of Thermus thermophilus gyrase B in complex with novobiocin. Acta Cryst. D Biol. Cryst. 2002, 58, 1376–1378. [Google Scholar] [CrossRef]
- Lamour, V.; Hoermann, L.; Jeltsch, J.M.; Oudet, P.; Moras, D. An open conformation of the Thermus thermophilus gyrase B ATP-binding domain. J. Biol. Chem. 2002, 277, 18947–18953. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.J.; Flatman, R.H.; Mitchenall, L.A.; Stevenson, C.E.; Le, T.B.; Clarke, T.A.; McKay, A.R.; Fiedler, H.P.; Buttner, M.J.; Lawson, D.M.; et al. A crystal structure of the bifunctional antibiotic simocyclinone D8, bound to DNA gyrase. Science 2009, 326, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Hearnshaw, S.J.; Edwards, M.J.; Stevenson, C.E.; Lawson, D.M.; Maxwell, A. A new crystal structure of the bifunctional antibiotic simocyclinone D8 bound to DNA gyrase gives fresh insight into the mechanism of inhibition. J. Mol. Biol. 2014, 426, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Flatman, R.H.; Eustaquio, A.; Li, S.M.; Heide, L.; Maxwell, A. Structure-activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis. Antimicrob. Agents Chemother. 2006, 50, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Schimana, J.; Fiedler, H.P.; Groth, I.; Sussmuth, R.; Beil, W.; Walker, M.; Zeeck, A. Simocyclinones, novel cytostatic angucyclinone antibiotics produced by Streptomyces antibioticus Tu 6040. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 2000, 53, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, A.A.; Patel, M.R.; Jacobson, B.A.; Escobedo, M.; Ellis, K.; Oppegard, L.M.; Hiasa, H.; Kratzke, R.A. Anti-proliferative effects of simocyclinone D8 (SD8), a novel catalytic inhibitor of topoisomerase II. Investig. New Drugs 2010, 28, 20–25. [Google Scholar] [CrossRef]
- Lu, J.; Patel, S.; Sharma, N.; Soisson, S.M.; Kishii, R.; Takei, M.; Fukuda, Y.; Lumb, K.J.; Singh, S.B. Structures of kibdelomycin bound to Staphylococcus aureus GyrB and ParE showed a novel U-shaped binding mode. Chem. Biol. 2014, 9, 2023–2031. [Google Scholar] [CrossRef]
- Agrawal, A.; Roue, M.; Spitzfaden, C.; Petrella, S.; Aubry, A.; Hann, M.; Bax, B.; Mayer, C. Mycobacterium tuberculosis DNA gyrase ATPase domain structures suggest a dissociative mechanism that explains how ATP hydrolysis is coupled to domain motion. Biochem. J. 2013, 456, 263–273. [Google Scholar] [CrossRef]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef]
- Yoshida, H.; Bogaki, M.; Nakamura, M.; Yamanaka, L.M.; Nakamura, S. Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Bogaki, M.; Nakamura, M.; Nakamura, S. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob. Agents Chemother. 1990, 34, 1271–1272. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Germe, T.; Voros, J.; Jeannot, F.; Taillier, T.; Stavenger, R.A.; Bacque, E.; Maxwell, A.; Bax, B.D. A new class of antibacterials, the imidazopyrazinones, reveal structural transitions involved in DNA gyrase poisoning and mechanisms of resistance. Nucleic Acids Res. 2018, 46, 4114–4128. [Google Scholar] [CrossRef] [PubMed]
- Black, M.T.; Stachyra, T.; Platel, D.; Girard, A.M.; Claudon, M.; Bruneau, J.M.; Miossec, C. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. [Google Scholar] [CrossRef] [PubMed]
- Kolaric, A.; Germe, T.; Hrast, M.; Stevenson, C.E.M.; Lawson, D.M.; Burton, N.P.; Voros, J.; Maxwell, A.; Minovski, N.; Anderluh, M. Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds. Nat. Commun. 2021, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, W.; Tiffany, C.; Scangarella-Oman, N.; Perry, C.; Hossain, M.; Ashton, T.; Dumont, E. Efficacy, Safety, and Tolerability of Gepotidacin (GSK2140944) in the Treatment of Patients with Suspected or Confirmed Gram-Positive Acute Bacterial Skin and Skin Structure Infections. Antimicrob. Agents Chemother. 2017, 61, e02095. [Google Scholar] [CrossRef]
- Biedenbach, D.J.; Bouchillon, S.K.; Hackel, M.; Miller, L.A.; Scangarella-Oman, N.E.; Jakielaszek, C.; Sahm, D.F. In Vitro Activity of Gepotidacin, a Novel Triazaacenaphthylene Bacterial Topoisomerase Inhibitor, against a Broad Spectrum of Bacterial Pathogens. Antimicrob. Agents Chemother. 2016, 60, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Weidlich, D.; Klostermeier, D. Single-Molecule Confocal FRET Microscopy to Dissect Conformational Changes in the Catalytic Cycle of DNA Topoisomerases. Methods Enzym. 2016, 581, 317–351. [Google Scholar] [CrossRef]
- Gubaev, A.; Klostermeier, D. Potassium ions are required for nucleotide-induced closure of gyrase N-gate. J. Biol. Chem. 2012, 287, 10916–10921. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.C.; Berger, J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 2007, 450, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Huang, N.L.; Lin, J.H.; Wu, C.C.; Wang, Y.R.; Yu, Y.J.; Gilson, M.K.; Chan, N.L. Structural insights into the gating of DNA passage by the topoisomerase II DNA-gate. Nat. Commun. 2018, 9, 3085. [Google Scholar] [CrossRef]
- Stelljes, J.T.; Weidlich, D.; Gubaev, A.; Klostermeier, D. Gyrase containing a single C-terminal domain catalyzes negative supercoiling of DNA by decreasing the linking number in steps of two. Nucleic Acids Res. 2018, 46, 6773–6784. [Google Scholar] [CrossRef]
- Gubaev, A.; Weidlich, D.; Klostermeier, D. DNA gyrase with a single catalytic tyrosine can catalyze DNA supercoiling by a nicking-closing mechanism. Nucleic Acids Res. 2016, 44, 10354–10366. [Google Scholar] [CrossRef]
- Sugino, A.; Peebles, C.L.; Kreuzer, K.N.; Cozzarelli, N.R. Mechanism of action of nalidixic acid: Purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl. Acad. Sci. USA 1977, 74, 4767–4771. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Micrococcus luteus DNA gyrase: Active components and a model for its supercoiling of DNA. Proc. Natl. Acad. Sci. USA 1978, 75, 2098–2102. [Google Scholar] [CrossRef]
- Klostermeier, D. Why two? On the role of (a-)symmetry for negative DNA supercoiling by gyrase. Int. J. Mol. Sci. 2018, 19, 1489. [Google Scholar] [CrossRef] [PubMed]
- Schoeffler, A.J.; May, A.P.; Berger, J.M. A domain insertion in Escherichia coli GyrB adopts a novel fold that plays a critical role in gyrase function. Nucleic Acids Res. 2010, 38, 7830–7844. [Google Scholar] [CrossRef] [PubMed]
- Petrella, S.; Capton, E.; Raynal, B.; Giffard, C.; Thureau, A.; Bonnete, F.; Alzari, P.M.; Aubry, A.; Mayer, C. Overall Structures of Mycobacterium tuberculosis DNA Gyrase Reveal the Role of a Corynebacteriales GyrB-Specific Insert in ATPase Activity. Structure 2019, 27, 579–589. [Google Scholar] [CrossRef]
- Weidlich, D.; Klostermeier, D. Functional interactions between gyrase subunits are optimized in a species-specific manner. J. Biol. Chem. 2020, 295, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Tretter, E.M.; Berger, J.M. Mechanisms for defining the supercoiling setpoint of DNA gyrase orthologs I. A non-conserved acidic C-terminal tail modulates E. coli gyrase activity. J. Biol. Chem. 2012, 287, 18636–18644. [Google Scholar] [CrossRef]
- Lanz, M.A.; Farhat, M.; Klostermeier, D. The acidic C-terminal tail of the GyrA subunit moderates the DNA supercoiling activity of Bacillus subtilis gyrase. J. Biol. Chem. 2014, 289, 12275–12285. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Graybosch, D.M.; Huetsch, J.C.; Verdine, G.L. A superhelical spiral in the Escherichia coli DNA gyrase A C-terminal domain imparts unidirectional supercoiling bias. J. Biol. Chem. 2005, 280, 26177–26184. [Google Scholar] [CrossRef] [PubMed]
- Tretter, E.M.; Berger, J.M. Mechanisms for Defining Supercoiling Setpoint By DNA Gyrase Orthologs II. The shape of the GyrA CTD is not a sole determinant for controlling supercoiling efficiency. J. Biol. Chem. 2012, 287, 18645–18654. [Google Scholar] [CrossRef]
- Stracy, M.; Wollman, A.J.M.; Kaja, E.; Gapinski, J.; Lee, J.E.; Leek, V.A.; McKie, S.J.; Mitchenall, L.A.; Maxwell, A.; Sherratt, D.J.; et al. Single-molecule imaging of DNA gyrase activity in living Escherichia coli. Nucleic Acids Res. 2019, 47, 210–220. [Google Scholar] [CrossRef]
- Yaginuma, H.; Kawai, S.; Tabata, K.V.; Tomiyama, K.; Kakizuka, A.; Komatsuzaki, T.; Noji, H.; Imamura, H. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 2014, 4, 6522. [Google Scholar] [CrossRef]
- Cayley, S.; Lewis, B.A.; Guttman, H.J.; Record, M.T., Jr. Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J. Mol. Biol. 1991, 222, 281–300. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Trach, S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [Google Scholar] [CrossRef]
- Kolesov, G.; Wunderlich, Z.; Laikova, O.N.; Gelfand, M.S.; Mirny, L.A. How gene order is influenced by the biophysics of transcription regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 13948–13953. [Google Scholar] [CrossRef] [PubMed]
- Strahilevitz, J.; Hooper, D.C. Dual targeting of topoisomerase IV and gyrase to reduce mutant selection: Direct testing of the paradigm by using WCK-1734, a new fluoroquinolone, and ciprofloxacin. Antimicrob. Agents Chemother. 2005, 49, 1949–1956. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klostermeier, D. Towards Conformation-Sensitive Inhibition of Gyrase: Implications of Mechanistic Insight for the Identification and Improvement of Inhibitors. Molecules 2021, 26, 1234. https://doi.org/10.3390/molecules26051234
Klostermeier D. Towards Conformation-Sensitive Inhibition of Gyrase: Implications of Mechanistic Insight for the Identification and Improvement of Inhibitors. Molecules. 2021; 26(5):1234. https://doi.org/10.3390/molecules26051234
Chicago/Turabian StyleKlostermeier, Dagmar. 2021. "Towards Conformation-Sensitive Inhibition of Gyrase: Implications of Mechanistic Insight for the Identification and Improvement of Inhibitors" Molecules 26, no. 5: 1234. https://doi.org/10.3390/molecules26051234
APA StyleKlostermeier, D. (2021). Towards Conformation-Sensitive Inhibition of Gyrase: Implications of Mechanistic Insight for the Identification and Improvement of Inhibitors. Molecules, 26(5), 1234. https://doi.org/10.3390/molecules26051234