
Anharmonic Vibrational Frequencies of Water Borane and Associated Molecules


Abstract
:1. Introduction
2. Computational Details
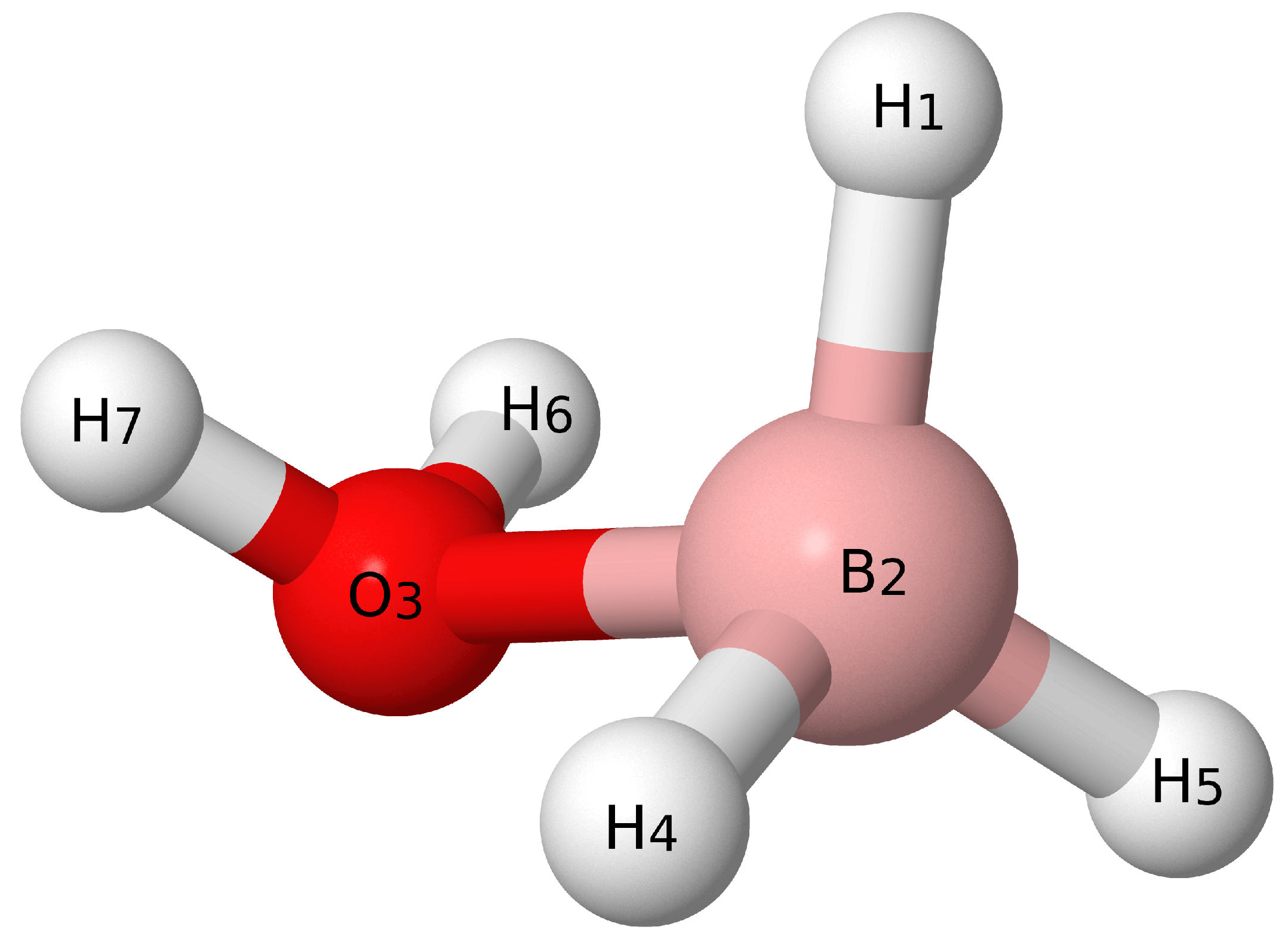
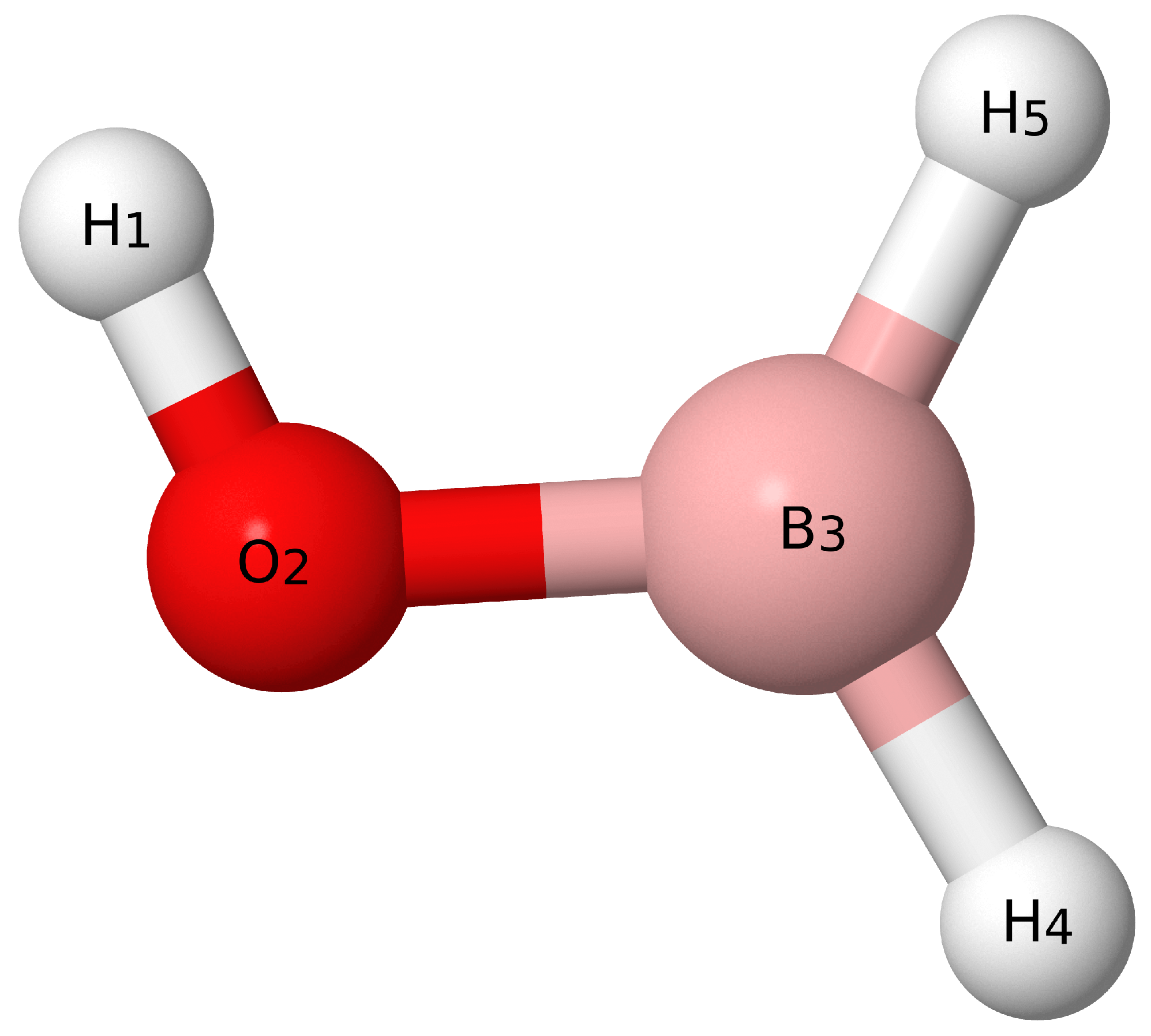
3. Results and Discussion
3.1. Benchmarks
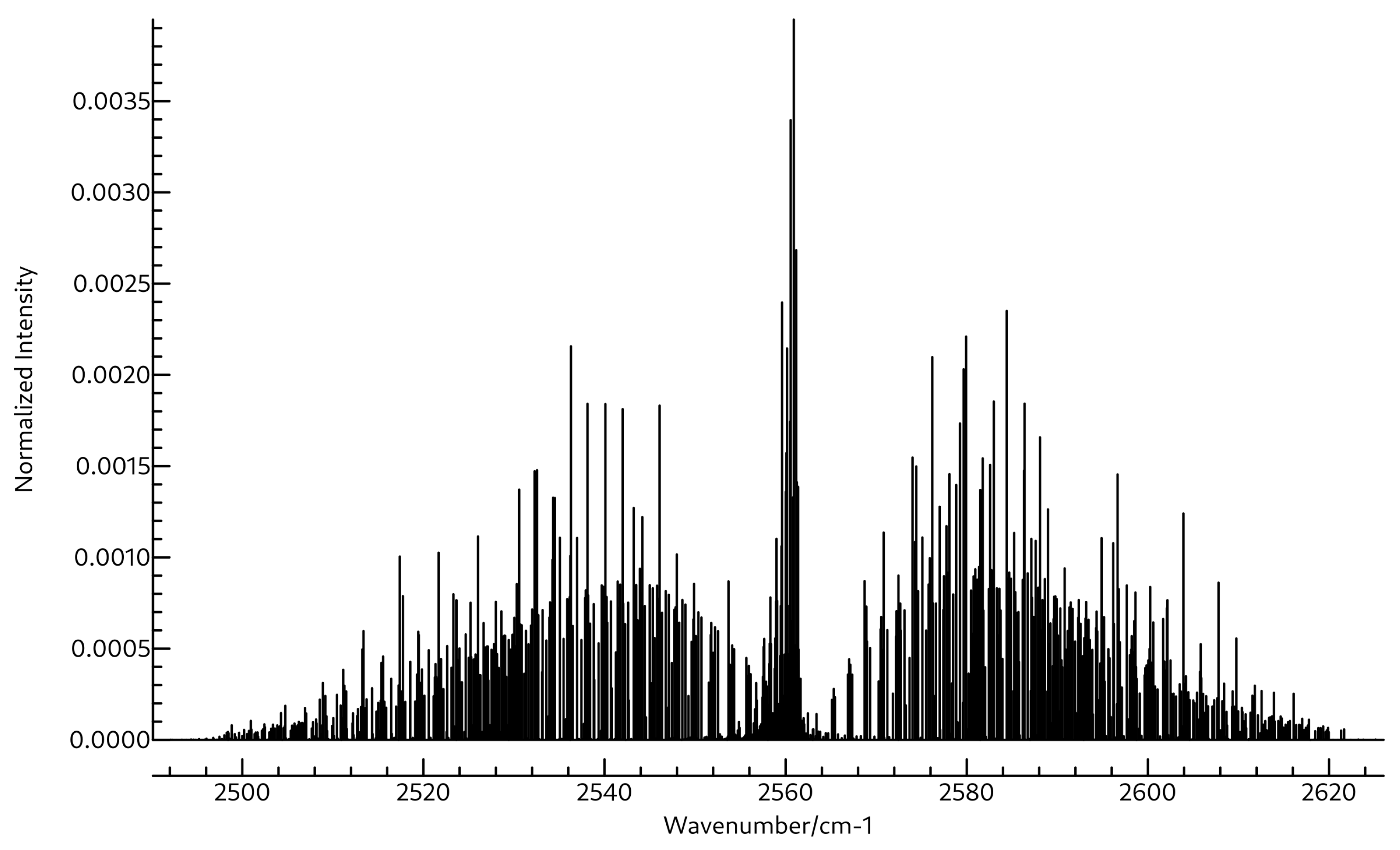
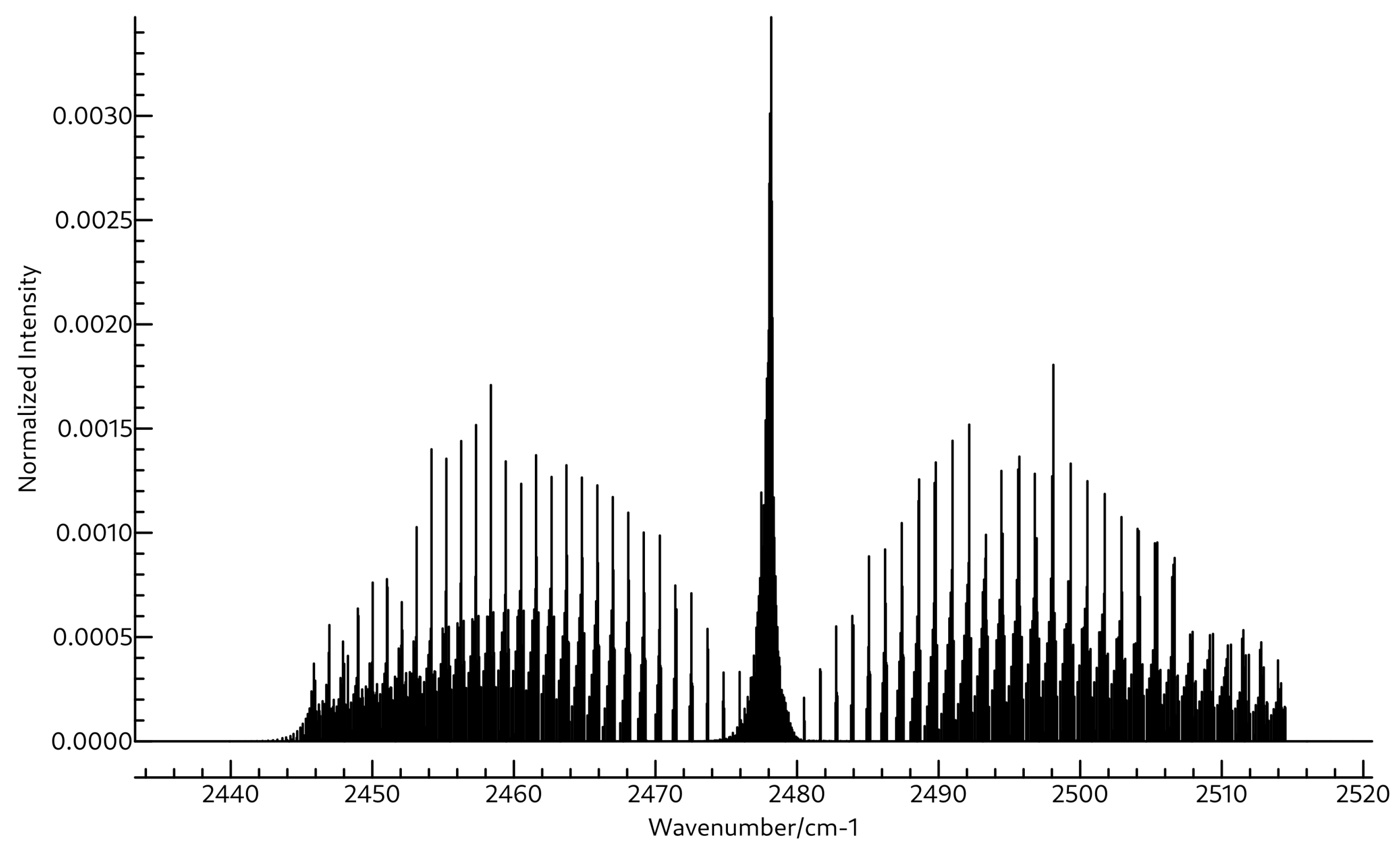
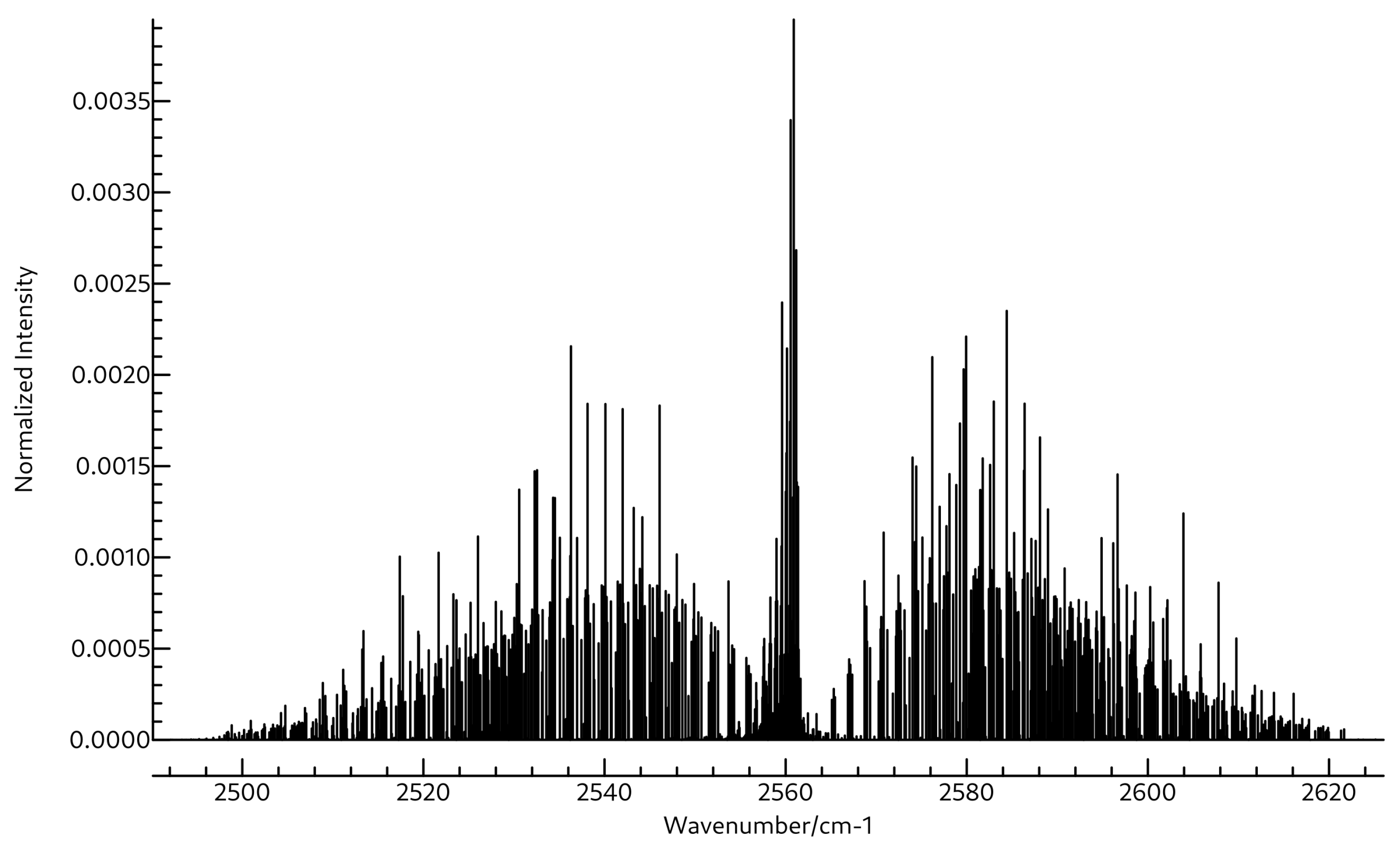
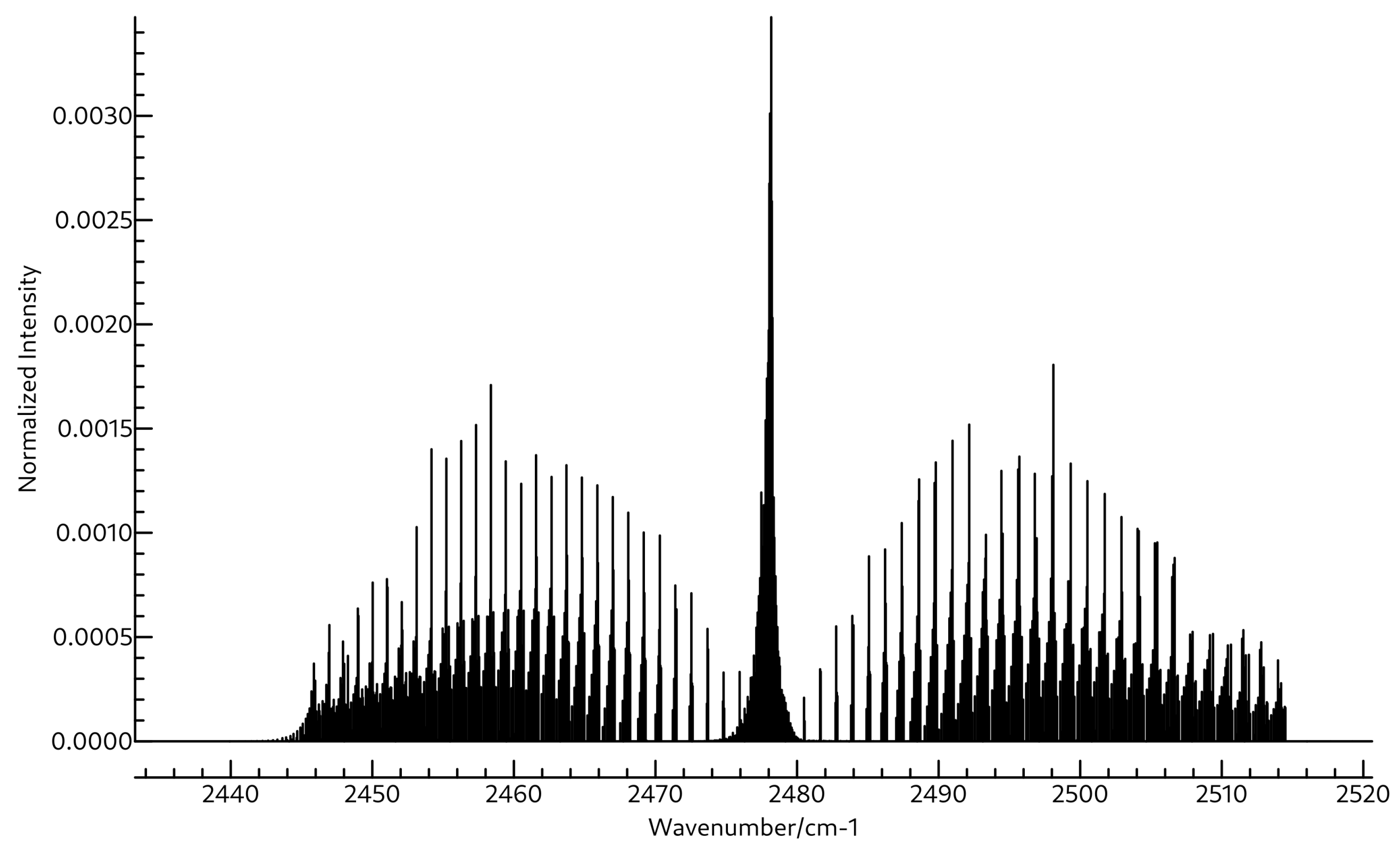
3.2. Spectroscopic Data
3.3. B Isotopologues
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephens, F.H.; Pons, V.; Baker, R.T. Ammonia-borane: The Hydrogen Source par excellence. Dalton Trans. 2007, 25, 2613–2626. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Chen, J. Ammonia Borane as an Efficient and Lightweight Hydrogen Storage Medium. Energy Environ. Sci. 2008, 4, 479–483. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.P.M.; Manners, I. Ammonia-Borane and Related Compounds as Dihydrogen Sources. Chem. Rev. 2010, 110, 4079–4124. [Google Scholar] [CrossRef] [PubMed]
- Sams, R.L.; Xantheas, S.S.; Blake, T.A. Vapor Phase Infrared Spectroscopy and Ab Initio Fundamental Anharmonic Frequencies of Ammonia Borane. J. Phys. Chem. A 2012, 116, 3124–3136. [Google Scholar] [CrossRef]
- Weiss, H.G.; Shapiro, I. Mechanism of the Hydrolysis of Diborane in the Vapor Phase. J. Am. Chem. Soc. 1953, 75, 1221–1224. [Google Scholar] [CrossRef]
- Nanayakkara, S.; Freindorf, M.; Tao, Y.; Kraka, E. Modeling Hydrogen Release from Water with Borane and Alane Catalysts: A Unified Reaction Valley Approach. J. Phys. Chem. A 2020, 124, 8978–8993. [Google Scholar] [CrossRef]
- Swinnen, S.; Nguygen, V.S.; Sakai, S.; Nguyen, M.T. Calculations suggest facile hydrogen release from water using boranes and alanes as catalysts. Chem. Phys. Lett. 2009, 472, 175–180. [Google Scholar] [CrossRef]
- Hirscher, M.; Autrey, T.; Orimo, S.I. Hydrogen Energy. Chem. Phys. Chem. 2019, 20, 1157. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.W.; Yuen, H.C.; Yan, S.R.; Huang, W.L. The Multiple Selections of Fostering Applications of Hydrogen Energy by Integrating Economic and Industrial Evaluation of Different Regions. Int. J. Hydrogen Energy 2019, 44, 29390–29398. [Google Scholar] [CrossRef]
- Martin, A.; Agnoletti, M.F.; Brangier, E. Users in the Design of Hydrogen Energy Systems: A Systematic Review. Int. J. Hydrogen Energy 2020, 45, 11889–11900. [Google Scholar] [CrossRef]
- Maghami, M.R.; Hassani, R.; Gomes, C.; Hizam, H.; Othman, M.L.; Behmanesh, M. Hybrid Energy Management with Respect to a Hydrogen Energy System and Demand Response. Int. J. Hydrogen Energy 2020, 45, 1499–1509. [Google Scholar] [CrossRef]
- Dreux, K.M.; McNamara, L.E.; Kelly, J.T.; Wright, A.M.; Hammer, N.I.; Tschumper, G.S. Probing Dative and Dihydrogen Bonding in Ammonia Borane with Electronic Structure Computations and Raman under Nitrogen Spectroscopy. J. Phys. Chem. A 2017, 121, 5884–5893. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, B.R.; Valencia, E.M.; Rushing, S.C.; Tschumper, G.S.; Fortenberry, R.C. Anharmonic Vibrational Frequencies of Ammonia Borane (BH3NH3). J. Chem. Phys. 2021, 154, 041104. [Google Scholar] [CrossRef]
- Hess, N.J.; Bowden, M.E.; Parvanov, V.M.; Mundy, C.; Kathmann, S.M.; Schenter, G.K.; Autrey, T. Spectroscopic Studies of the Phase Transition in Ammonia Borane: Raman Spectroscopy of Single Crystal NH3BH3 as a Function of Temperature from 88 to 330K. J. Chem. Phys. 2008, 128, 034508. [Google Scholar] [CrossRef]
- Paolone, A.; Teocoli, F.; Sanna, S.; Palumbo, O.; Autrey, T. Temperature Dependence of the Infrared Spectrum of Ammonia Borane: Librations, Rotations, and Molecular Vibrations. J. Phys. Chem. C 2013, 117, 729–734. [Google Scholar] [CrossRef]
- Doerksen, E.S.; Fortenberry, R.C. A Coincidence Between Bond Strength, Atomic Abundance, and the Composition of Rocky Materials. ACS Earth Space Chem. 2020, 4, 812–816. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Replogle, E. Highly Correlated Systems: Structure, Binding Energy and Harmonic Vibrational Frequencies of Ozone. Chem. Phys. Lett. 1989, 158, 207–212. [Google Scholar] [CrossRef]
- Adler, T.B.; Knizia, G.; Werner, H.J. A Simple and Efficient CCSD(T)-F12 Approximation. J. Chem. Phys. 2007, 127, 221106. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G.; Adler, T.B.; Werner, H.J. Simplified CCSD(T)-F12 Methods: Theory and Benchmarks. J. Chem. Phys. 2009, 130, 054104. [Google Scholar] [CrossRef]
- Fortenberry, R.C.; Lee, T.J. Computational Vibrational Spectroscopy for the Detection of Molecules in Space. Ann. Rep. Comput. Chem. 2019, 15, 173–202. [Google Scholar]
- Huang, X.; Valeev, E.F.; Lee, T.J. Comparison of One-Particle Basis Set Extrapolation to Explicitly Correlated Methods for the Calculation of Accurate Quartic Force Fields, Vibrational Frequencies, and Spectroscopic Constants: Application to H2O, N2H+, NO2+, and C2H2. J. Chem. Phys. 2010, 133, 244108. [Google Scholar] [CrossRef]
- Agbaglo, D.; Lee, T.J.; Thackston, R.; Fortenberry, R.C. A Small Molecule with PAH Vibrational Properties and a Detectable Rotational Spectrum: c-(C)C3H2, Cyclopropenylidenyl Carbene. Astrophys. J. 2019, 871, 236. [Google Scholar] [CrossRef]
- Agbaglo, D.; Fortenberry, R.C. The Performance of CCSD(T)-F12/aug-cc-pVTZ for the Computation of Anharmonic Fundamental Vibrational Frequencies. Int. J. Quantum Chem. 2019, 119, e25899. [Google Scholar] [CrossRef]
- Agbaglo, D.; Fortenberry, R.C. The Performance of Explicitly Correlated Wavefunctions [CCSD(T)-F12b] in the Computation of Anharmonic Vibrational Frequencies. Chem. Phys. Lett. 2019, 734, 136720. [Google Scholar] [CrossRef]
- Westbrook, B.R.; Fortenberry, R.C. Anharmonic Frequencies of (MO)2 & Related Hydrides for M = Mg, Al, Si, P, S, Ca, & Ti and Heuristics for Predicting Anharmonic Corrections of Inorganic Oxides. J. Phys. Chem. A 2020, 124, 3191–3204. [Google Scholar] [PubMed]
- Valiev, R.R.; Nasibullin, R.T.; Cherepanov, V.N.; Baryshnikov, G.V.; Sundholm, D.; Ågren, H.; Minaev, B.F.; Kurtén, T. First-principles calculations of anharmonic and deuteration effects on the photophysical properties of polyacenes and porphyrinoids. Phys. Chem. Chem. Phys. 2020, 22, 22314–22323. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.B.; Westbrook, B.R.; Fortenberry, R.C.; Lee, T.J. Highly-Accurate Quartic Force Fields for the Prediction of Anharmonic Rotational Constants and Fundamental Vibrational Frequencies. Spectrochim. Acta A 2021, 248, 119184. [Google Scholar] [CrossRef]
- Watrous, A.G.; Westbrook, B.R.; Fortenberry, R.C. F12-TZ-cCR: A Methodology for Faster and Still Highly-Accurate Quartic Force Fields. J. Phys. Chem. A 2021. submitted. [Google Scholar] [CrossRef]
- Kawaguchi, K. Fourier Transform Infrared Spectroscopy of BH3 with the First Identification of the ν4 Band. J. Mol. Spectrosc. 2020, 373, 111352. [Google Scholar] [CrossRef]
- Kawashima, Y.; Endo, Y.; Hirota, E. Microwave Spectrum, Molecular Structure, and Force Field of HBO. J. Mol. Spectrosc. 1989, 133, 116–127. [Google Scholar] [CrossRef]
- Kawashima, Y.; Takeo, H.; Matsumura, C. Microwave Spectrum of Borinic Acid BH2OH. J. Chem. Phys. 1981, 74, 5430–5435. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D.A.; et al. MOLPRO, Version 2020.1, a Package of ab Initio Programs. 2020. Available online: http://www.molpro.net (accessed on 14 September 2021).
- Peterson, K.A.; Adler, T.B.; Werner, H.J. Systematically Convergent Basis Sets for Explicitly Correlated Wavefunctions: The Atoms H, He, B-Ne, and Al-Ar. J. Chem. Phys. 2008, 128, 084102. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.G.; Mazumder, S.; Peterson, K.A. Correlation Consistent Basis Sets for Molecular Core-Valence Effects with Explicitly Correlated Wave Functions: The Atoms B-Ne and Al-Ar. J. Chem. Phys. 2010, 132, 054108. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.; Kroll, N. Quantum Electrodynamical Corrections to the Fine Structure of Helium. Ann. Phys. 1974, 82, 89–155. [Google Scholar] [CrossRef]
- Jansen, G.; Hess, B.A. Revision of the Douglas-Kroll Hamiltonian. Phys. Rev. A 1989, 39, 6016–6017. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Bowman, J.M.; Fortenberry, R.C.; Mancini, J.S.; Lee, T.J.; Crawford, T.D.; Klemperer, W.; Francisco, J.S. The Structure, Anharmonic Vibrational Frequencies, and Intensities of NNHNN+. J. Phys. Chem. A 2015, 119, 11623–11631. [Google Scholar] [CrossRef]
- Finney, B.; Fortenberry, R.C.; Francisco, J.S.; Peterson, K.A. A Spectroscopic Case for SPSi Detection: The Third-Row in a Single Molecule. J. Chem. Phys. 2016, 145, 124311. [Google Scholar] [CrossRef]
- Westbrook, B.R.; Patel, D.J.; Dallas, J.D.; Swartzfager, G.C.; Lee, T.J.; Fortenberry, R.C. Fundamental Vibrational Frequencies and Spectroscopic Constants of Substituted Cyclopropenylidene (c-C3HX, X = F, Cl, CN). J. Phys. Chem. A 2021, in press. [Google Scholar] [CrossRef]
- Watrous, A.G.; Westbrook, B.R.; Davis, M.C.; Fortenberry, R.C. Vibrational and Rotational Spectral Data for Possible Interstellar Detection of AlH3OH2, SiH3OH, and SiH3NH2. Mon. Not. R. Astron. Soc. 2021, 508, 2613–2619. [Google Scholar] [CrossRef]
- Watrous, A.G.; Davis, M.C.; Fortenberry, R.C. Pathways to Detection of Strongly-Bound Inorganic Species: The Vibrational and Rotational Spectral Data of AlH2OH, HMgOH, AlH2NH2, and HMgNH2. Front. Astron. Space Sci. 2021, 8, 1–11. [Google Scholar] [CrossRef]
- Allen, W.D.; Császár, A.G.; Szalay, V.; Mills, I.M.; Horner, D.A. INTDER 2005 Is a General Program Written by W. D. Allen and Coworkers, Which Performs Vibrational Analysis and Higher-Order Non-Linear Transformations. 2005. Available online: https://r410berry.com/static/media/INTDER05_Manual.09dde4b6.pdf (accessed on 15 September 2021).
- Mills, I.M. Vibration-Rotation Structure in Asymmetric- and Symmetric-Top Molecules. In Molecular Spectroscopy—Modern Research; Rao, K.N., Mathews, C.W., Eds.; Academic Press: New York, NY, USA, 1972; pp. 115–140. [Google Scholar]
- Gaw, J.F.; Willets, A.; Green, W.H.; Handy, N.C. SPECTRO: A Program for the Derivation of Spectrscopic Constants from Provided Quartic Force Fields. In Advances in Molecular Vibrations and Collision Dynamics; Bowman, J.M., Ratner, M.A., Eds.; JAI Press, Inc.: Greenwich, CT, USA, 1991; pp. 170–185. [Google Scholar]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure; During, J.R., Ed.; Elsevier: Amsterdam, The Netherlands, 1977; pp. 1–89. [Google Scholar]
- Papousek, D.; Aliev, M.R. Molecular Vibration-Rotation Spectra; Elsevier: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Martin, J.M.L.; Taylor, P.R. Accurate ab Initio Quartic Force Field for trans-HNNH and Treatment of Resonance Polyads. Spectrochim. Acta A 1997, 53, 1039–1050. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Lee, T.J.; Taylor, P.R.; François, J.P. The Anharmonic Force Field of Ethylene, C2H4, by Means of Accurate ab Initio Calculations. J. Chem. Phys. 1995, 103, 2589–2602. [Google Scholar] [CrossRef] [Green Version]
- Western, C.M. PGOPHER, A Program for Simulating Rotational, Vibrational and Electronic Spectra. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 221–242. [Google Scholar] [CrossRef] [Green Version]

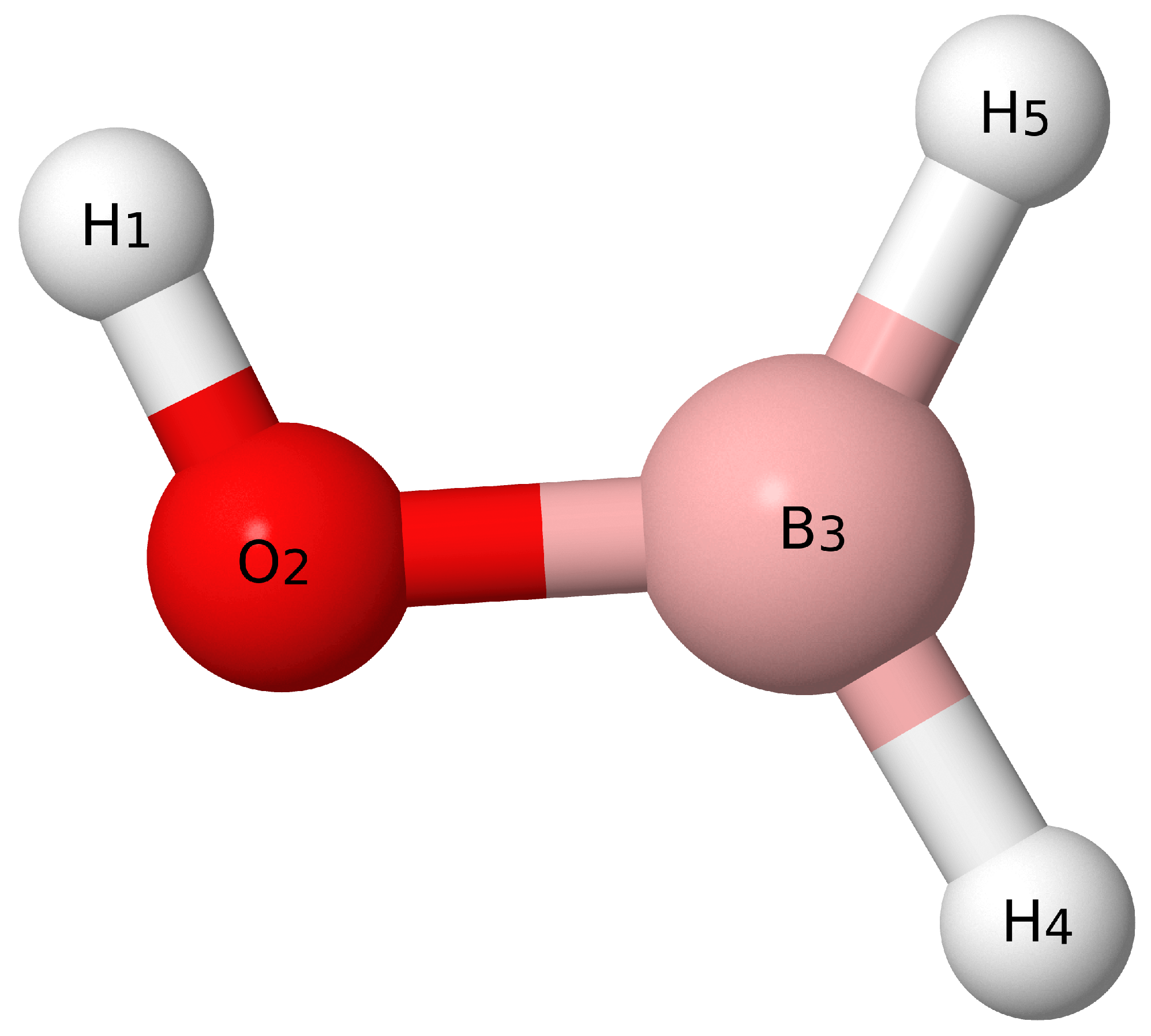
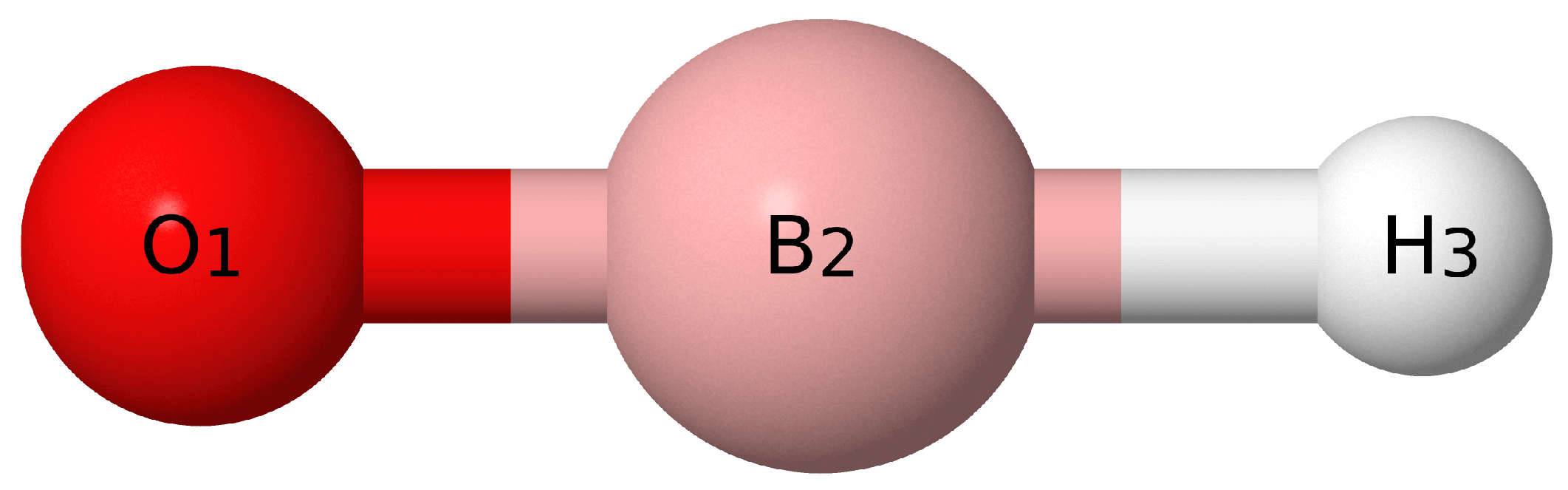
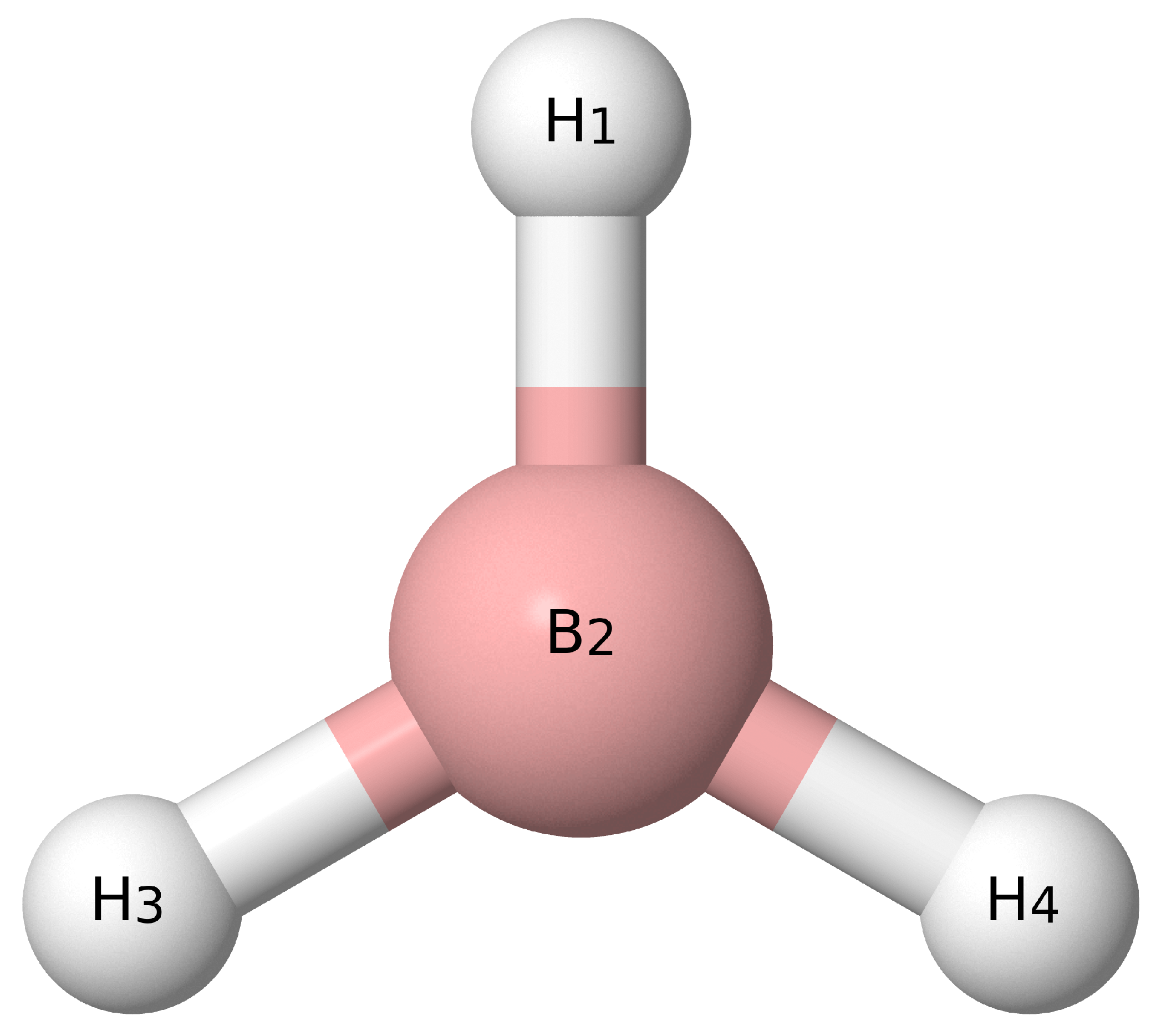


{kind=link}
{kind=link}
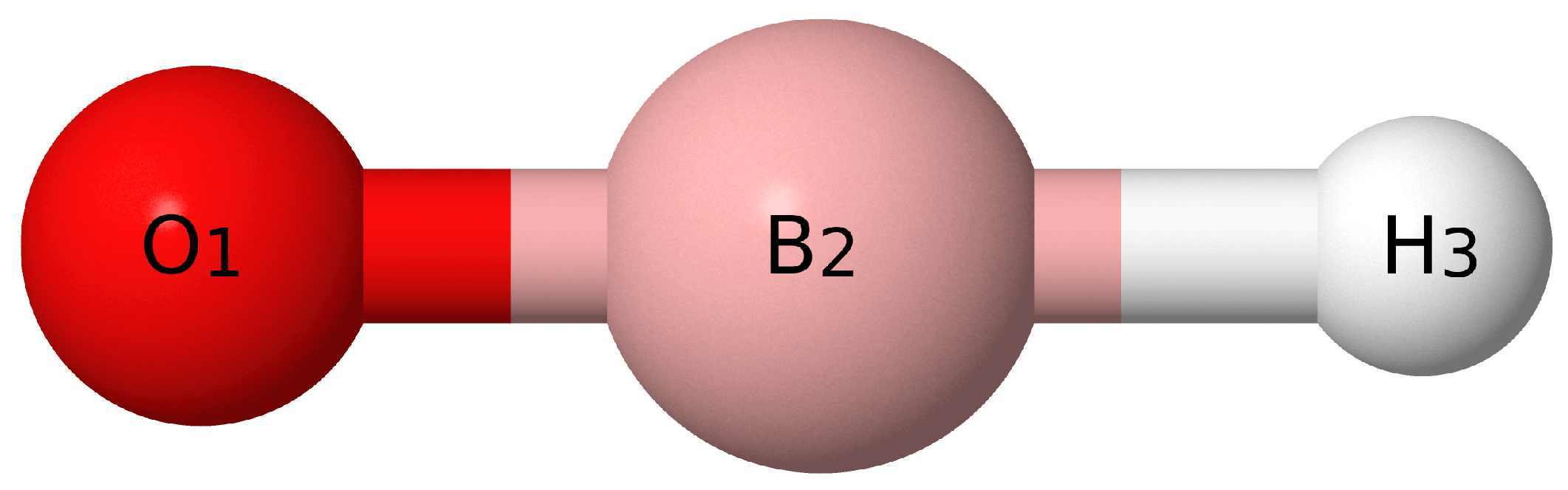{kind=link}
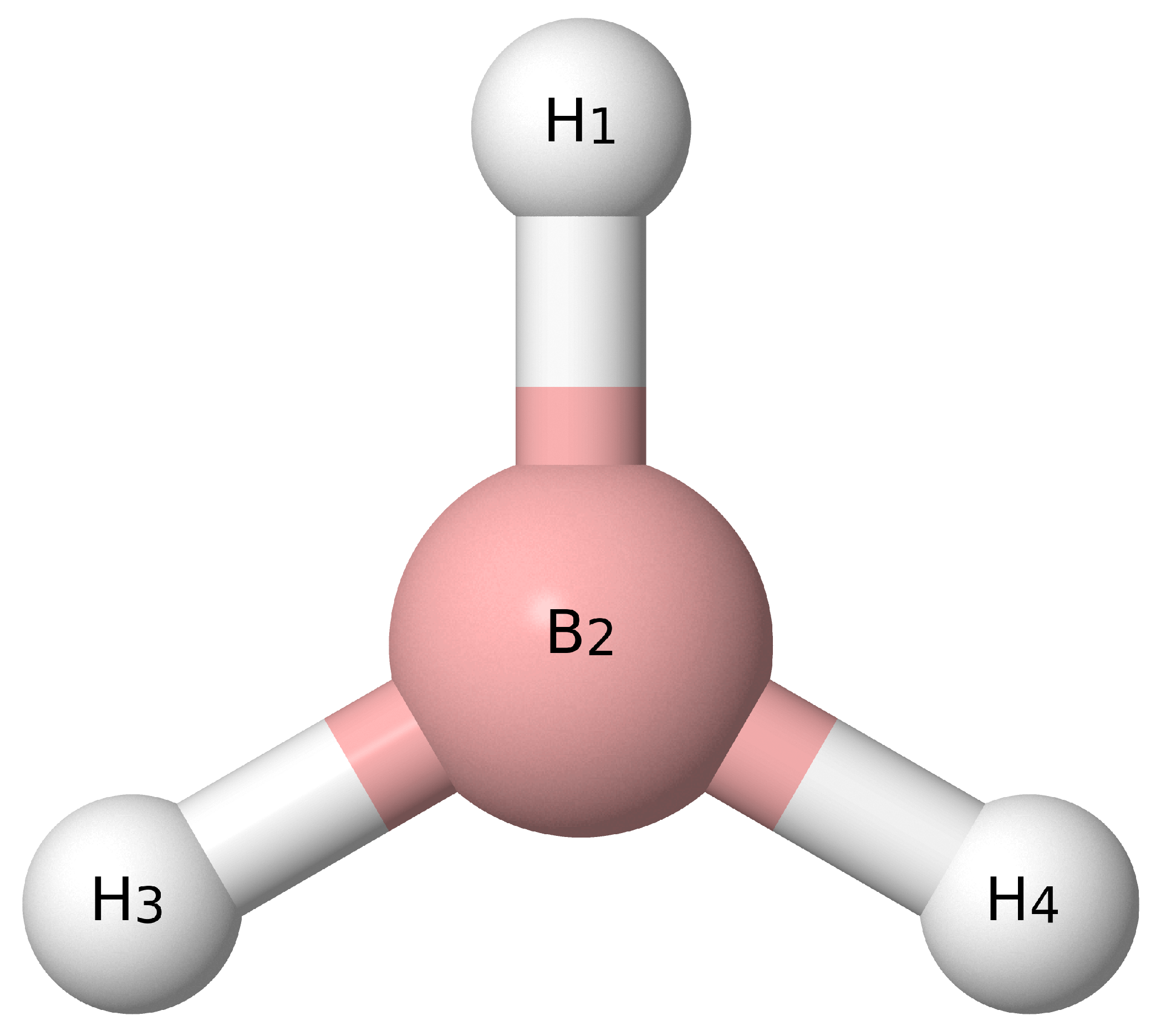{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SICs | Description | Symmetry | f | F12-TZ | F12-TZ-cCR | Expt. | |
|---|---|---|---|---|---|---|---|
| 0.932 − 0.068 | B-H stretch | 4 | 2890.3 | 2897.6 | |||
| 0.932 + 0.068 | B-O stretch | 35 | 1837.8 | 1845.8 | |||
| 0.500 + 0.500 | H-B-O bend | 12 | 764.6 | 764.9 | |||
| ZPVE | 3098.4 | 3111.3 | |||||
| 0.932 − 0.068 | B-H stretch | 4 | 2779.5 | 2795.2 | |||
| 0.932 + 0.068 | B-O stretch | 33 | 1822.8 | 1829.5 | 1825.5610 | ||
| 0.500 + 0.500 | H-B-O bend | 11 | 749.4 | 756.1 | 754.4163 | ||
| 39,198.2 | 39,411.0 | 39,400.668 | |||||
| 39,026.3 | 39,238.6 | 39,224.247 | |||||
| 38,757.3 | 38,967.4 | ||||||
| 38,767.3 | 38,978.0 | ||||||
| 39,118.3 | 39,331.9 | ||||||
| 2.74 |
| SICs | Description | Symmetry | f | F12-TZ | F12-TZ-cCR | Expt. | |
|---|---|---|---|---|---|---|---|
| 0.334 − 0.167+ 0.501 | antisymm. stretch | 133 | 2700.3 | 2708.5 | |||
| 0.667 + 0.333 | symm. stretch | 0 | 2567.6 | 2575.0 | |||
| 0.501 + 0.501 | in-plane rock | 17 | 1218.4 | 1221.4 | |||
| 1.000 | out-of-plane wag | 90 | 1156.9 | 1159.9 | |||
| ZPVE | 5724.1 | 5740.6 | |||||
| 0.334 − 0.167 + 0.501 | antisymm. stretch | 135 | 2594.0 | 2601.9 | 2601.5779 | ||
| 0.667 + 0.333 | symm. stretch | 0 | 2498.4 | 2505.8 | |||
| 0.501 + 0.501 | in-plane rock | 17 | 1194.9 | 1197.9 | 1196.0217 | ||
| 1.000 | out-of-plane wag | 86 | 1144.9 | 1148.7 | 1147.49087 | ||
| 236,255.9 | 237,314.5 | ||||||
| 118,127.8 | 118,657.3 | ||||||
| 235,210.3 | 236,268.5 | 236,071.4 | |||||
| 115,831.7 | 116,349.1 | 116,283.5 | |||||
| 233,018.6 | 234,064.7 | 234,104.0 | |||||
| 114,910.4 | 115,422.2 | 115,411.0 | |||||
| 233,201.4 | 234,248.8 | ||||||
| 114,827.3 | 115,339.3 | ||||||
| 238,431.6 | 239,513.3 | 23,9112.4 | |||||
| 114,665.5 | 115,177.1 | 11,5130.0 | |||||
| 233,068.4 | 234,113.6 | 23,4801.4 | |||||
| 116,418.6 | 116,940.3 | 116,643.1 | |||||
| 17.016 | 17.160 | 18.199 | |||||
| −29.579 | −29.834 | −32.746 | |||||
| 13.676 | 13.795 | 15.46 | |||||
| 3.476 | 3.521 | 3.639 | |||||
| −12.606 | −12.771 | −14.48 | |||||
| 14.853 | 15.047 | 17.898 | |||||
| −5.711 | −5.786 | −7.081 | |||||
| 0.00 |
| SICs | Description | Symmetry | f | F12-TZ | F12-TZ-cCR | Expt. | |
|---|---|---|---|---|---|---|---|
| 1.000 | O-H stretch | 83 | 3867.8 | 3869.8 | |||
| 0.739 − 0.260 | B-H antisymm. stretch | 172 | 2673.2 | 2679.9 | |||
| 0.739 + 0.259 | B-H symm. stretch | 104 | 2572.8 | 2579.4 | |||
| 0.568 − 0.247 − 0.182 | B-O stretch | 151 | 1376.7 | 1382.0 | |||
| 0.577 − 0.273 + 0.099 − 0.050 | H-O-B bend | 6 | 1190.9 | 1194.0 | |||
| 0.412 + 0.380 + 0.167 | H-B-O bend | 112 | 1188.2 | 1192.4 | |||
| 0.559 − 0.441 | out-of-plane wag | 51 | 1061.9 | 1065.1 | |||
| 0.381 + 0.314 − 0.308 | in-plane rock | 57 | 894.0 | 897.8 | |||
| 0.559 + 0.441 | torsion | 83 | 785.4 | 789.3 | |||
| ZPVE | 7708.4 | 7725.5 | |||||
| 1.000 | O-H stretch | 77 | 3681.1 | 3683.0 | |||
| 0.739 − 0.260 | B-H antisymm. stretch | 169 | 2555.6 | 2561.6 | |||
| 0.739 + 0.259 | B-H symm. stretch | 76 | 2456.2 | 2462.4 | |||
| 0.568 − 0.247 − 0.182 | B-O stretch | 152 | 1347.7 | 1352.8 | |||
| 0.577 − 0.273 + 0.099 − 0.050 | H-O-B bend | 22 | 1154.1 | 1158.3 | |||
| 0.412 + 0.380 + 0.167 | H-B-O bend | 98 | 1167.8 | 1171.1 | |||
| 0.559 − 0.441 | out-of-plane wag | 50 | 1048.1 | 1051.4 | |||
| 0.381 + 0.314 − 0.308 | in-plane rock | 59 | 880.7 | 882.8 | |||
| 0.559 + 0.441 | torsion | 82 | 753.5 | 751.0 | |||
| 172,890.0 | 173,612.5 | ||||||
| 30,552.4 | 30,704.1 | ||||||
| 25,964.1 | 26,090.0 | ||||||
| 171,969.4 | 172,687.1 | 17,2621.1 | |||||
| 30,319.5 | 30,469.7 | 30,470.22 | |||||
| 25,689.0 | 25,813.1 | 25,812.73 | |||||
| 170,127.7 | 170,835.2 | ||||||
| 30,294.4 | 30,444.4 | ||||||
| 25,631.6 | 25,755.3 | ||||||
| 170,798.0 | 171,510.7 | ||||||
| 30,278.4 | 30,428.3 | ||||||
| 25,646.5 | 25,770.4 | ||||||
| 170,001.1 | 170,709.4 | ||||||
| 30,306.0 | 30,456.1 | ||||||
| 25,638.0 | 25,761.7 | ||||||
| 172,528.1 | 173,243.9 | ||||||
| 30,328.9 | 30,477.1 | ||||||
| 25,515.1 | 25,637.8 | ||||||
| 175,303.8 | 176,002.0 | ||||||
| 30,366.7 | 30,511.9 | ||||||
| 25,594.4 | 25,712.3 | ||||||
| 172,698.2 | 173,646.6 | ||||||
| 30,278.2 | 30,437.8 | ||||||
| 25,601.6 | 25,730.7 | ||||||
| 170,201.5 | 170,730.6 | ||||||
| 30,086.2 | 30,232.3 | ||||||
| 25,724.7 | 25,849.0 | ||||||
| 176,655.6 | 177,443.9 | ||||||
| 30,274.3 | 30,423.6 | ||||||
| 25,610.4 | 25,733.9 | ||||||
| 167,569.6 | 168,210.5 | ||||||
| 30,196.4 | 30,346.5 | ||||||
| 25,689.4 | 25,813.5 | ||||||
| 1.51 | 1.506 |
| SICs | Description | Symmetry | f | F12-TZ | F12-TZ-cCR | |
|---|---|---|---|---|---|---|
| 1.000 | antisymm. O-H stretch | 151 | 3889.9 | 3892.5 | ||
| 1.001 | symm. O-H stretch | 56 | 3789.2 | 3792.0 | ||
| 1.002 | antisymm. B-H stretch | 215 | 2586.8 | 2593.6 | ||
| 0.623 − 0.378 | symm. B-H stretch | 218 | 2556.1 | 2562.9 | ||
| 0.624 + 0.378 | B-H breathing | 55 | 2482.1 | 2488.7 | ||
| 0.723 − 0.285 | H-O-H bend | 92 | 1653.8 | 1655.0 | ||
| 0.663 − 0.305 | B-H in-plane rock | 13 | 1208.4 | 1211.7 | ||
| 0.574 + 0.278 + 0.181 | B-H in-plane bend | 29 | 1200.4 | 1204.1 | ||
| 0.462 − 0.438 + 0.145 | B-H out-of-plane wag | 131 | 1194.6 | 1198.3 | ||
| 0.670 − 0.320 | torsion | 13 | 1025.9 | 1031.4 | ||
| 0.503 − 0.344 − 0.098 − 0.057 | O-H wag | 56 | 952.3 | 957.4 | ||
| 0.675 + 0.296 | antisymm. B-O-H bend | 3 | 647.3 | 651.4 | ||
| 0.629 + 0.311 + 0.083 + 0.057 | symm. B-O-H bend | 177 | 607.2 | 610.4 | ||
| 1.037 | B-O stretch | 84 | 467.0 | 472.1 | ||
| 0.695 + 0.299 | torsion | 39 | 156.4 | 158.1 | ||
| ZPVE | 11,981.3 | 12,050.8 | ||||
| 1.000 | antisymm. O-H stretch | 133 | 3700.9 | 3704.8 | ||
| 1.001 | symm. O-H stretch | 48 | 3612.6 | 3615.6 | ||
| 1.002 | antisymm. B-H stretch | 206 | 2476.3 | 2488.4 | ||
| 0.623 − 0.378 | symm. B-H stretch | 135 | 2444.9 | 2452.3 | ||
| 0.624 + 0.378 | B-H breathing | 94 | 2433.5 | 2441.2 | ||
| 0.723 − 0.285 | H-O-H bend | 60 | 1627.8 | 1634.3 | ||
| 0.663 − 0.305 | B-H in-plane rock | 12 | 1178.2 | 1179.4 | ||
| 0.574 + 0.278 + 0.181 | B-H in-plane bend | 23 | 1172.3 | 1175.5 | ||
| 0.462 − 0.438 + 0.145 | B-H out-of-plane wag | 139 | 1169.0 | 1176.8 | ||
| 0.670 − 0.320 | torsion | 13 | 944.9 | 971.9 | ||
| 0.503 − 0.344 − 0.098 − 0.057 | O-H wag | 34 | 898.9 | 927.2 | ||
| 0.675 + 0.296 | antisymm. B-O-H bend | 1 | 609.4 | 619.4 | ||
| 0.629 + 0.311 + 0.083 + 0.057 | symm. B-O-H bend | 179 | 543.9 | 528.4 | ||
| 1.037 | B-O stretch | 82 | 397.1 | 399.4 | ||
| 0.695 + 0.299 | torsion | 35 | 92.1 | 241.2 | ||
| 87,262.1 | 87,643.7 | |||||
| 17,819.1 | 17,935.7 | |||||
| 17,330.7 | 17,440.4 | |||||
| 86,509.6 | 86,912.3 | |||||
| 17,143.2 | 17,269.0 | |||||
| 16,699.3 | 16,816.4 | |||||
| 86,052.2 | 86,451.9 | |||||
| 17,149.6 | 17,274.8 | |||||
| 16,716.3 | 16,832.9 | |||||
| 85,973.9 | 86,373.0 | |||||
| 17,147.4 | 17,272.8 | |||||
| 16,701.5 | 16,818.2 | |||||
| 86,275.9 | 86,681.7 | |||||
| 17,212.6 | 17,338.0 | |||||
| 16,762.2 | 16,878.9 | |||||
| 85,874.1 | 86,271.5 | |||||
| 17,229.3 | 17,354.6 | |||||
| 16,768.6 | 16,885.2 | |||||
| 85,926.2 | 86,325.9 | |||||
| 17,193.6 | 17,319.1 | |||||
| 16,754.8 | 16,871.6 | |||||
| 86,341.9 | 86,745.4 | |||||
| 17,166.0 | 17,292.6 | |||||
| 16,679.8 | 16,796.9 | |||||
| 88,456.0 | 89,000.3 | |||||
| 17,140.2 | 17,265.6 | |||||
| 16,727.6 | 16,845.1 | |||||
| 84,427.7 | 84,908.1 | |||||
| 17,124.3 | 17,239.7 | |||||
| 16,675.4 | 16,786.5 | |||||
| 85,882.3 | 86,067.2 | |||||
| 17,111.5 | 17,248.4 | |||||
| 16,649.4 | 16,759.4 | |||||
| 87,344.8 | 87,759.8 | |||||
| 16,917.6 | 17,046.2 | |||||
| 16,453.6 | 16,586.5 | |||||
| 86,365.5 | 86,750.3 | |||||
| 16,885.5 | 17,012.9 | |||||
| 16,485.5 | 16,604.8 | |||||
| 87,566.4 | 87,974.7 | |||||
| 16,989.2 | 17,117.7 | |||||
| 16,514.9 | 16,634.0 | |||||
| 86,943.7 | 87,346.7 | |||||
| 16,960.9 | 17,088.5 | |||||
| 16,516.0 | 16,634.8 | |||||
| 86,280.7 | 86,678.2 | |||||
| 16,619.7 | 16,744.1 | |||||
| 16,201.6 | 16,317.7 | |||||
| 86,427.9 | 86,887.0 | |||||
| 16,951.3 | 17,088.7 | |||||
| 16,616.6 | 16,743.7 | |||||
| 4.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westbrook, B.R.; Fortenberry, R.C. Anharmonic Vibrational Frequencies of Water Borane and Associated Molecules. Molecules 2021, 26, 7348. https://doi.org/10.3390/molecules26237348
Westbrook BR, Fortenberry RC. Anharmonic Vibrational Frequencies of Water Borane and Associated Molecules. Molecules. 2021; 26(23):7348. https://doi.org/10.3390/molecules26237348
Chicago/Turabian StyleWestbrook, Brent R., and Ryan C. Fortenberry. 2021. "Anharmonic Vibrational Frequencies of Water Borane and Associated Molecules" Molecules 26, no. 23: 7348. https://doi.org/10.3390/molecules26237348
APA StyleWestbrook, B. R., & Fortenberry, R. C. (2021). Anharmonic Vibrational Frequencies of Water Borane and Associated Molecules. Molecules, 26(23), 7348. https://doi.org/10.3390/molecules26237348