
Hydrolysis and Enantiodiscrimination of (R)- and (S)-Oxazepam Hemisuccinate by Methylated β-Cyclodextrins: An NMR Investigation



Abstract
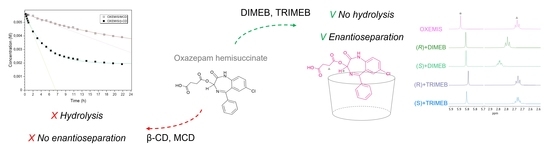
:
1. Introduction
2. Results and Discussion
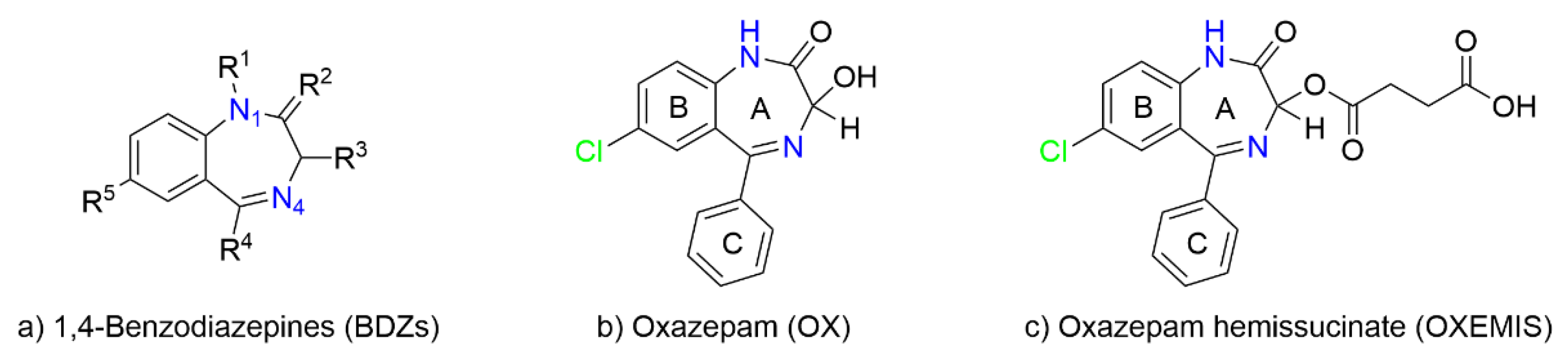
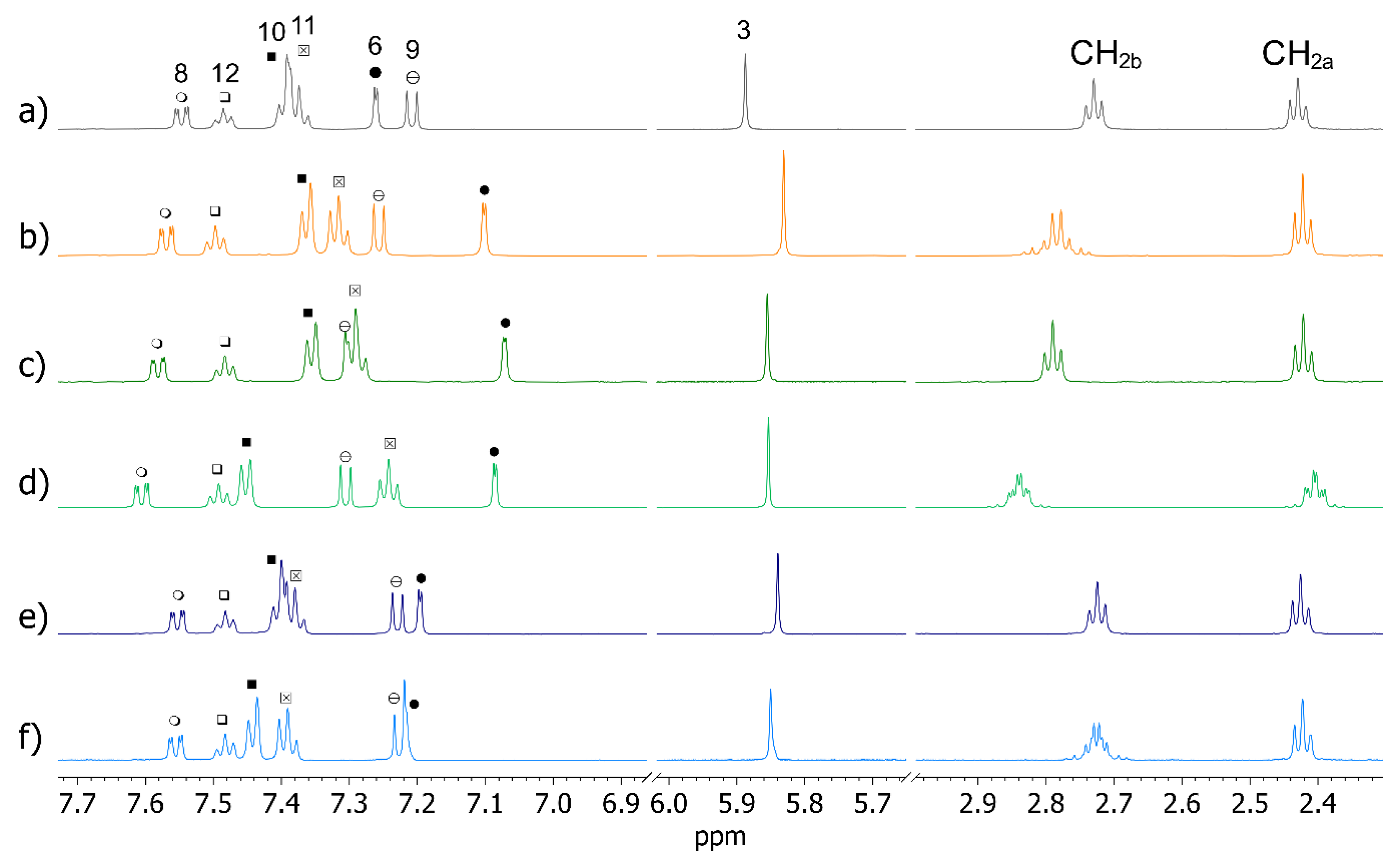
2.1. OXEMIS Characterization
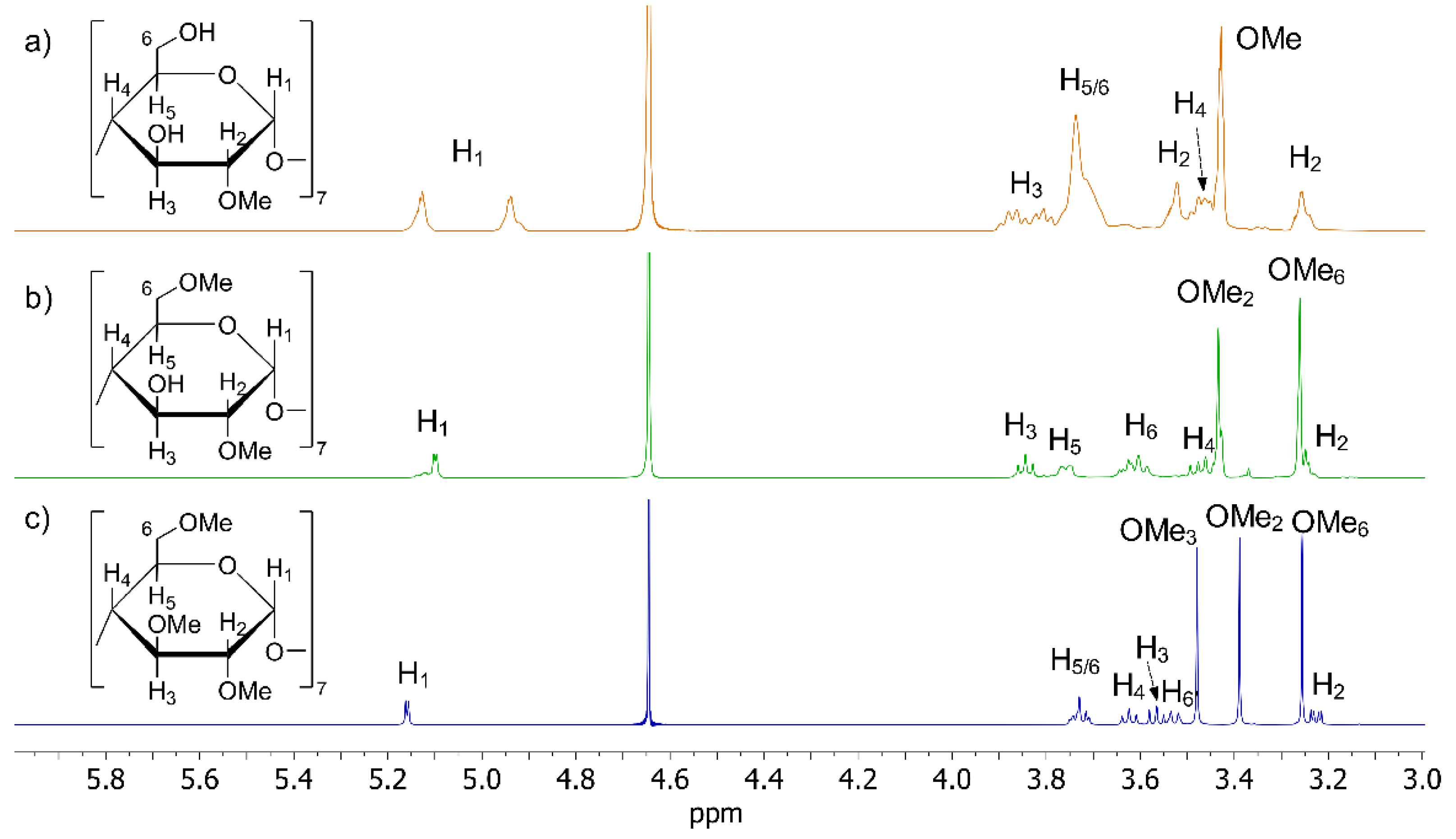
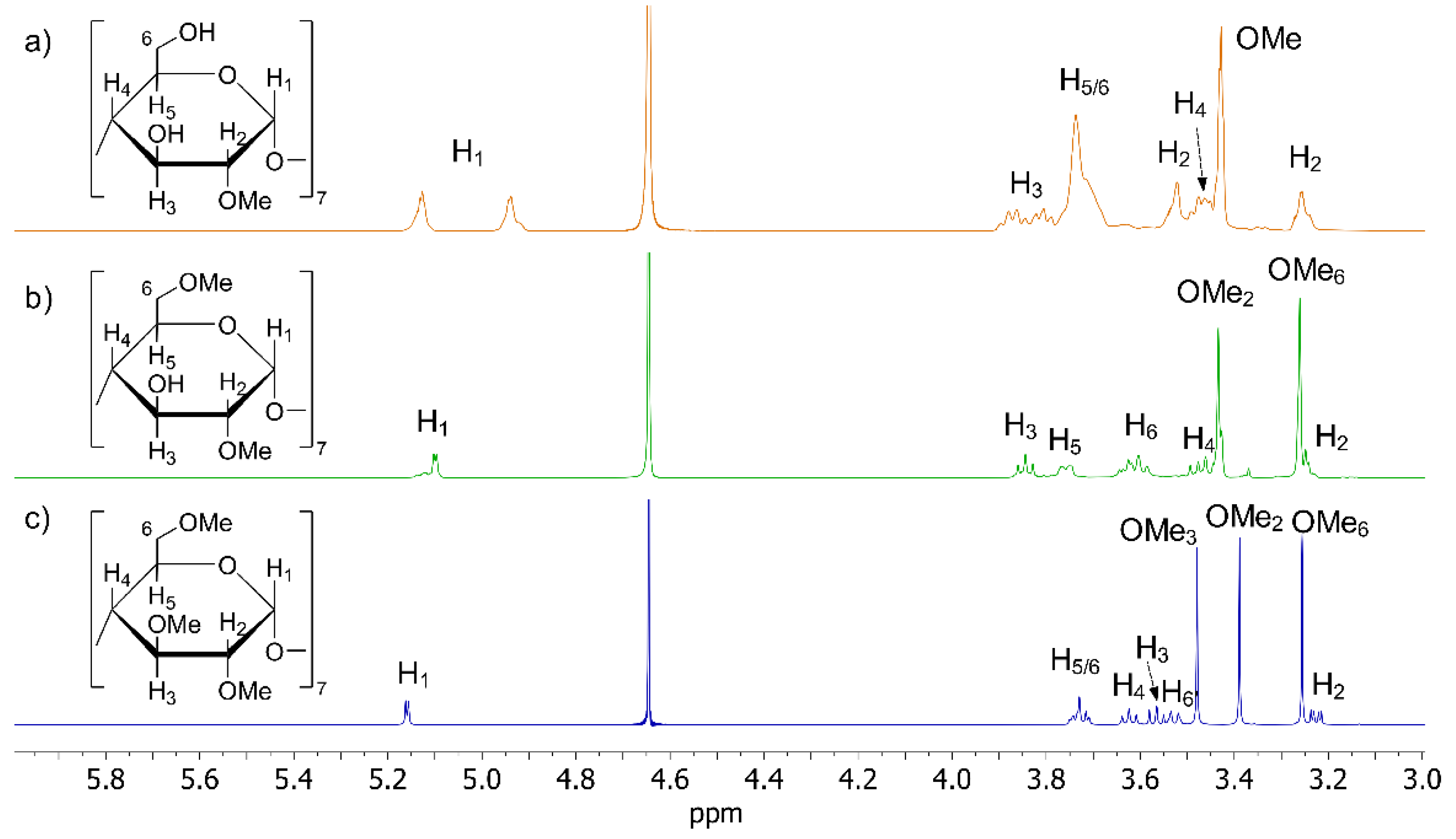
2.2. CDs Characterization
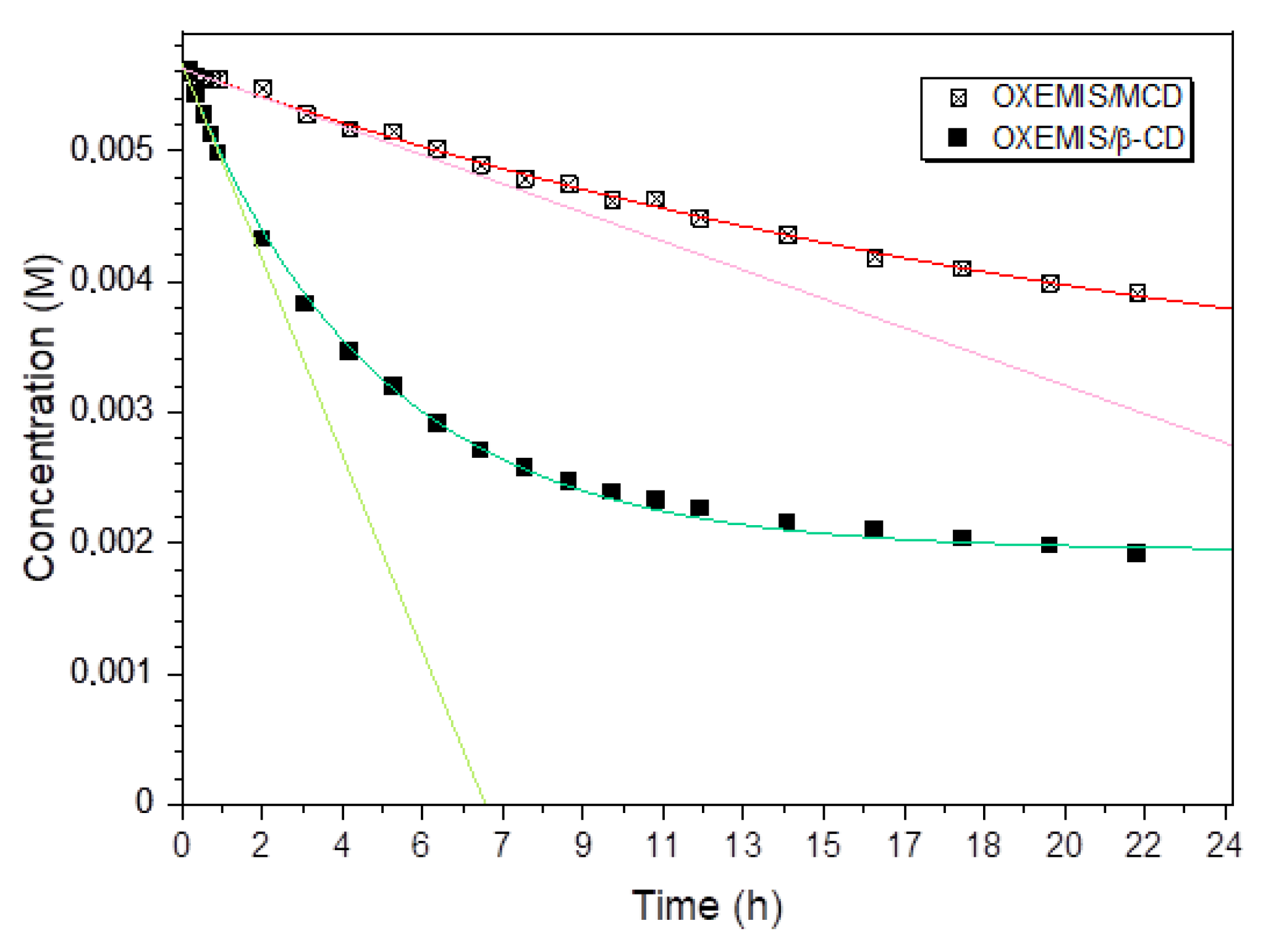
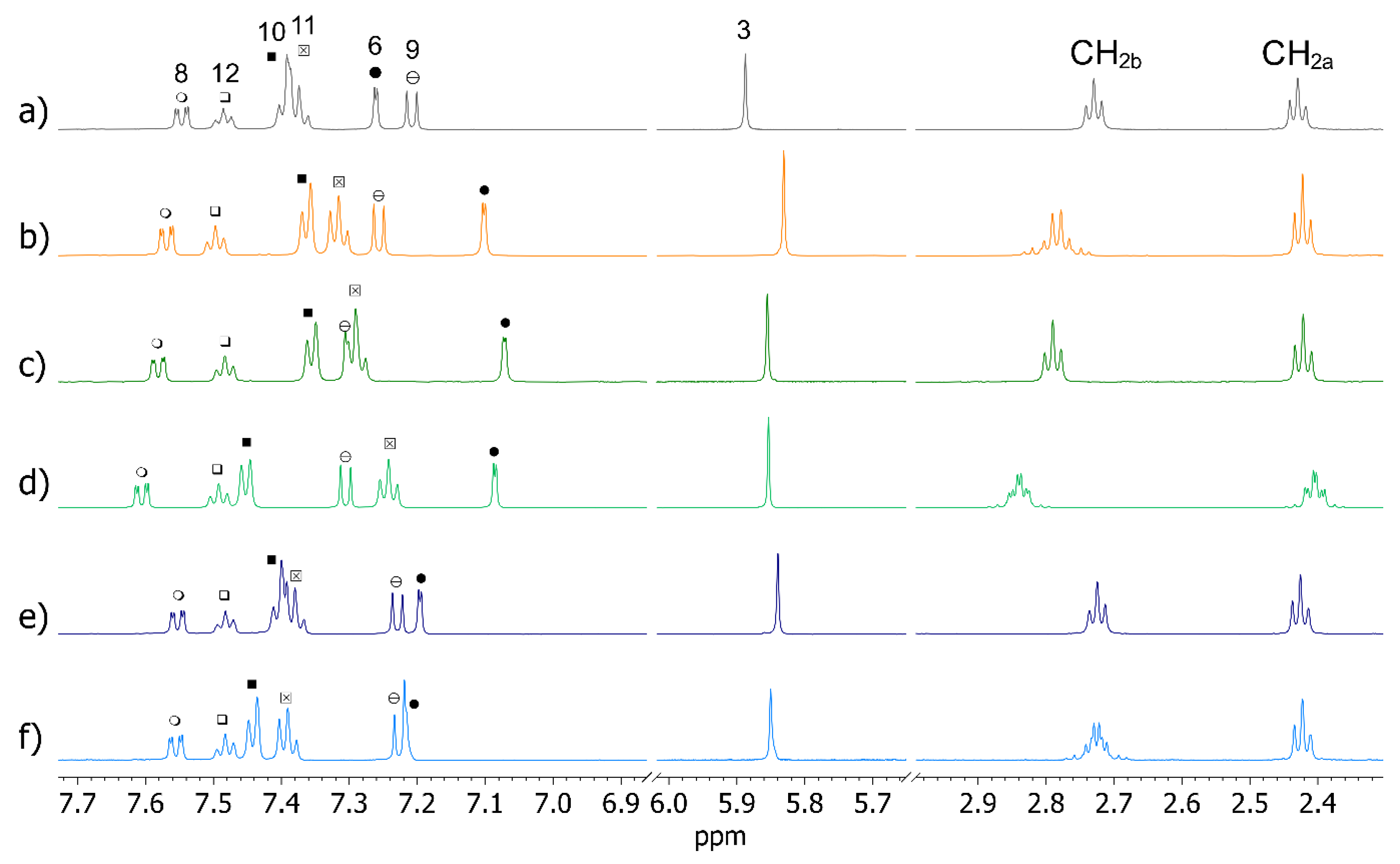
2.3. OXEMIS Hydrolysis by Cyclodextrins
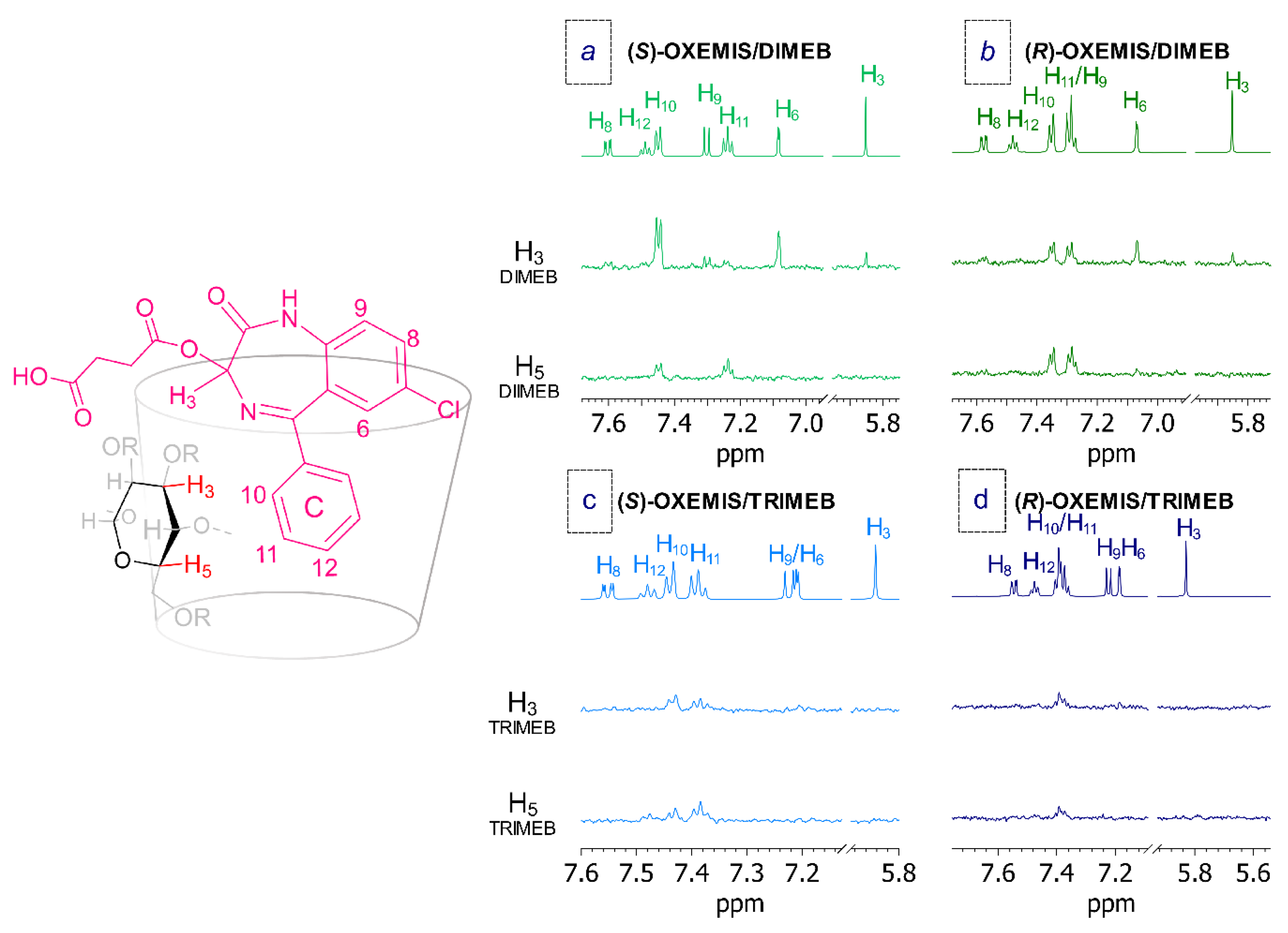
2.4. OXEMIS Enantiodiscrimination
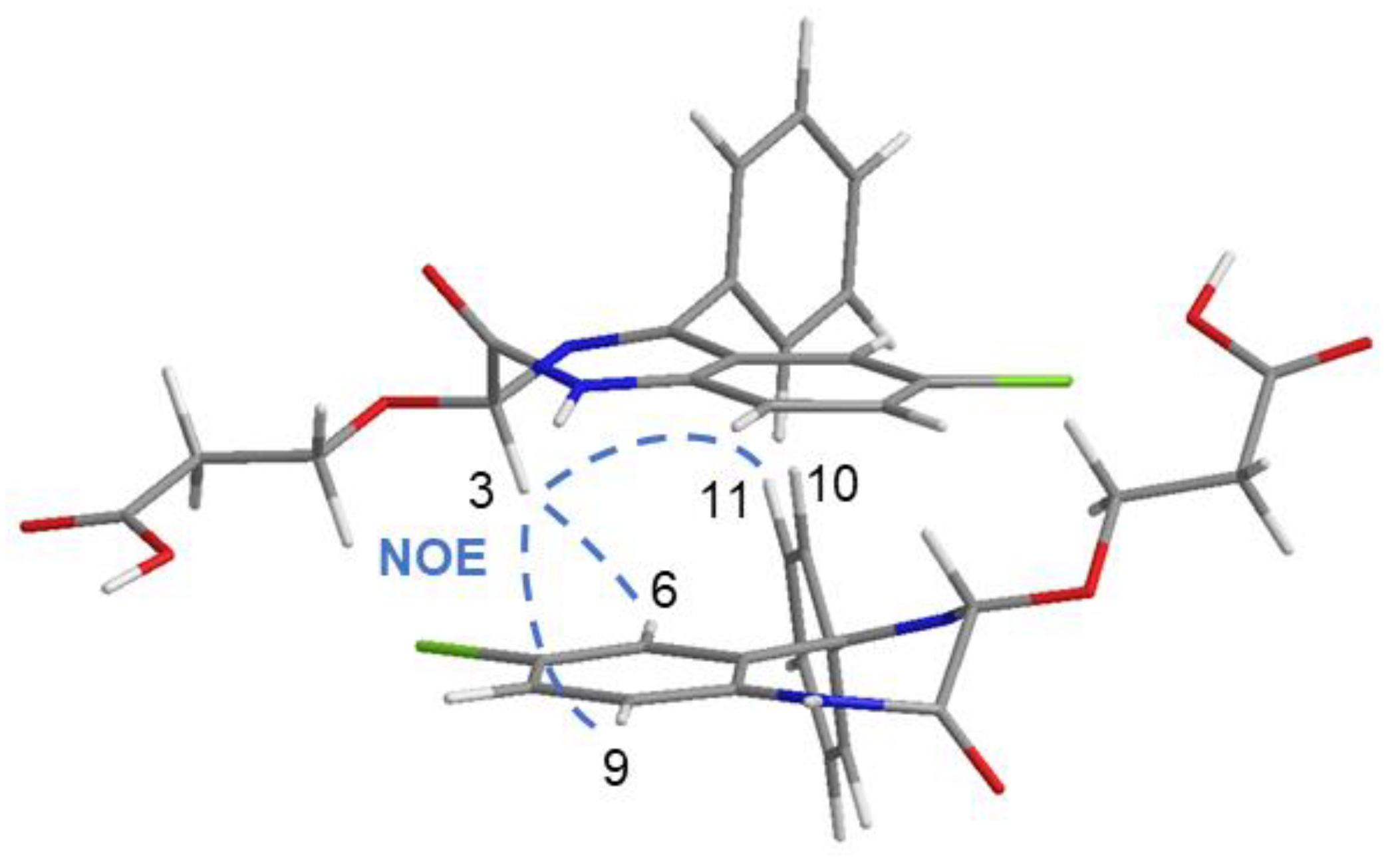
2.5. OXEMIS/CD Interaction Mechanism
3. Materials and Methods
3.1. Materials
3.2. NMR Experiments
3.3. NMR Samples Preparation for Stoichiometries and Association Constants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Griffin, C.E., III; Kaye, A.M.; Kaye, A.D. Benzodiazepine Pharmacology and Central Nervous System–Mediated Effects. Ochsner J. 2013, 13, 214–223. [Google Scholar]
- Schmitz, A. Benzodiazepine Use, Misuse, and Abuse: A Review. Ment. Health Clin. 2016, 6, 120–126. [Google Scholar] [CrossRef]
- Pham-Huy, C.; Villain-Pautet, G.; Hua, H.; Chikhi-Chorfi, N.; Galons, H.; Thevenin, M.; Claude, J.-R.; Warnet, J.-M. Separation of oxazepam, lorazepam, and temazepam enantiomers by HPLC on a derivatized cyclodextrin-bonded phase: Application to the determination of oxazepam in plasma. J. Biochem. Biophys. Methods 2002, 54, 287–299. [Google Scholar] [CrossRef]
- Court, M.H.; Duan, S.X.; Guillemette, C.; Journault, K.; Krishnaswamy, S.; Von Moltke, L.L.; Greenblatt, D.J. Stereoselective Conjugation of Oxazepam by Human UDP-Glucuronosyltransferases (UGTs): S-Oxazepam Is Glucuronidated by UGT2B15, while R-Oxazepam Is Glucuronidated by UGT2B7 and UGT1A9. Drug Metab. Dispos. 2002, 30, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.; Tang, B.K.; Grant, D.M.; Kalow, W. Interindividual variability in the glucuronidation of (S) oxazepam contrasted with that of (R) oxazepam. Pharmacogenetics 1995, 5, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Suthisisang, C.; Miners, J.O. The Glucuronidation of R- and S-Lorazepam: Human Liver Microsomal Kinetics, UDP-Glucuronosyltransferase Enzyme Selectivity, and Inhibition by Drugs. Drug Metab. Dispos. 2013, 41, 1273–1284. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.; Tang, B.K.; Kalow, W. (S)Oxazepam glucuronidation is inhibited by Ketoprofen and other substrates of UGT2B7. Pharmacogenetics 1995, 5, 43–49. [Google Scholar] [CrossRef]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar]
- Hok, L.; Božičević, L.; Sremec, H.; Šakić, D.; Vrček, V. Racemization of oxazepam and chiral 1,4-benzodiazepines. DFT study of the reaction mechanism in aqueous solution. Org. Biomol. Chem. 2019, 17, 1471–1479. [Google Scholar] [CrossRef]
- Yang, S.K.; Lu, X.-L. Racemization Kinetics of Enantiomeric Oxazepams and Stereoselective Hydrolysis of Enantiomeric Oxazepam 3-Acetates in Rat Liver Microsomes and Brain Homogenate. J. Pharm. Sci. 1989, 78, 789–795. [Google Scholar] [CrossRef]
- Maksay, G.; Tegyey, Z.; Kemeny, V.; Lukovits, I.; Otvos, L.; Palosi, E. Oxazepam esters. 1. Correlation between hydrolysis rates and brain appearance of oxazepam. J. Med. Chem. 1979, 22, 1436–1443. [Google Scholar] [CrossRef]
- Cassano, T.; Lopalco, A.; de Candia, M.; Laquintana, V.; Lopedota, A.; Cutrignelli, A.; Perrone, M.; Iacobazzi, R.M.; Bedse, G.; Franco, M.; et al. Oxazepam–Dopamine Conjugates Increase Dopamine Delivery into Striatum of Intact Rats. Mol. Pharm. 2017, 14, 3178–3187. [Google Scholar] [CrossRef]
- Bertucci, C.; Domenici, E.; Uccello-Barretta, G.; Salvadori, P. High-performance liquid chromatographic resolution of racemic 1,4-benzodiazepin-2-ones by means of a β-cyclodextrin silica bonded chiral stationary phase. J. Chromatogr. A 1990, 506, 617–625. [Google Scholar] [CrossRef]
- Szente, L.; Szemán, J. Cyclodextrins in Analytical Chemistry: Host–Guest Type Molecular Recognition. Anal. Chem. 2013, 85, 8024–8030. [Google Scholar] [CrossRef]
- Kfoury, M.; Landy, D.; Fourmentin, S. Characterization of Cyclodextrin/Volatile Inclusion Complexes: A Review. Molecules 2018, 23, 1204. [Google Scholar] [CrossRef] [Green Version]
- Morin-Crini, N.; Fourmentin, S.; Fenyvesi, E.; Lichtfouse, E.; Torri, G.; Fourmentin, M.; Crini, G. 130 years of cyclodextrin discovery for health, food, agriculture, and the industry: A review. Environ. Chem. Lett. 2021, 19, 2581–2617. [Google Scholar] [CrossRef]
- Lee, J.-U.; Lee, S.-S.; Lee, S.; Bin Oh, H. Noncovalent Complexes of Cyclodextrin with Small Organic Molecules: Applications and Insights into Host–Guest Interactions in the Gas Phase and Condensed Phase. Molecules 2020, 25, 4048. [Google Scholar] [CrossRef]
- Periasamy, R. Cyclodextrin-based molecules as hosts in the formation of supramolecular complexes and their practical ap-plications—A review. J. Carbohydr. Chem. 2021, 135–155. [Google Scholar] [CrossRef]
- Chankvetadze, B. Application of enantioselective separation techniques to bioanalysis of chiral drugs and their metabolites. TrAC Trends Anal. Chem. 2021, 143, 116332. [Google Scholar] [CrossRef]
- Alvarez-Rivera, G.; Bueno, M.; Vivas, D.B.; Cifuentes, A. Chiral analysis in food science. Trends Anal. Chem. 2019, 123, 115761. [Google Scholar] [CrossRef]
- Juvancz, Z.; Bodáné-Kendrovics, R.; Szente, L.; Maklári, D. Cyclodextrins as Dominant Chiral Selective Agents in the Capillary Separation Techniques. Period. Polytech. Chem. Eng. 2021, 65, 580–594. [Google Scholar] [CrossRef]
- Scriba, G.K. Chiral recognition in separation sciences: Part I: Polysaccharide and cyclodextrin selectors. Trends Anal. Chem. 2019, 120, 115639. [Google Scholar] [CrossRef]
- Hancu, G.; Papp, L.; Tóth, G.; Kelemen, H. The Use of Dual Cyclodextrin Chiral Selector Systems in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: An Overview. Molecules 2021, 26, 2261. [Google Scholar] [CrossRef]
- Guo, C.; Xiao, Y. Negatively charged cyclodextrins: Synthesis and applications in chiral analysis-A review. Carbohydr. Polym. 2020, 256, 117517. [Google Scholar] [CrossRef]
- Gunjal, P.; Singh, S.K.; Kumar, R.; Kumar, R.; Gulati, M. Role of Chromatograph-based Analytical Techniques in Quantification of Chiral Compounds: An Update. Curr. Anal. Chem. 2021, 17, 355–373. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Li, L.; Xiao, Y.; Wang, Y. Recent advances in cyclodextrins-based chiral-recognizing platforms. Trends Anal. Chem. 2019, 121, 115691. [Google Scholar] [CrossRef]
- Wenzel, T.J. Differentiation of Chiral Compounds Using NMR Spectroscopy, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; ISBN 9781119324768. [Google Scholar]
- Balzano, F.; Uccello-Barretta, G.; Aiello, F. Chiral Analysis by NMR Spectroscopy: Chiral Solvating Agents. In Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 367–427. ISBN 9780444640277. [Google Scholar]
- Chin, T.-F.; Chung, P.-H.; Lach, J.L. Influence of Cyclodextrins on Ester Hydrolysis. J. Pharm. Sci. 1968, 57, 44–48. [Google Scholar] [CrossRef]
- Popielec, A.; Loftsson, T. Effects of cyclodextrins on the chemical stability of drugs. Int. J. Pharm. 2017, 531, 532–542. [Google Scholar] [CrossRef]
- Trotta, F.; Moraglio, G.; Rapposelli, A. The effect of cyclodextrins on the hydrolysis of carboxylic acid esters. J. Incl. Phenom. Macrocycl. Chem. 1994, 20, 353–361. [Google Scholar] [CrossRef]
- Cesari, A.; Piras, A.M.; Zambito, Y.; Barretta, G.U.; Balzano, F. 2-Methyl-β-cyclodextrin grafted ammonium chitosan: Synergistic effects of cyclodextrin host and polymer backbone in the interaction with amphiphilic prednisolone phosphate salt as revealed by NMR spectroscopy. Int. J. Pharm. 2020, 587, 119698. [Google Scholar] [CrossRef]
- Homer, J.; Perry, M.C. Molecular complexes: Part 18—A nuclear magnetic resonance adaptation of the continuous variation (job) method of stoicheiometry determination. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1986, 82, 533–543. [Google Scholar] [CrossRef]
- Foster, R.; Fyfe, C.A. Interaction of electron acceptors with bases: Part 15—Determination of association constants of organic charge-transfer complexes by n.m.r. spectroscopy. Trans. Faraday Soc. 1965, 61, 1626–1631. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-CD | MCD 1 | DIMEB | TRIMEB | |
|---|---|---|---|---|
| H1 | 4.94 | 4.94/5.13 | 5.11 | 5.16 |
| H2 | 3.52 | 3.53/3.26 | 3.26 | 3.22 |
| H3 | 3.84 | 3.81/3.88 | 3.85 | 3.56 |
| H4 | 3.46 | 3.45/3.48 | 3.47 | 3.62 |
| H5 | 3.74 | 3.73/3.70 | 3.77 | 3.74 |
| H6/6′ | 3.75 | 3.74/3.74 | 3.63 | 3.53/3.72 |
| OMe2 | - | 3.44 | 3.44 | 3.39 |
| OMe3 | - | - | 3.27 | 3.48 |
| OMe6 | - | - | - | 3.25 |
| OXEMIS/MCD | OXEMIS/DIMEB | OXEMIS/TRIMEB | |||||
|---|---|---|---|---|---|---|---|
| Ring | δ | Δδ | Δδ(S) | Δδ(R) | Δδ(S) | Δδ(R) | |
| A | H3 | 5.89 | −0.06 | −0.03 | −0.03 | −0.05 | −0.04 |
| B | H6 | 7.25 | −0.16 | −0.18 | −0.19 | −0.04 | −0.06 |
| B | H8 | 7.54 | 0.02 | 0.06 | 0.03 | 0.00 | 0.00 |
| B | H9 | 7.20 | 0.05 | 0.10 | 0.09 | 0.01 | 0.02 |
| C | H10 | 7.40 | −0.03 | 0.06 | −0.04 | 0.05 | 0.01 |
| C | H11 | 7.37 | −0.05 | −0.13 | −0.08 | 0.02 | 0.01 |
| C | H12 | 7.49 | 0.01 | 0.01 | −0.01 | −0.01 | −0.01 |
| - | CH2a | 2.43 | −0.01 | −0.03 | −0.01 | −0.01 | 0.00 |
| - | CH2b | 2.73 | 0.05 | 0.11 | 0.06 | −0.01 | −0.01 |
| DIMEB/OXEMIS | TRIMEB/OXEMIS | |||
|---|---|---|---|---|
| Proton | Δδ(S) | Δδ(R) | Δδ(S) | Δδ(R) |
| H1 | −0.02 | −0.02 | −0.01 | −0.01 |
| H2 | −0.03 | 0.00 | −0.01 | −0.01 |
| H3 | −0.09 | −0.07 | −0.02 | −0.01 |
| H4 | −0.03 | −0.01 | −0.01 | −0.01 |
| H5 | −0.16 | −0.09 | −0.03 | −0.01 |
| H6/6′ | −0.02 | −0.06 | 0.00 | 0.00 |
| OMe2 | 0.00 | 0.00 | −0.01 | −0.01 |
| OMe3 | −0.01 | −0.01 | −0.03 | −0.03 |
| OMe6 | - | - | 0.00 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cesari, A.; Balzano, F.; Uccello Barretta, G.; Recchimurzo, A. Hydrolysis and Enantiodiscrimination of (R)- and (S)-Oxazepam Hemisuccinate by Methylated β-Cyclodextrins: An NMR Investigation. Molecules 2021, 26, 6347. https://doi.org/10.3390/molecules26216347
Cesari A, Balzano F, Uccello Barretta G, Recchimurzo A. Hydrolysis and Enantiodiscrimination of (R)- and (S)-Oxazepam Hemisuccinate by Methylated β-Cyclodextrins: An NMR Investigation. Molecules. 2021; 26(21):6347. https://doi.org/10.3390/molecules26216347
Chicago/Turabian StyleCesari, Andrea, Federica Balzano, Gloria Uccello Barretta, and Alessandra Recchimurzo. 2021. "Hydrolysis and Enantiodiscrimination of (R)- and (S)-Oxazepam Hemisuccinate by Methylated β-Cyclodextrins: An NMR Investigation" Molecules 26, no. 21: 6347. https://doi.org/10.3390/molecules26216347
APA StyleCesari, A., Balzano, F., Uccello Barretta, G., & Recchimurzo, A. (2021). Hydrolysis and Enantiodiscrimination of (R)- and (S)-Oxazepam Hemisuccinate by Methylated β-Cyclodextrins: An NMR Investigation. Molecules, 26(21), 6347. https://doi.org/10.3390/molecules26216347