
Rational Design 2-Hydroxypropylphosphonium Salts as Cancer Cell Mitochondria-Targeted Vectors: Synthesis, Structure, and Biological Properties

 , , ,
, , ,  , ,
, , 
Abstract
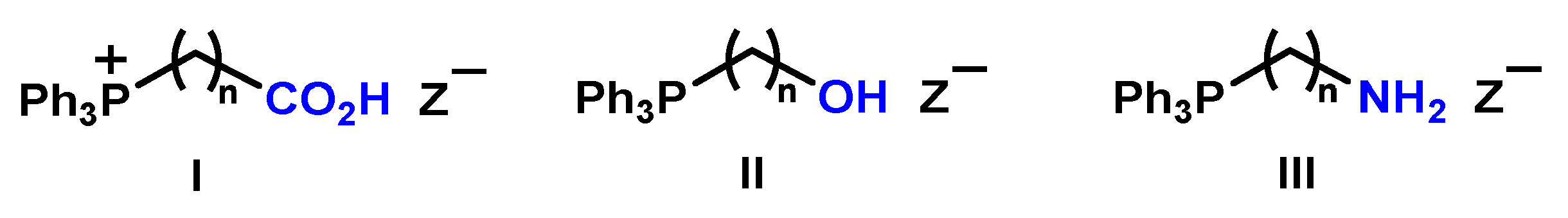
1. Introduction
2. Results and Discussion
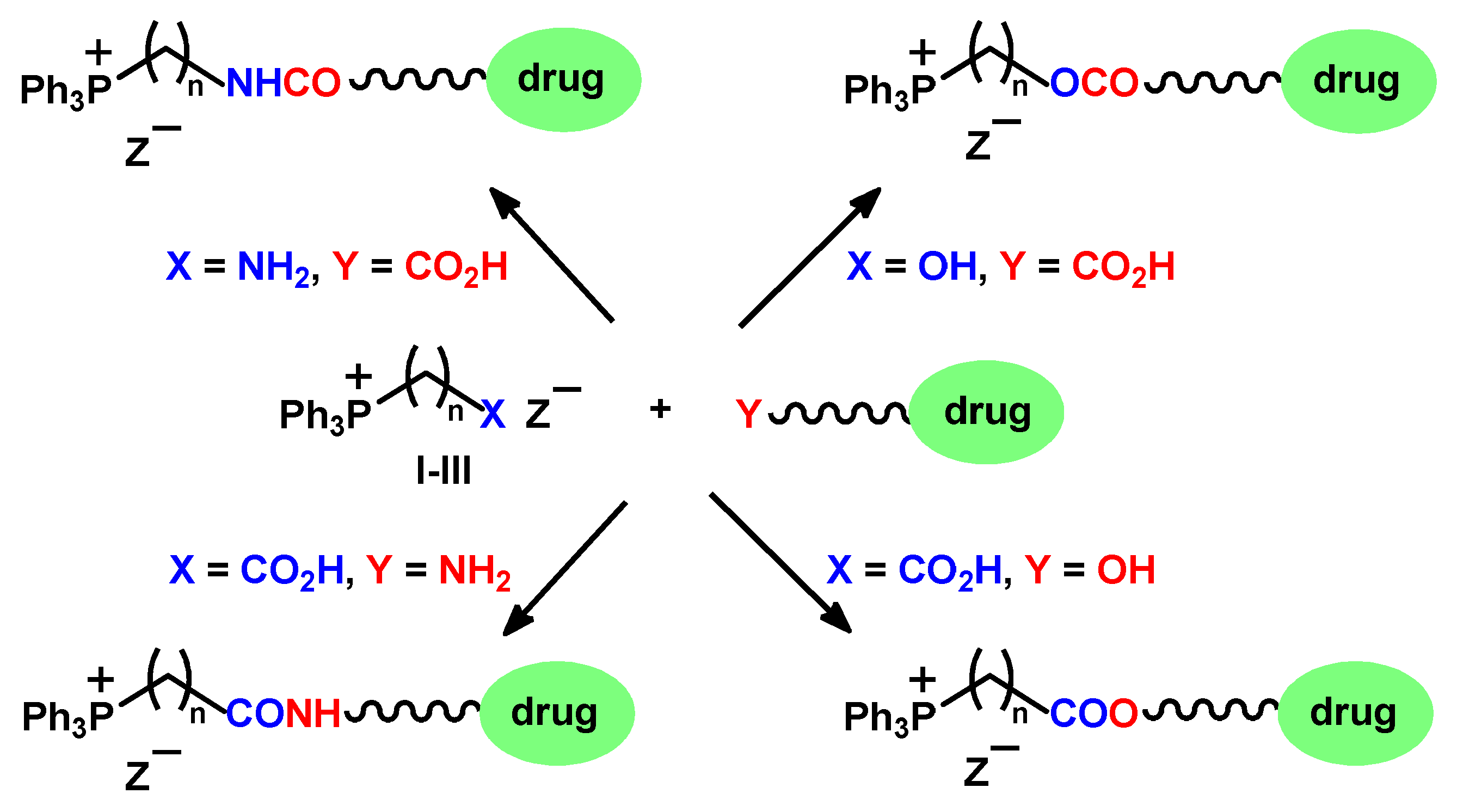
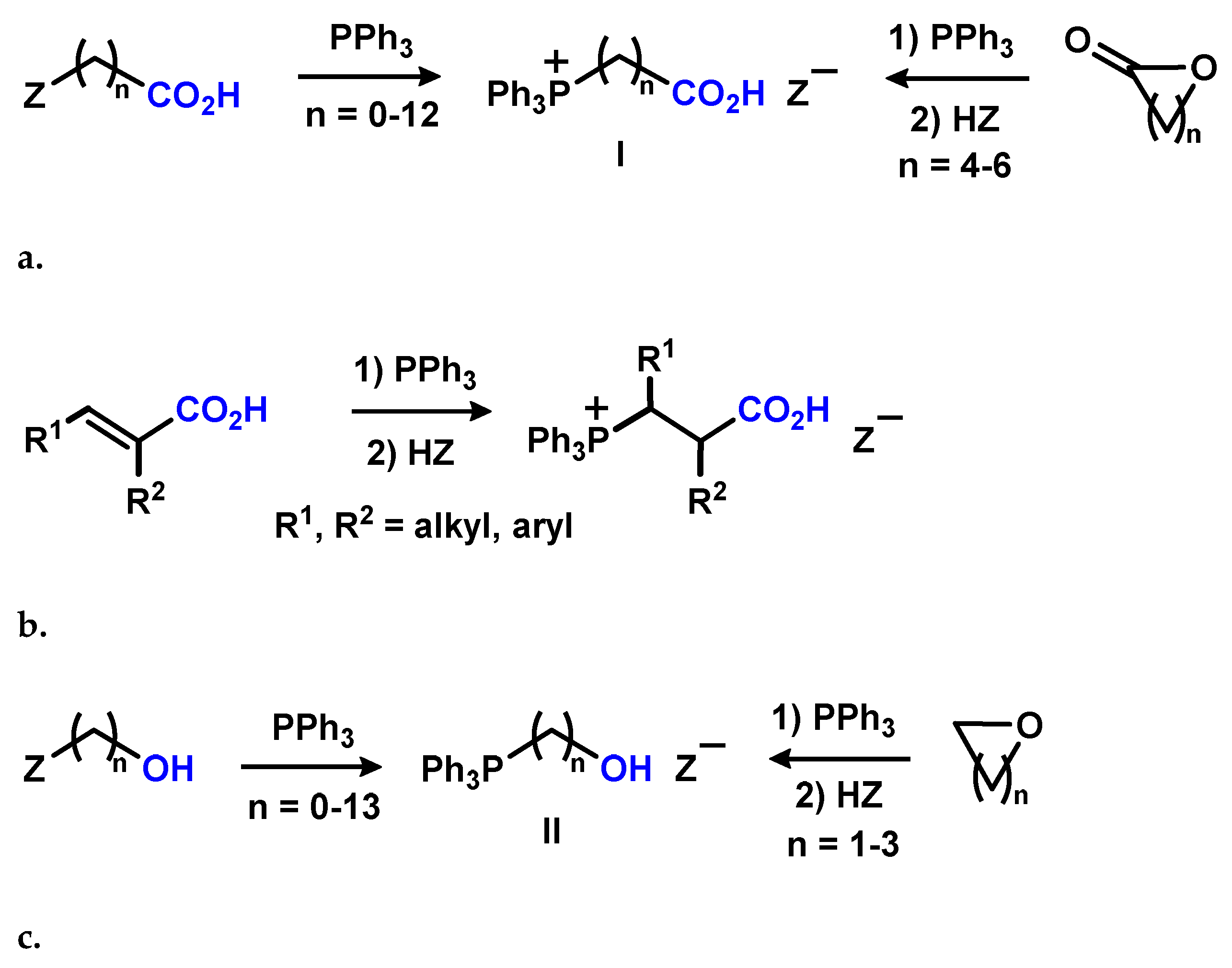
2.1. Chemistry
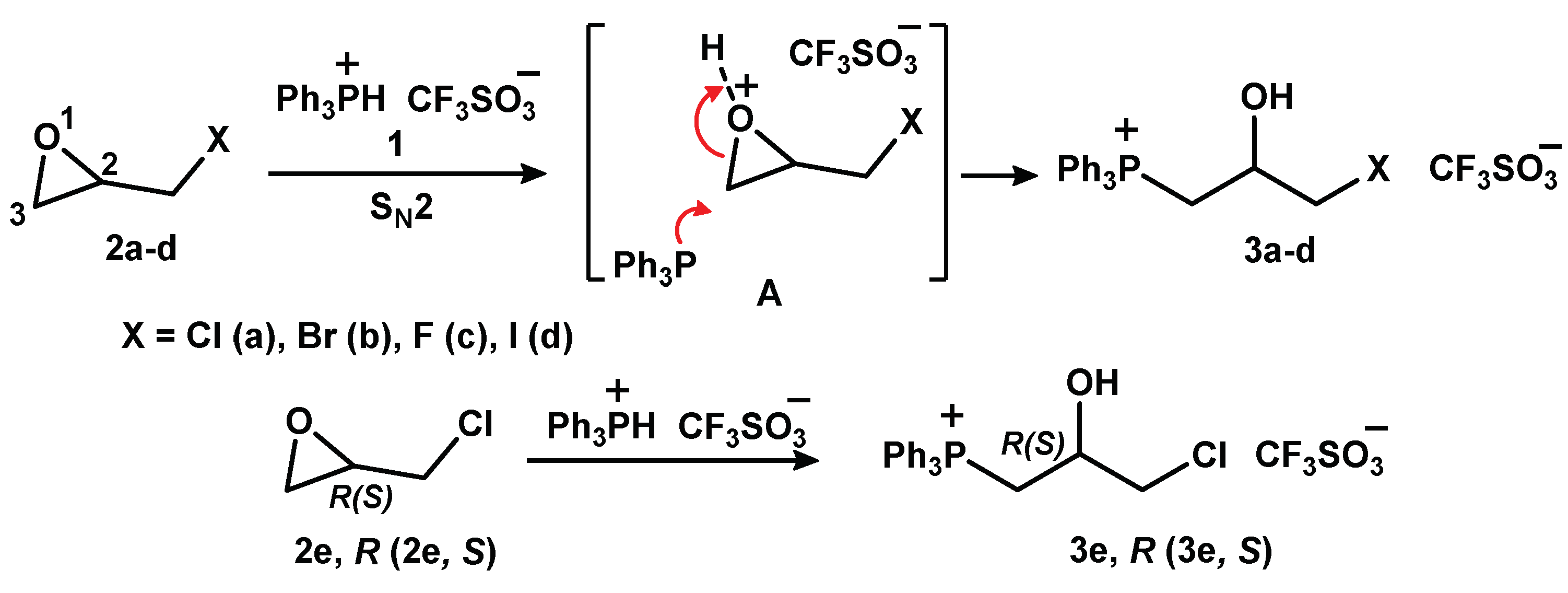
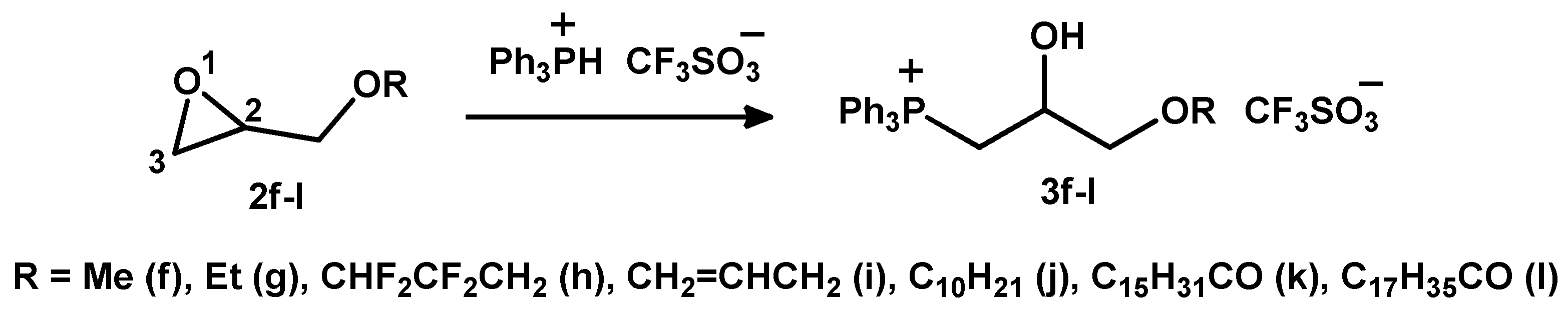
2.1.1. Reaction of Triphenylphosphonium Triflate with Halomethyloxiranes
2.1.2. Reaction of Triphenylphosphonium Triflate with Alkyl- and Acyl Glycidyl Ether
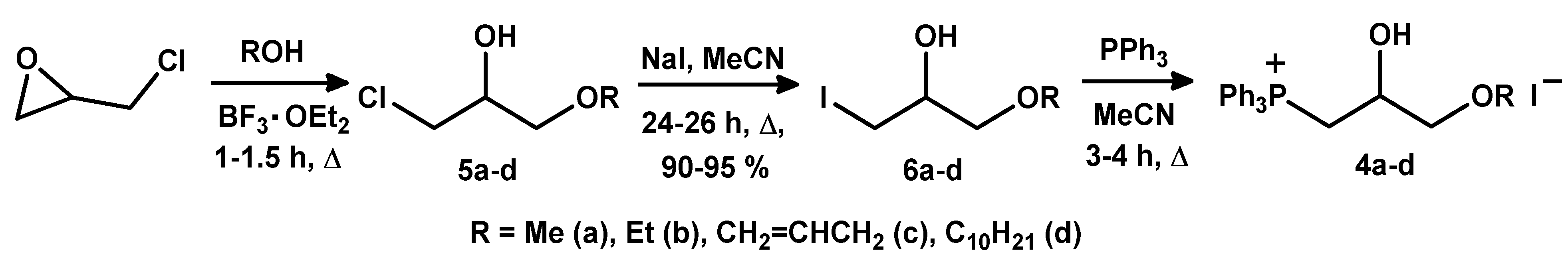
2.1.3. Synthesis of 3-Alkoxy(Iodo)-2-hydroxypropyl Triphenylphosphonium Iodides
2.1.4. Liposomal Systems Based on Amphiphilic Triflates of Acyloxypropylphosphonium and L-α-Phosphatidylcholine
2.2. Biology
2.2.1. Cytotoxicity
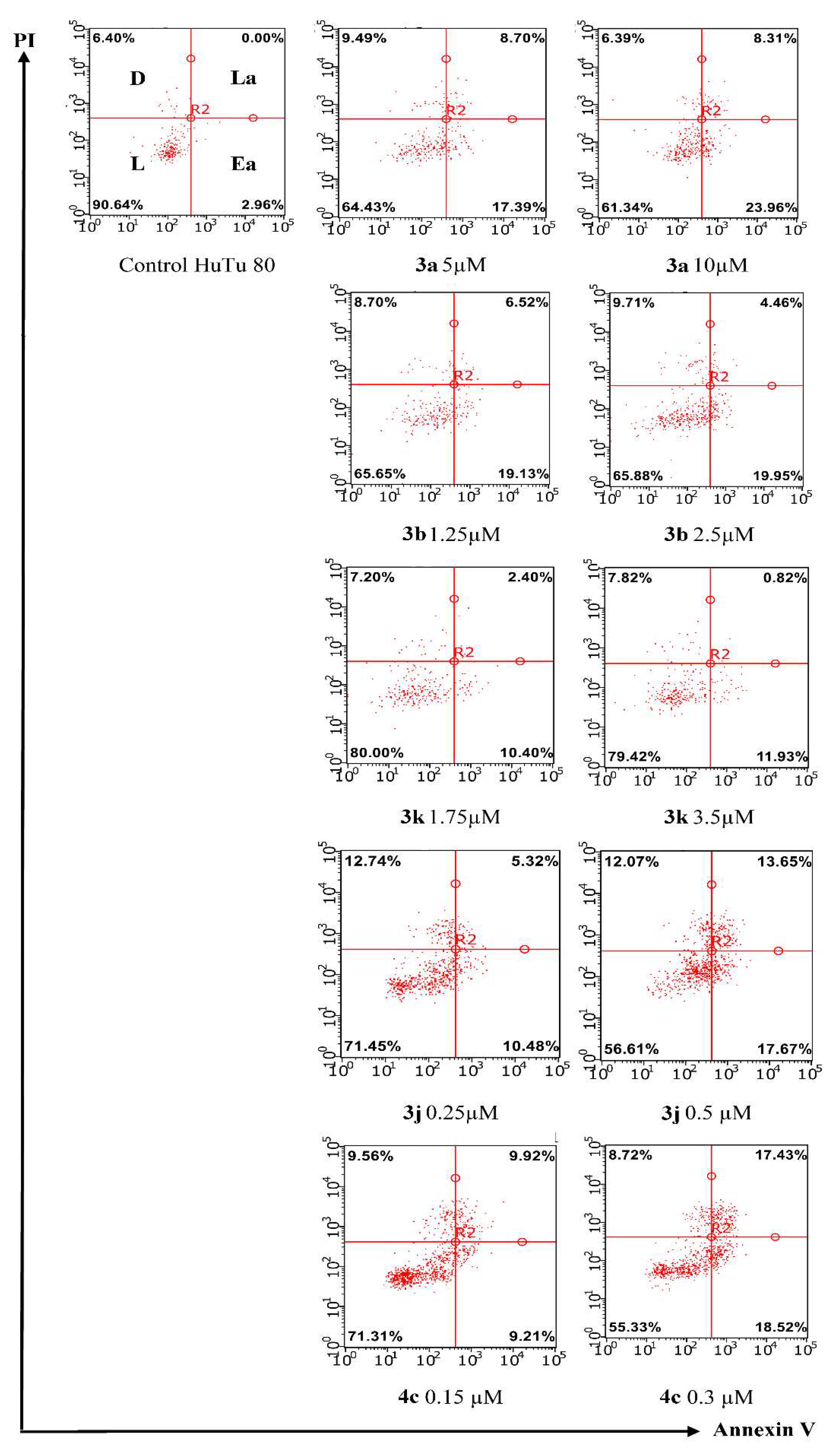
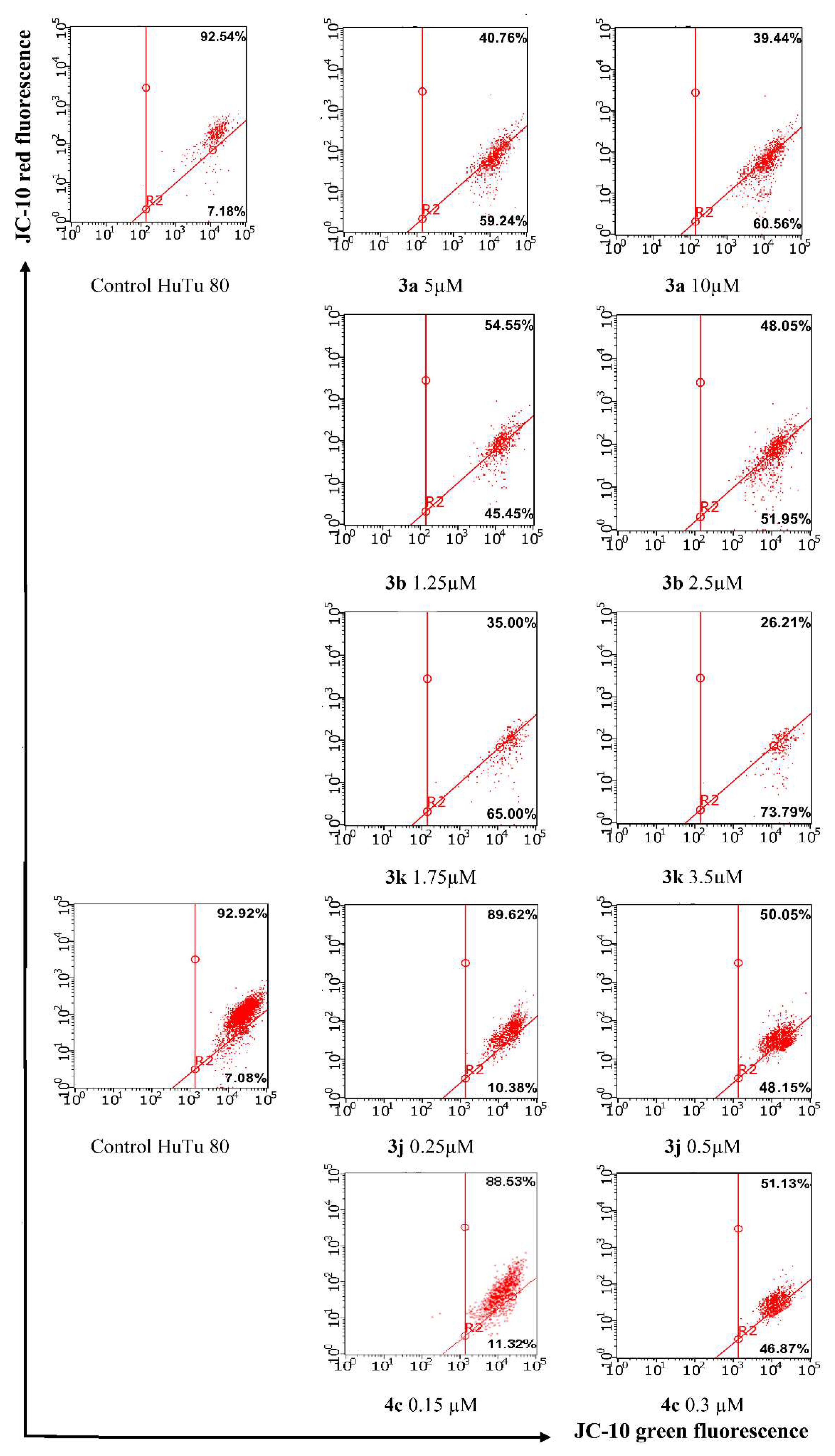
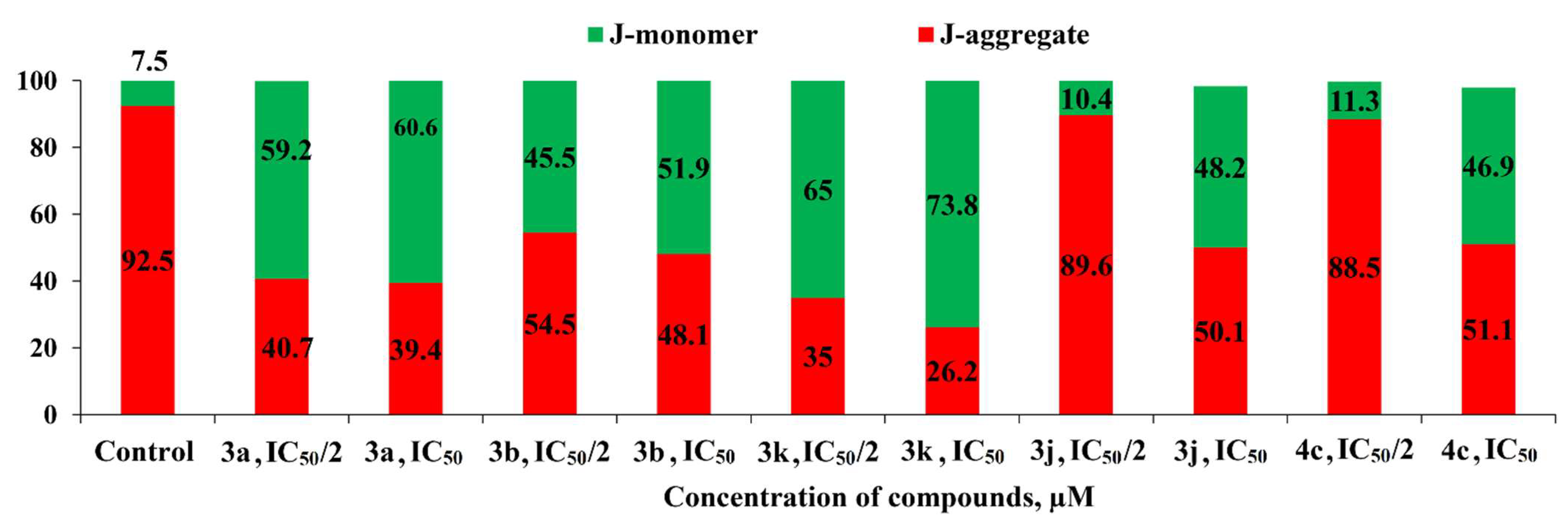
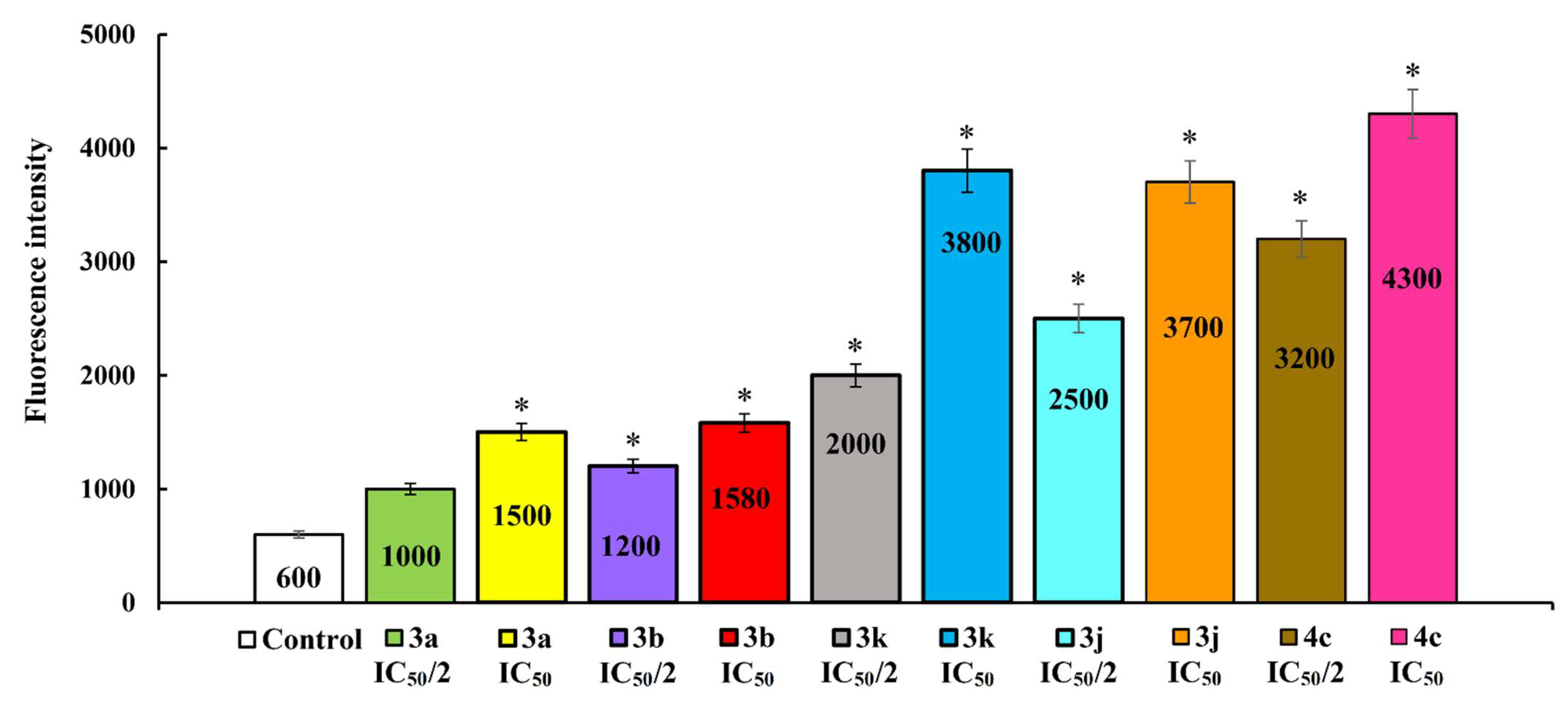
2.2.2. Induction of Apoptotic Effects by Lead Compounds
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of 1-Chloro-3-alkoxypropan-2-ol (5a–d)
3.2.2. General Procedure for the Synthesis of 2-(Alkoxymethyl)oxirane (2f–j)
3.2.3. General Procedure for the Synthesis of 3-Alkoxy-1-iodopropan-2-ols (6a–d)
3.2.4. General Procedure for the Synthesis of Triphenylphosphonium Salts (3a–e, 3f–j)
3.2.5. General Procedure for the Synthesis of (3-Alkoxy-2-hydroxypropyl)triphenylphosphonium Iodide (4a–d)
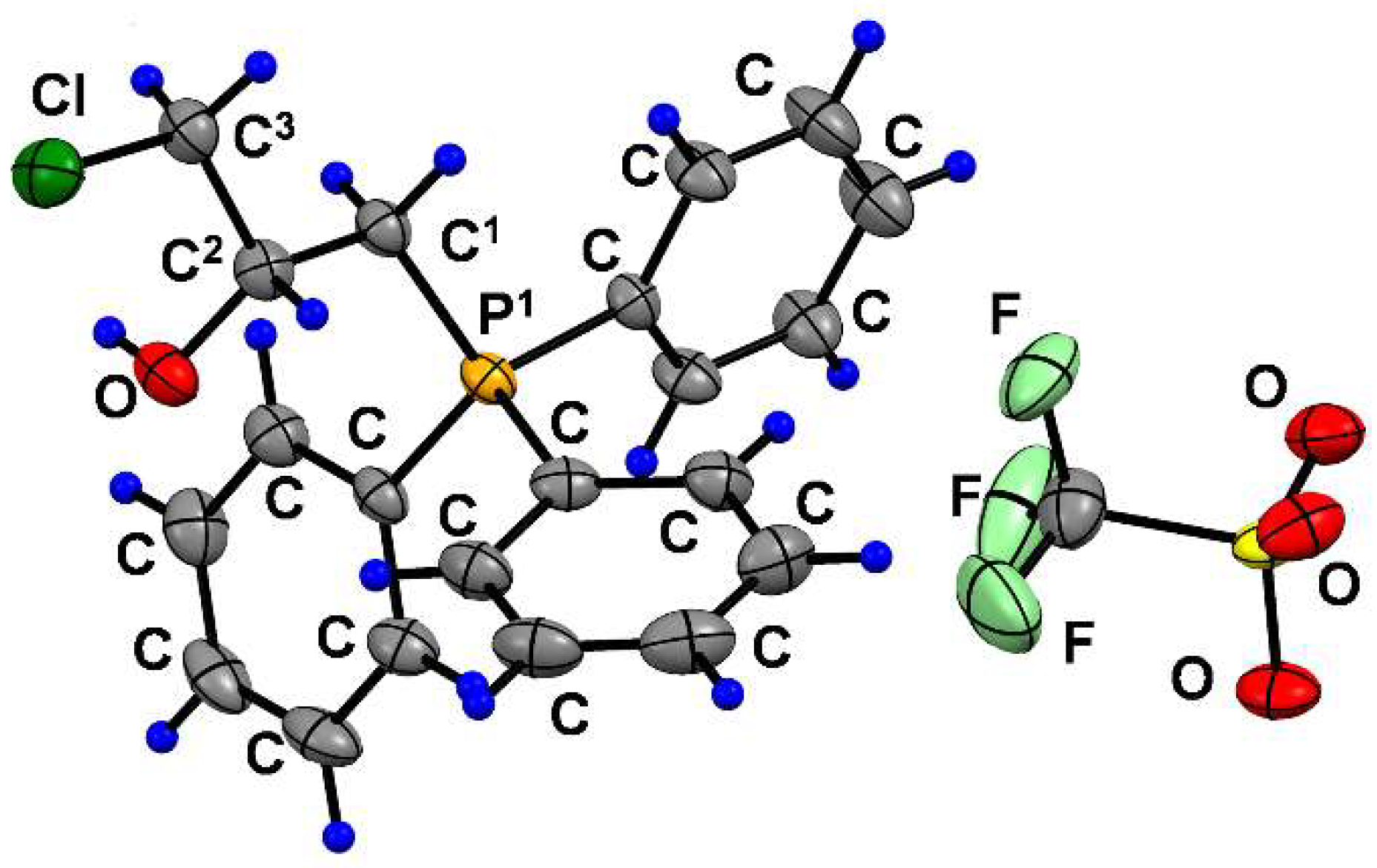
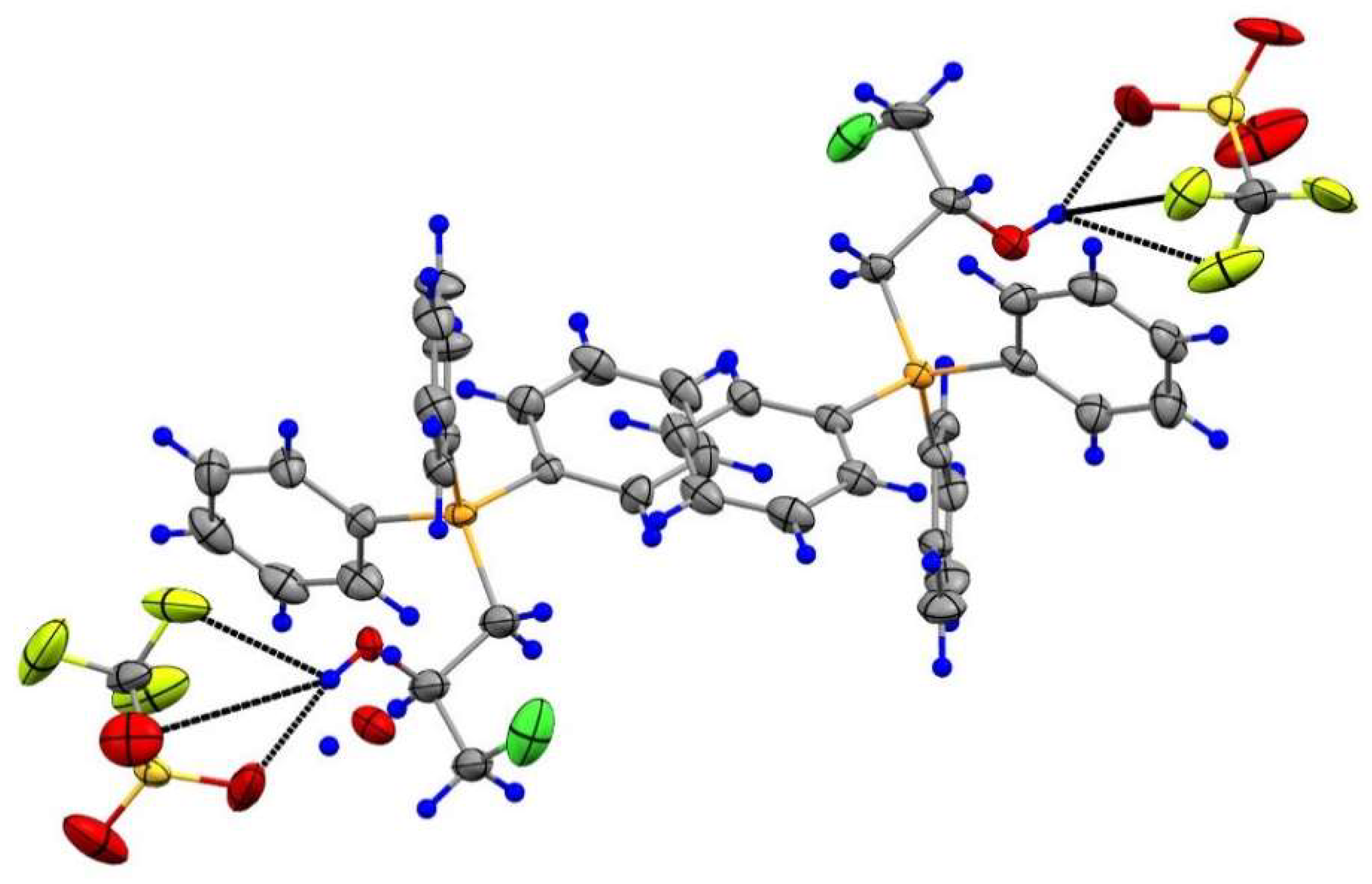
3.2.6. Synthesis of 3d and 7
3.2.7. Preparation and Characterization Phosphorus-Containing Amphiphile Liposomes
Chemicals
Preparation and Characterization
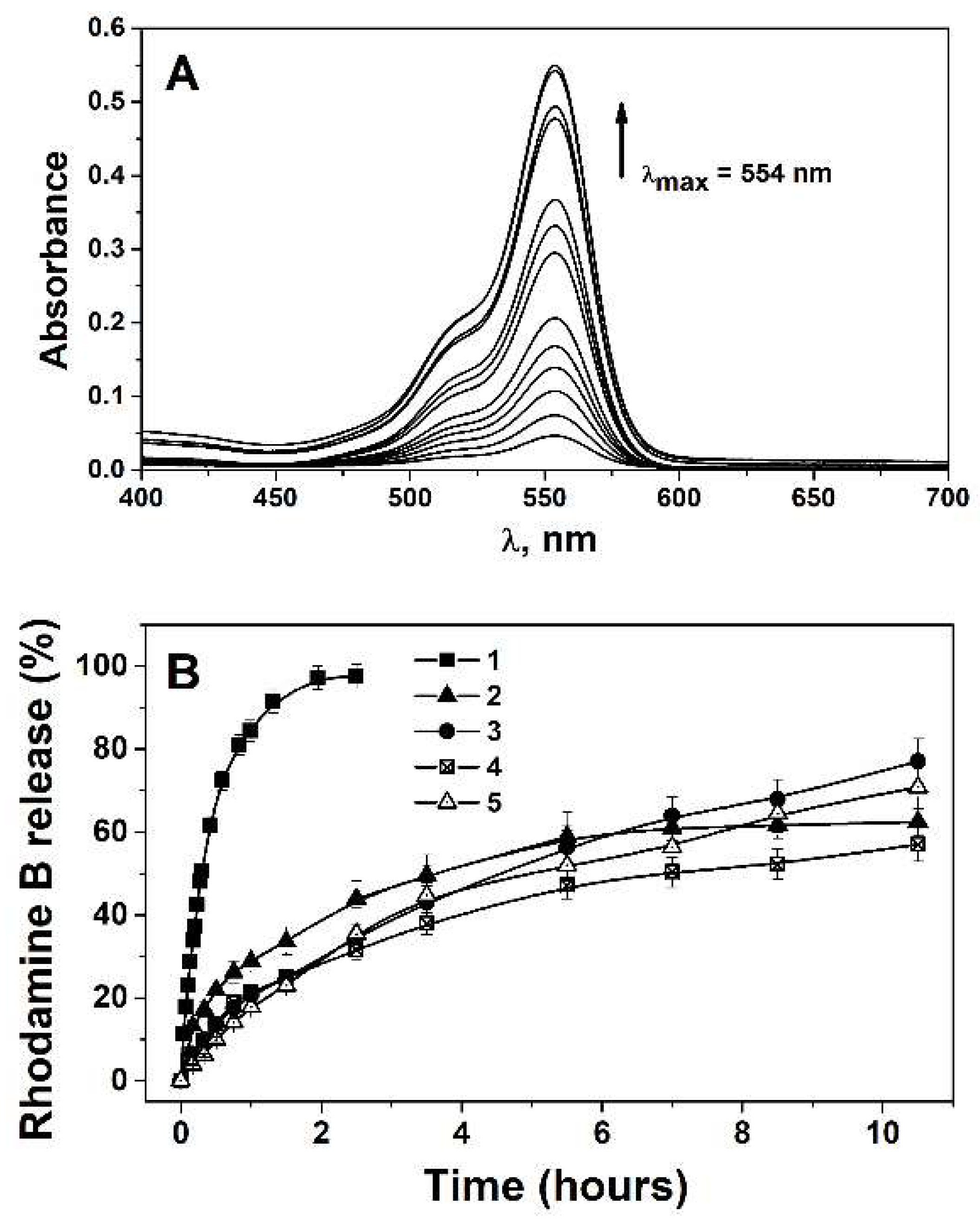
In Vitro Rhodamine B Release Profile
Encapsulation Efficiency and Loading Capacity
3.3. Biology
3.3.1. Cell Toxicity Assay (MTT-Test)
3.3.2. Induction of Apoptotic Effects by Test Compounds (Flow Cytometry Assay)
Cell Culture
Cell Apoptosis Analysis
Mitochondrial Membrane Potential
Detection of Intracellular ROS
Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Moura, I.M.R.; Tranquilino, A.; Sátiro, B.G.; Silva, R.O.; de Oliveira-Silva, D.; Oliveirsa, R.A.; Menezes, P.H. Unusual application for phosphonium salts and phosphoranes: Synthesis of chalcogenides. J. Org. Chem. 2021, 86, 5954–5964. [Google Scholar] [CrossRef]
- Fink, J. Hydraulic Fracturing Chemicals and Fluids Technology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Wakelyn, P.J. Environmentally friendly flame resistant textiles. In Advances in Fire Retardant Materials, 1st ed.; Horrocks, A.R., Price, D., Eds.; Woodhead Publishing Limited: Sawston, UK; CRC Press: Boca Raton, FL, USA, 2008; pp. 188–212. [Google Scholar]
- Corbridge, D.E.C. Phosphorus: An Outline of Its Chemistry, Biochemistry, and Technology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 1995; p. 1220. [Google Scholar]
- Li, H.; Liu, H.; Guo, H. Recent Advances in Phosphonium Salt Catalysis. Adv. Synth. Catal. 2021, 363, 2023–2036. [Google Scholar] [CrossRef]
- Jean, S.R.; Ahmed, M.; Lei, E.K.; Wisnovsky, S.P.; Kelley, S.O. Peptide-mediated delivery of chemical probes and therapeutics to mitochondria. Acc. Chem. Res. 2016, 49, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 2011, 15, 30231–33038. [Google Scholar] [CrossRef]
- Murphy, M. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Ross, M.F.; Prime, T.A.; Abakumova, I.; James, A.M.; Porteous, C.M.; Smith, R.A.; Murphy, M.P. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 2008, 411, 633–645. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Pourahmad, J.; Salimi, A.; Seydi, E. Mitochondrial targeting for drug development. In Toxicology Studies—Cells, Drugs and Environment, 1st ed.; Andreazza, A.C., Scola, G., Eds.; InTech: Rijeka, Croatia, 2015; pp. 61–82. [Google Scholar]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Asin-Cayuela, J.; Manas, A.R.; James, A.M.; Smith, R.A.; Murphy, M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004, 571, 9–16. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Coulter, C.M.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Filipovska, A.; Kelso, G.F.; Brown, S.E.; Beer, S.M.; Smith, R.A.; Murphy, M.P. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic: Insights into the interaction of ebselen with mitochondria. J. Biol. Chem. 2005, 280, 24113–24126. [Google Scholar] [CrossRef]
- Finichiu, P.G.; Larsen, D.S.; Evans, C.; Larsen, L.; Bright, T.P.; Robb, E.L.; Trnka, J.; Prime, T.A.; James, A.M.; Smith, R.A.; et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free Radic. Biol. Med. 2015, 89, 668–678. [Google Scholar] [CrossRef]
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H.; et al. Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in mitopark transgenic mice. Antioxid. Redox Signal. 2017, 27, 1048–1066. [Google Scholar] [CrossRef] [PubMed]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, J.A.; Khairullina, Z.Z.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Volynkina, I.A.; Skvortsov, D.A.; Makarov, G.I.; Abad, E.; Murayama, S.Y.; et al. Triphenilphosphonium analogs of chloramphenicol as dual-acting antimicrobial and antiproliferating agents. Antibiotics 2021, 10, 489. [Google Scholar] [CrossRef]
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J. Med. Chem. 2013, 56, 9170–9179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, W.Y.; Ma, X.; Ju, R.J.; Li, X.Y.; Li, N.; Sun, M.G.; Shi, J.F.; Zhang, C.X.; Lu, W.L. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials 2013, 34, 3626–3638. [Google Scholar] [CrossRef]
- Liu, H.N.; Guo, N.N.; Wang, T.T.; Guo, W.W.; Lin, M.T.; Huang-Fu, M.Y.; Vakili, M.R.; Xu, W.H.; Chen, J.J.; Wei, Q.C.; et al. Mitochondrial Targeted Doxorubicin-Triphenylphosphonium Delivered by Hyaluronic Acid Modified and pH Responsive Nanocarriers to Breast Tumor: In Vitro and in Vivo Studies. Mol. Pharm. 2018, 15, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef]
- Kang, S.; Sunwoo, K.; Jung, Y.; Hur, J.K.; Park, K.H.; Kim, J.S.; Kim, D. Membrane-targeting triphenylphosphonium functionalized ciprofloxacin for methicillin-resistant Staphylococcus aureus (MRSA). Antibiotics 2020, 9, 758. [Google Scholar] [CrossRef]
- Sunwoo, K.; Won, M.; Ko, K.P.; Choi, M.; Arambula, J.F.; Chi, S.G.; Sessler, J.L.; Verwilst, P.; Kim, J.S. Mitochondrial relocation of a common synthetic antibiotic: A non-genotoxic approach to cancer therapy. Chemistry 2020, 6, 1408–1419. [Google Scholar] [CrossRef]
- Cochrane, E.J.; Hulit, J.; Lagasse, F.P.; Lechertier, T.; Stevenson, B.; Tudor, C.; Trebicka, D.; Sparey, T.; Ratcliffe, A.J. Impact of Mitochondrial Targeting Antibiotics on Mitochondrial Function and Proliferation of Cancer Cells. ACS Med. Chem. Lett. 2021, 12, 579–584. [Google Scholar] [CrossRef]
- Ózsvári, B.; Magalhães, L.G.; Latimer, J.; Kangasmetsa, J.; Sotgia, F.; Lisanti, M.P. A myristoyl amide derivative of doxycycline potently targets cancer stem cells (CSCs) and prevents spontaneous metastasis, without retaining antibiotic activity. Front. Oncol. 2020, 10, 1528. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. A novel triphenylphosphonium carrier to target mitochondria without uncoupling oxidative phosphorylation. J. Med. Chem. 2021, 64, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Millard, M.; Pathania, D.; Shabaik, Y.; Taheri, L.; Deng, J.; Neamati, N. From broad-spectrum biocides to quorum sensing disruptors and mussel repellents: Antifouling profile of alkyl triphenylphosphonium salts. PLoS ONE 2010, 5, e0123652. [Google Scholar]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.; Murphy, M.P.; Darley-Usmar, V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013, 1, 86–93. [Google Scholar] [CrossRef]
- Trnka, J.; Elkalaf, M.; Anděl, M. Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak. PLoS ONE 2015, 10, e0121837. [Google Scholar] [CrossRef]
- Elkalaf, M.; Tuma, P.; Weiszenstein, M.; Polák, J.; Trnka, J. Mitochondrial probe methyltriphenylphosphonium (TPMP) inhibits the krebs cycle enzyme 2-Oxoglutarate dehydrogenase. PLoS ONE 2016, 11, e0161413. [Google Scholar] [CrossRef]
- Filipczaka, N.; Pana, J.; Yalamartya, S.S.K.; Torchilin, V.P. Recent advancements in liposome technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, W.; Zeng, D.; Huang, Q.; Xie, J.; Wen, H.; Li, J.; Yu, X.; Qin, L.; Zhou, Y. pH-activated, mitochondria-targeted, and redox-responsive delivery of paclitaxel nanomicelles to overcome drug resistance and suppress metastasis in lung cancer. J. Nanobiotech. 2021, 19, 152. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, H.I.; Chang, W.S.; Choi, J.S. Triphenylphosphonium-conjugated glycol chitosan microspheres for mitochondria-targeted drug delivery. Int. J. Biol. Macromol. 2021, 167, 35–45. [Google Scholar] [CrossRef]
- Oladimeji, O.; Akinyelu, J.; Daniels, A.; Singh, M. Modified gold nanoparticles for efficient delivery of betulinic acid to cancer cell mitochondria. Int. J. Mol. Sci. 2021, 22, 5072. [Google Scholar] [CrossRef]
- Denney, D.B.; Smith, L.C. Preparation and Reactions of Some Phosphobetaines. J. Org. Chem. 1962, 27, 3404–3408. [Google Scholar] [CrossRef]
- Wei-Li, D.; Bi, J.; Sheng-Lian, L.; Xu-Biao, L.; Xin-Man, T.; Chak-Tong, A. Functionalized phosphonium-based ionic liquids as efficient catalysts for the synthesis of cyclic carbonate from epoxides and carbon dioxide. Appl. Catal. A Gen. 2014, 470, 183–188. [Google Scholar] [CrossRef]
- Hudson, R.F.; Chopard, P.A. Structure et réactions du composé d’addition: Triphénylphosphine–anhydride maléique. Helv. Chim. Acta 1963, 46, 2178–2185. [Google Scholar] [CrossRef]
- Findlay, J.A.; Kwan, D. Metabolites from a Scytalidium Species. Can. J. Chem. 1973, 51, 3299–3301. [Google Scholar] [CrossRef]
- Hayashi, M.; Wakatsuka, H.; Kori, S. Trans-Δ2-Prostaglandins. U.S. Patent 3931296, 6 January 1976. [Google Scholar]
- Bundy, G.L. Phenyl-Substituted Prostaglandin-f Type Analogs. U.S. Patent 3987087, 19 October 1976. [Google Scholar]
- Narayanan, K.S.; Berlin, K.D. Novel synthesis of .omega.-(diphenylphosphinyl)alkylcarboxylic acids from triphenyl-.omega.-carboxyalkylphosphonium salts. J. Org. Chem. 1980, 45, 2240–2243. [Google Scholar] [CrossRef]
- Hann, M.M.; Sammes, P.G.; Kennewell, P.D.; Taylor, J.B. On the double bond isostere of the peptide bond: Preparation of an enkephalin analogue. J. Chem. Soc. Perkin Trans. 1 1982, 1, 307–314. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B.; Duhl-Emswiler, B.A. Stereochemistry of the Wittig reaction. Effect of nucleophilic groups in the phosphonium ylide. J. Am. Chem. Soc. 1985, 107, 217–226. [Google Scholar] [CrossRef]
- Carr, G.; Whittaker, D. Lactone formation in superacidic media. J. Chem. Soc. Perkin Trans. II 1987, 12, 1877–1880. [Google Scholar] [CrossRef]
- Cheskis, B.A.; Shpiro, N.A.; Moiseenkov, A.M. Effective synthesis of femrrulactone II based on the use of 2-carhoxyethyltriphenylphosphonium bromide. Russ. Chem. Bull. 1993, 42, 760–763. [Google Scholar] [CrossRef]
- Wakita, H.; Yoshiwara, H.; Nishiyama, H.; Nagase, H. Total Synthesis of Optically Active m-Phenyline PGI2 Derivative: Beraprost. Heterocycles 2000, 53, 1085–1110. [Google Scholar]
- Wiech, N.L.; Lan-Hargest, H.-Y. Histone Deacetylase Inhibitors. U.S. Patent 2006160902, 20 July 2006. [Google Scholar]
- Ahmed, R.; Altieri, A.; D’Souza, D.M.; Leigh, D.A.; Mullen, K.M.; Papmeyer, M.; Slawin, A.M.Z.; Wong, J.K.Y.; Woollins, J.D. Phosphorus-Based Functional Groups as Hydrogen Bonding Templates for Rotaxane Formation. J. Am. Chem. Soc. 2011, 133, 12304–12310. [Google Scholar] [CrossRef]
- Bakhtiyarova, Y.V.; Aksunova, A.F.; Galkina, I.V.; Galkin, V.I.; Lodochnikova, O.A.; Kataeva, O.N. Crystal structure of new carboxylate phosphabetaines and phosphonium salts conjugated with them. Russ. Chem. Bull. 2016, 65, 1313–1318. [Google Scholar] [CrossRef]
- Xu, J.; Zeng, F.; Wu, H.; Wu, S. A Mitochondria-targeted and NO-based Anticancer Nanosystem with Enhanced Photo-controllability and Low Dark-toxicity. J. Mater. Chem. B 2015, 3, 4904–4912. [Google Scholar] [CrossRef]
- Sodano, F.; Rolando, B.; Spyrakis, F.; Failla, M.; Lazzarato, L.; Gazzano, E.; Riganti, C.; Fruttero, R.; Gasco, A.; Sortino, S. Tuning the Hydrophobicity of a Mitochondria-Targeted NO Photodonor. ChemMedChem 2018, 13, 1238–1245. [Google Scholar] [CrossRef]
- Brooks, A.F.; Ichiishi, N.; Jackson, I.M.; Lee, S.J.; Sanford, M.S.; Scott, P.J.H.; Thompson, S. Synthesis of [18F]-γ-fluoro-α,β-unsaturated esters and ketones via vinylogous 18F-fluorination of α-diazoacetates with [18F]AgF. Synthesis 2019, 51, 4401–4407. [Google Scholar]
- Drikermann, D.; Mößel, R.S.; Al-Jammal, W.K.; Vilotijevic, I. Synthesis of Allylboranes via Cu(I)-catalyzed B-H Insertion of Vinyldiazoacetates into Phosphine-Borane Adducts. Org. Lett. 2020, 22, 1091–1095. [Google Scholar] [CrossRef]
- Testai, L.; Sestito, S.; Martelli, A.; Gorica, E.; Flori, L.; Calderone, V.; Rapposelli, S. Synthesis and pharmacological characterization of mitochondrial KATP channel openers with enhanced mitochondriotropic effects. Bioorg. Chem. 2021, 107, 104572. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Wang, S.; Ma, Y.-Y.; Zhao, D.-G.; Zhan, R.; Huang, H. Asymmetric Construction of Cyclobutanes via Direct Vinylogous Michael Addition/Cyclization of β,γ-Unsaturated Amides. Org. Lett. 2020, 22, 7135–7140. [Google Scholar] [CrossRef]
- Schweizer, E.E.; Wehmann, A.T. Reactions of phosphorus compounds. Part XXII. A reinvestigation of the reactions of activated alkynes with triphenylphosphine hydrobromide. An investigation of the reactions with bases of the vinylphosphonium salts prepared. J. Chem. Soc. C 1970, 1901–1905. [Google Scholar] [CrossRef]
- Bundy, G.L.; Nelson, N.A. Cis-4,5-didehydro-15 or 16-Alkylated 11-Deoxy-PGE1 Analogs. U.S. Patent 3987072, 19 October 1976. [Google Scholar]
- Kato, K.; Ohkawa, S.; Terao, S.; Terashita, Z.; Nishikawa, K. Thromboxane synthetase inhibitors (TXSI). Design, synthesis, and evaluation of a novel series of .omega.-pyridylalkenoic acids. J. Med. Chem. 1985, 28, 287–294. [Google Scholar] [CrossRef]
- Caldwell, C.; Chapman, K.T.; Hale, J.; Kim, D.; Lynch, C.; Maccoss, M.; Mills, S.G.; Rosauer, K.; Willoughby, C.; Berk, S. Pyrrolidine Modulators of Chemokine Receptor Activity. U.S. Patent 6265434, 24 July 2001. [Google Scholar]
- Mukku, V.J.R.V.; Maskey, R.P.; Monecke, P.; Grün-Wollny, I.; Laatsch, H. 5-(2-Methylphenyl)-4-pentenoic Acid from a Terrestrial Streptomycete. Z. Naturforsch. B 2002, 57, 335–337. [Google Scholar] [CrossRef]
- Wube, A.A.; Hüfner, A.; Thomaschitz, C.; Blunder, M.; Kollroser, M.; Bauer, R.; Bucar, F. Design, synthesis and antimycobacterial activities of 1-methyl-2-alkenyl-4(1H)-quinolones. Bioorg. Med. Chem. 2011, 19, 567–579. [Google Scholar] [CrossRef]
- Luo, D.; Sharma, H.; Yedlapudi, D.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Novel multifunctional dopamine D2/D3 receptors agonists with potential neuroprotection and anti-alpha synuclein protein aggregation properties. Bioorg. Med. Chem. 2016, 24, 5088–5102. [Google Scholar] [CrossRef]
- Chevalier, A.; Zhang, Y.; Khdour, O.M.; Hecht, S.M. Selective Functionalization of Antimycin A Through an N-Transacylation Reaction. Org. Lett. 2016, 18, 2395–2398. [Google Scholar] [CrossRef]
- Yalla, R.; Raghavan, S. Synthesis of solandelactone F, constanolactone A and an advanced intermediate towards solandelactone E from a common synthetic intermediate. Org. Biomol. Chem. 2019, 17, 4572–4592. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, G.; Yang, G.; Zhang, X.; Zhao, J.; Zhou, S. Stepwise dual targeting and dual responsive polymer micelles for mitochondrion therapy. J. Control. Release 2020, 322, 157–169. [Google Scholar]
- Deng, H.; Discordia, R.; Feng, X.; Jin, Z.; Leigh, C.; Moshos, K.; Sun, G. Cannabinoids and Uses Thereof. W.O. Patent 2021113669, 10 June 2021. [Google Scholar]
- Plattner, J.J.; Bhalerao, U.T.; Rapoport, H. Synthesis of dl-Sirenin. J. Am. Chem. Soc. 1969, 91, 4933–4934. [Google Scholar] [CrossRef]
- Uijttewaal, A.P.; Jonkers, F.L.; Van der Gen, A. Reactions of esters with phosphorus ylides. 3. Direct conversion into branched olefins. J. Org. Chem. 1979, 44, 3157–3168. [Google Scholar] [CrossRef]
- Bergman, N.A.; Jansson, M.; Chiang, Y.; Kresge, A.J.; Yin, Y. Synthesis and kinetic studies of a simple prostacyclin model. J. Org. Chem. 1987, 52, 4449–4453. [Google Scholar] [CrossRef]
- Misra, R.N.; Hüfner Sher, P.M.; Stein, P.D.; Hall, S.E.; Floyd, D.; Barrish, J.C. 7-Oxabicycloheptyl Substituted Heterocyclic Amide or Ester Prostaglandin Analogs Useful in the Treatment of Thrombotic and Vasospastic Disease. U.S. Patent 5100889, 31 March 1992. [Google Scholar]
- Provent, C.; Chautemps, P.; Gellon, G.; Pierre, J.-L. Double Wittig reactions with 4-carboxybutylidene triphenylphosphorane as the key step in the synthesis of benzene derivatives metadisubstituted with ωω’-difunctionalized six-carbon chains. Tetrahedron Lett. 1996, 37, 1393–1396. [Google Scholar] [CrossRef]
- Rey, M.D.L.A.; Martinez-Perez, J.A.; Fernandez-Gacio, A.; Halkes, K.; Fall, Y.; Granja, J.; Mourino, A. New Synthetic Strategies to Vitamin D Analogues Modified at the Side Chain and D Ring. Synthesis of 1alpha,25-Dihydroxy-16-ene-vitamin D(3) and C-20 Analogues(1). J. Org. Chem. 1999, 64, 3196–3206. [Google Scholar]
- Henry-Riyad, H.; Tidwell, T.T. Cyclization of 5-hexenyl radicals from nitroxyl radical additions to 4-pentenylketenes and from the acyloin reaction. Canad. J. Chem. 2003, 81, 697–704. [Google Scholar] [CrossRef]
- Aotsuka, T.; Kumazawa, K.; Wagatsuma, N.; Ishitani, K. Novel 1,8-Naphthyridin-2(1H)-one Derivatives. U.S. Patent 2003036651, 4 December 2003. [Google Scholar]
- Henschke, J.P.; Liu, Y.; Huang, X.; Chen, Y.; Meng, D.; Xia, L.; Wei, X.; Xie, A.; Li, D.; Huang, Q.; et al. The Manufacture of a Homochiral 4-Silyloxycyclopentenone Intermediate for the Synthesis of Prostaglandin Analogues. Org. Proc. Res. Dev. 2012, 16, 1905–1916. [Google Scholar] [CrossRef]
- Wang, K. Preparation Method of Optically Pure Dextro Cloprostenol Sodium. Patent CN 104513186, 5 October 2016. [Google Scholar]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar]
- Cui, D.; Yue, C.; Yang, Y. Mitochondrion-Targeting Nano-Drug Delivery System and Preparation Method and Application Thereof. Patent CN 105833289, 9 April 2019. [Google Scholar]
- Hamri, S.; Jouha, J.; Oumessaoud, A.; Pujol, M.D.; Khouili, M.; Guillaumet, G. Convenient approach for the synthesis of ONO-LB-457, a potent leukotriene B4 receptor antagonist. Tetrahedron 2021, 77, 131740. [Google Scholar] [CrossRef]
- Bogdanov, A.A.; Chen, C.-W.; Khairullina, Z.Z.; Konevega, A.L.; Lukianov, D.A.; Makarov, G.I.; Osterman, I.A.; Paleskava, A.; Pavlova, J.A.; Polikanov, Y.S.; et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics 2021, 10, 390. [Google Scholar]
- Castellanos, L.; Gateau-Olesker, A.; Panne-Jacolot, F.; Cleophax, J.; Gero, S.D. Synthèse d’analogues de derives dioxaprostanoïques à partir du D et DU L-xylose. Tetrahedron 1981, 37, 1691–1696. [Google Scholar] [CrossRef]
- Dobner, B.; Nuhn, P.; Burkhardt, U. Synthese mittelständig methylverzweigter Fettsäuren. Z. Chem. 1987, 27, 63–64. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Cruz, H.; Hill, C.A.; De Voss, J.J.; Garson, M. Identification and Total Synthesis of Novel Fatty Acids from the Siphonarid Limpet Siphonaria denticulate. J. Nat. Prod. 2001, 64, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Momán, E.; Nicoletti, D.; Mouriňo, A. Synthesis of novel analogues of 1α,25-dihydroxyvitamin D3 with side chains at C-18. J. Org. Chem. 2004, 69, 4615–4625. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Azcon, J.; Galve, R.; Sanchez-Baeza, F.; Marco, M.-P. Development of An Enzyme-Linked Immunosorbent Assay (ELISA) for the Determination of the Linear Alkylbenzene Sulfonates (LAS) and long chain Sulfophenhyl Carboxylates (SPCs) using Antibodies Generated by pseudo-Heterologous Immunization. Anal. Chem. 2006, 78, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Hardouin, C.; Kelso, M.J.; Romero, F.A.; Rayl, T.J.; Leung, D.; Hwang, I.; Cravatt, B.F.; Boger, D.L. Structure–Activity Relationships of α-Keto Oxazole Inhibitors of Fatty Acid Amide Hydrolase. J. Med. Chem. 2007, 50, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L. Tricyclic Inhibitors of Fatty Acid Amide Hydrolase. Patent WO 2008150492, 11 December 2008. [Google Scholar]
- Zhao, G.; Yang, C.; Li, B.; Xia, W. A new phenylethyl alkyl amide from the Ambrostoma quadriimpressum Motschulsky. Beilstein J. Org. Chem. 2011, 7, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, K.; Mainkar, P.S. PPAR Modulators. U.S. Patent 2011178112, 21 July 2011. [Google Scholar]
- Wang, Y.; Wang, B.; Liao, H.; Song, X.; Wu, H.; Wang, H.; Shen, H.; Ma, X.; Tan, M. Liposomal nanohybrid cerasomes for mitochondria-targeted drug delivery. J. Mater. Chem. B 2015, 3, 7291–7299. [Google Scholar] [CrossRef]
- Guo, R.; Huang, J.; Huang, H.; Zhao, X. Organoselenium-Catalyzed Synthesis of Oxygen- and Nitrogen-Containing Heterocycles. Org. Lett. 2016, 18, 504–507. [Google Scholar] [CrossRef]
- Kalathil, A.A.; Kumar, A.; Banik, B.; Ruiter, T.A.; Pathak, R.K.; Dhar, S. New formulation of old aspirin for better delivery. Chem. Comm. 2016, 52, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Alamudi, S.H.; Satapathy, R.; Su, D. Background-Free Fluorescent Probes for Live Cell Imaging. Patent WO 201778623, 30 March 2011. [Google Scholar]
- Cheng, J. Method for Preparing 6-Bromotriphenylphosphonio-n-Caproic Acid. Patent CN 106632474, 7 September 2018. [Google Scholar]
- Kalathil, A.A.; Banik, B.; Kumar, A.; Dhar, S. Modification of Drugs for Incorporation into Nanoparticles. U.S. Patent 2017087167, 30 March 2017. [Google Scholar]
- Dhar, S.; Pathak, R. Precise Delivery of Therapeutic Agents to Cell Mitochondria for Anti-Cancer Therapy. U.S. Patent 10004809, 26 June 2018. [Google Scholar]
- Pantelia, A.; Daskalaki, I.; Cuquerella, M.C.; Rotas, G.; Miranda, M.A.; Vougioukalakis, G.C. Synthesis and Chemiluminescent Properties of Amino-Acylated luminol Derivatives Bearing Phosphonium Cations. Molecules 2019, 24, 3957. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Z.; Chen, M.; Xie, S.; Wang, T.; Li, X. Synergistic antitumor efficacy of hybrid micelles with mitochondrial targeting and stimuli-responsive drug release. J. Mater. Chem. B 2019, 7, 1415–1426. [Google Scholar] [CrossRef]
- Bundy, G.L. 9-Deoxy-PGF2 Analogs. U.S. Patent 4033989, 5 July 1977. [Google Scholar]
- Bundy, G.L.; Nelson, N.A. 2a,2b-Dihomo-11-deoxy-17(substituted phenyl)-18,19,20-trinor-PGE2 Compounds and Their Corresponding Esters. U.S. Patent 4029693, 14 July 1977. [Google Scholar]
- Youngdale, G.A. 2a,2b-Dihomo-16,16-dimethyl-PGF2 Analogs. U.S. Patent 4067891, 10 January 1978. [Google Scholar]
- Smith, H.W. 13,14-Didehydro-PGA1 Compounds. U.S. Patent 4086258, 25 April 1978. [Google Scholar]
- Nelson, N.A. 15-Epi-15-methyl-16-phenoxy-PGE Compounds. U.S. Patent 4154950, 15 May 1979. [Google Scholar]
- Allais, A.; Clemence, F.; Meier, J.; Deraedt, R. Novel Carboxylic Acids, Benzoyl Phenyl Alkanoic Acids and Use Thereof. U.S. Patent 4337353, 29 June 1982. [Google Scholar]
- Johnson, A.T.; Jiao, G.-S. Hydroxamic Acid Derivatives of 3-Phenyl Propionic Acids Useful as Therapeutic Agents for Treating Anthrax Poisoning. U.S. Patent 2008188566, 5 October 2010. [Google Scholar]
- Kim, S.; Jiao, G.-S.; Moayeri, M.; Crown, D.; Cregar-Hernandez, L.; McKasson, L.; Margosiak, S.A.; Leppla, S.H.; Johnson, A.T. Antidotes to anthrax lethal factor intoxication. Part 2: Structural modifications leading to improved in vivo efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 2030–2033. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Migglautsch, A.K.; Willim, M.; Schweda, B.; Glieder, A.; Breinbauer, R.; Winkler, M. Aliphatic hydroxylation and epoxidation of capsaicin by cytochrome P450 CYP505X. Tetrahedron 2018, 74, 6199–6204. [Google Scholar] [CrossRef]
- Li, S.; Quan, X.; Xu, J.; Li, J.; Xie, J. Preparation and Application of Quaternary Phosphonium Salt Modified Dendriform Molecule. Patent CN 104844650, 29 December 2017. [Google Scholar]
- Suzuki, Y.; Takahashi, Y. Cationic Lipid. Patent EP 3476832, 1 May 2019. [Google Scholar]
- Chun, J.; Cincilla, G.; Del Valle, C.R.; Devesa, I.; Ferrer-Montiel, A.V.; González-Gil, I.; Hernández-Torres, G.; Khiar-Fernández, N.; Kihara, Y.; López-Rodríguez, M.L.; et al. A novel agonist of the type 1 lysophosphatidic acid receptor (LPA1), UCM-05194, shows efficacy in neuropathic pain amelioration. J. Med. Chem. 2019, 63, 2372–2390. [Google Scholar]
- Dawson, M.I.; Vasser, M. Synthesis of prostaglandin synthetase substrate analogs. 1. (Z)-14-Hydroxy-12,13-methano-8-nonadecenoic acid. J. Org. Chem. 1977, 42, 2783–2785. [Google Scholar] [CrossRef]
- Guthrie, R.W.; Kierstead, R.W. Pyridine Compounds Which Are Useful in Treating a Disease State Characterized by an Excess of Platelet Activating Factors. U.S. Patent 4927838, 22 May 1990. [Google Scholar]
- Nanda, S.; Yadav, J.S. Asymmetric synthesis of unnatural (Z, Z, E)-octadecatrienoid and eicosatrienoid by lipoxygenase-catalyzed oxygenation. Tetrahedron Asymm. 2003, 14, 1799–1806. [Google Scholar] [CrossRef]
- Mor, M.; Lodola, A.; Rivara, S.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Piersanti, G.F.; Clapper, J.R.; King, A.R.; et al. Synthesis and Quantitative Structure−Activity Relationship of Fatty Acid Amide Hydrolase Inhibitors: Modulation at the N-Portion of Biphenyl-3-yl Alkylcarbamates. J. Med. Chem. 2008, 51, 3487–3498. [Google Scholar] [CrossRef][Green Version]
- Sparey, T.; Ratcliffe, A.; Cochrane, E.; Stevenson, B.; Hallett, D.; Lagasse, F.; Lassalle, G.; Froidbise, A. Azithromycin Derivatives Containing a Phosphonium Ion as Anticancer Agents. Patent WO 2018193113, 25 October 2018. [Google Scholar]
- Linnebank, P.R.; Ferreira, S.F.; Kluwer, A.M.; Reek, J.N.H. Regioselective Hydroformylation of Internal and Terminal Alkenes via Remote Supramolecular Control. Chem. Eur. J. 2020, 26, 8214–8219. [Google Scholar] [CrossRef]
- Sparey, T.; Ratcliffe, A.; Cochrane, E.; Stevenson, B.; Hallett, D.; Lagasse, F.; Lassalle, G.; Froidbise, A. Phosphonium-Ion Tethered Tetracycline Drugs for Treatment of Cancer. U.S. Patent 2020123182, 23 April 2020. [Google Scholar]
- Lou, H.; Wang, X.; Liu, J. Triazole Compounds, Preparation Method and Application of Triazole Compounds in Antifungal Drugs. Patent CN 111647019, 11 September 2020. [Google Scholar]
- Rao, A.V.R.; Reddy, E.R.; Purandare, A.V.; Varaprasad, C.V.N.S. Stereoselective synthesis of unsaturated C-18 hydroxy fatty acids the self defensive substances. Tetrahedron 1987, 43, 4385–4394. [Google Scholar] [CrossRef]
- Buchanan, G.W.; Smits, R.; Munteanu, E. Synthesis and 19F NMR study of RF-oleic acid-F13. J. Fluor. Chem. 2003, 123, 255–259. [Google Scholar] [CrossRef]
- Duffy, P.E.; Quinn, S.M.; Roche, H.M.; Evans, P. Synthesis of trans-vaccenic acid and cis-9-trans-11-conjugated linoleic acid. Tetrahedron 2006, 62, 4838–4843. [Google Scholar] [CrossRef]
- Thurnhofer, S.; Saskia, W. Synthesis of (S)-(+)-enantiomers of food-relevant (n-5)-monoenoic and saturated anteiso-fatty acids by a Wittig reaction. Tetrahedron 2007, 63, 1140–1145. [Google Scholar] [CrossRef]
- Andjelkovic, M.; Ceccarelli, S.M.; Chomienne, O.; Kaplan, G.L.; Mattei, P.; Tilley, J.W. 4-Dimethylaminobutyric Acid Derivatives. U.S. Patent 2009270505, 29 October 2009. [Google Scholar]
- Kumari, A.; Gholap, S.P.; Fernandes, R.A. Tandem IBX-Promoted Primary Alcohol Oxidation/Opening of Intermediate β, γ-Diolcarbonate Aldehydes to (E)-γ-Hydroxy-α, β-enals. Chem. Asian J. 2019, 14, 2278–2290. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.; Whiteman, M.; Perry, A. Hydrogen Sulfide Releasing Compounds and Their Use. Patent WO 201345951, 4 April 2013. [Google Scholar]
- Le Trionnaire, S.; Perry, A.; Szczesny, B.; Szabo, C.; Winyard, P.G.; Whatmore, J.L.; Wood, M.E.; Whiteman, M. The synthesis and functional evaluation of a mitochondria-targeted hydrogen sulfide donor,(10-oxo-10-(4-(3-thioxo-3 H-1,2-dithiol-5-yl) phenoxy) decyl) triphenylphosphonium bromide (AP39). MedChemComm 2014, 5, 728–736. [Google Scholar] [CrossRef]
- Sparey, T.; Ratcliffe, A.; Hallett, D.; Cochrane, E.; Lassalle, G.; Froidbise, A.; Stevenson, B. Triphenylphosphonium-Tethered Tetracycyclines for Use in Treating Cancer. Patent WO 2018193114, 25 October 2018. [Google Scholar]
- Baran, J.S.; Lowrie, H.S. Metabolites of Pentanedioic Acid Derivatives. U.S. Patent 5055613, 8 October 1991. [Google Scholar]
- Müller, S.; Schmidt, R.R. Synthesis of Unique Ceramides and Cerebrosides Occurring in Human Epidermis. Helv. Chim. Acta 1993, 76, 616–630. [Google Scholar] [CrossRef]
- Skulachev, M.V.; Skulachev, V.P.; Zamyatin, A.A.; Efremov, E.S.; Tashlitsky, V.N.; Yaguzhinsky, L.S.; Korshunova, G.A.; Sumbatyan, N.V.; Antonenko, Y.N.; Severina, I.I.; et al. Pharmaceutical Substances on the Basis of Mitochondria-Addressed Antioxidants. U.S. Patent 2012259110, 12 January 2016. [Google Scholar]
- Martín-Rodríguez, A.J.; Babarro, J.M.F.; Lahoz, F.; Sansón, M.; Martín, V.S.; Norte, M.; Fernández, J.J. From broad-spectrum biocides to quorum sensing disruptors and mussel repellents: Antifouling profile of alkyl triphenylphosphonium salts. PLoS ONE 2015, 10, e0123652. [Google Scholar] [CrossRef] [PubMed]
- Opálka, L.; Kováčik, A.; Sochorová, M.; Roh, J.; Kuneš, J.; Lenčo, J.; Vávrová, K. Scalable Synthesis of Human Ultralong Chain Ceramides. Org. Lett. 2015, 17, 5456–5459. [Google Scholar] [CrossRef]
- Kluba, C.A.; Bauman, A.; Valverde, I.E.; Vomstein, S.; Mindt, T.L. Dual-targeting conjugates designed to improve the efficacy of radiolabeled peptides. Org. Biomol. Chem. 2012, 10, 7594–7602. [Google Scholar] [CrossRef]
- Leavens, W.J.; Lane, S.J.; Carr, R.M.; Lockie, A.M.; Waterhouse, I. Derivatization for liquid chromatography/electrospray mass spectrometry: Synthesis of tris(trimethoxyphenyl)phosphonium compounds and their derivatives of amine and carboxylic acids. Rapid Commun. Mass Spectr. 2002, 16, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H. Zur Reaktion von Triphenylphosphin mit Olefinen. Chem. Ber. 1961, 94, 1331–1336. [Google Scholar] [CrossRef]
- Romanov, S.; Aksunova, A.; Bakhtiyarova, Y.; Shulaeva, M.; Pozdeev, O.; Egorova, S.; Galkina, I.; Galkin, V. Tertiary phosphines in reactions with substituted cinnamic acids. J. Organomet. Chem. 2020, 910, 121130. [Google Scholar] [CrossRef]
- Bestmann, H.J.; Mott, L.; Lienert, J. Reaktionen mit Triphenylphosphin sowie dessen Hydrobromid und Dibromid, III. Äther und Esterspaltungen mit Triphenylphosphinhydrobromid. Justus Liebigs Ann. Chem. 1967, 709, 105–112. [Google Scholar] [CrossRef]
- Kiuchi, F.; Nakamura, N.; Saito, M.; Komagome, K.; Hiramatsu, H.; Takimoto, N.; Akao, N.; Kondo, K.; Tsuda, Y. Synthesis and Nematocidal Activity of Aralkyl- and Aralkenylamides Related to Piperamide on Second-Stage Larvae of Toxocara canis. Chem. Pharm. Bull. 1997, 45, 685–696. [Google Scholar] [CrossRef][Green Version]
- Sancéau, J.-Y.; Maltais, R.; Poirier, D.; Marette, A. Total Synthesis of the Antidiabetic (Type 2) Lipid Mediator Protectin DX/PDX. J. Org. Chem. 2019, 84, 495–505. [Google Scholar] [CrossRef]
- Virieux, D.; Volle, J.-N.; Pirat, J.-L. Product class 12: Alkylphosphonium salts. Sci. Synth. 2009, 42, 503–594. [Google Scholar]
- Büttner, H.; Steinbauer, J.; Werner, T. Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide by Using Bifunctional One-Component Phosphorus-Based Organocatalysts. ChemSusChem 2015, 8, 2655–2669. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, M.; Liu, J.-T. Copper-Catalyzed Diastereoselective Synthesis of Trifluoromethylated Tetrahydrofurans. Adv. Synth. Cat. 2016, 358, 1322–1327. [Google Scholar] [CrossRef]
- Atkinson, J.; Stuart, J.; Kagan, V.E.; Stoyanovsky, D.A.; Epperly, M.W.; Greenberger, J.S.; Bayir, H. Mitochondria-Targeted Inhibitors of Cytochrome C Peroxidase for Protection from Apoptosis. U.S. Patent 9365597, 14 June 2016. [Google Scholar]
- Lei, H.; Atkinson, J. Synthesis of phytyl-and chroman-derivatized photoaffinity labels based on α-tocopherol. J. Org. Chem. 2000, 65, 2560–2567. [Google Scholar] [CrossRef] [PubMed]
- Horiike, M.; Tanouchi, M.; Hirano, C. Synthesis of insect sex pheromones and their homologues;(Z)-6-Alkenyl acetates from the Wittig reaction. Agric. Biol. Chem. 1978, 42, 1963–1965. [Google Scholar]
- Cripe, T.A.; Connor, D.S.; Vinson, P.K.; Burckett-St, L.J.C.; Willman, K.W. Polyoxyalkylene Surfactants. U.S. Patent 6093856, 25 July 2000. [Google Scholar]
- Culcasi, M.; Casano, G.; Lucchesi, C.; Mercier, A.; Clement, J.-L.; Pique, V.; Michelet, L.; Krieger-Liszkay, A.; Robin, M.; Pietri, S. Synthesis and biological characterization of new aminophosphonates for mitochondrial pH determination by 31P NMR spectroscopy. J. Med. Chem. 2013, 56, 2487–2499. [Google Scholar] [CrossRef] [PubMed]
- Muratore, A.; Plessis, C.; Chanot, J.-J. Novel Cycloalkane Aldehydes, Method for Preparing Same, and Use Thereof in the Perfume Industry. U.S. Patent 9051531, 9 June 2015. [Google Scholar]
- He, J.; Baldwin, J.E.; Lee, V. Studies towards the Synthesis of the Antibiotic Tetrodecamycin. Synlett 2018, 29, 1117–1121. [Google Scholar]
- Xue, J.; Zhao, X.; Huang, Y. Double-Targeted Phthalocyanine Anti-Cancer Photosensitizer and Preparation Method Thereof. Patent CN 109081852, 25 December 2018. [Google Scholar]
- Horiike, M.; Hirano, C. A Facile Synthesis of the Sex Pheromones (Z)-7-Dodecen-1-yl Acetate and Its Homologues. Agric. Biol. Chem. 1980, 44, 2229–2230. [Google Scholar] [CrossRef]
- Horiike, M.; Tanouchi, M.; Hirano, C. A convenient method for synthesizing (Z)-alkenols and their acetates. Agric. Biol. Chem. 1980, 44, 257–261. [Google Scholar] [CrossRef]
- Jara, J.A.; Castro-Castillo, V.; Saavedra-Olavarría, J.; Peredo, L.; Pavanni, M.; Jana, F.; Letelier, M.E.; Parra, E.; Becker, M.I.; Morello, A.; et al. Antiproliferative and uncoupling effects of delocalized, lipophilic, cationic gallic acid derivatives on cancer cell lines. Validation in vivo in singenic mice. J. Med. Chem. 2014, 57, 2440–2454. [Google Scholar] [CrossRef]
- Bruns, H.; Thiel, V.; Voget, S.; Patzelt, D.; Daniel, R.; Wagner-Doebler, I.; Schulz, S. N-acylated alanine methyl esters (NAMEs) from Roseovarius tolerans, structural analogs of quorum-sensing autoinducers, N-acylhomoserine lactones. Chem. Biodiver. 2013, 10, 1559–1573. [Google Scholar] [CrossRef]
- Bruns, H.; Herrmann, J.; Müller, R.; Wang, H.; Wagner-Döbler, I.; Schulz, S. Oxygenated N-acyl alanine methyl esters (NAMEs) from the marine bacterium Roseovarius tolerans EL-164//Journal of natural products. J. Nat. Prod. 2018, 81, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Sun, X.; Yuan, C.; Ma, S.; Zhou, Y.; Bian, Q.; Wang, M. Method for Synthesizing Grassland Spodoptera Litura Sex Pheromone Active Ingredients. Patent CN 111269114, 12 June 2020. [Google Scholar]
- Baker, S.R.; Ross, W.J. Sulfur Containing Alkenylenyl Substituted Benzoic Acids and Phenyl Tetrazoles and Their Use as Anti-allergic Agents. U.S. Patent 4675335, 23 June 1987. [Google Scholar]
- Lermer, L.; Neeland, E.G.; Ounsworth, J.P.; Sims, R.J.; Tischler, S.A.; Weiler, L. The synthesis of β-keto lactones via cyclization of β-keto ester dianions or the cyclization of Meldrum’s acid derivatives. Can. J. Chem. 1992, 70, 1427–1445. [Google Scholar] [CrossRef]
- Okuda, K.; Hasui, K.; Abe, M.; Matsumoto, K.; Shindo, M. Molecular design, synthesis, and evaluation of novel potent apoptosis inhibitors inspired from bongkrekic acid. Chem. Res. Toxicol. 2012, 25, 2253–2260. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Shao, W.; Zhu, C.; Wen, G.; Yue, X.; Wang, R.; Quan, J.; Du, J.; Bu, X. Mitochondria-targeted approach: Remarkably enhanced cellular bioactivities of TPP2a as selective inhibitor and probe toward TrxR. ACS Chem. Biol. 2016, 11, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Catalán, M.; Castro-Castillo, V.; Gajardo-de la Fuente, J.; Aguilera, J.; Ferreira, J.; Ramires-Fernandez, R.; Olmedo, I.; Molina-Berríos, A.; Palominos, C.; Valencia, M.; et al. Continuous flow synthesis of lipophilic cations derived from benzoic acid as new cytotoxic chemical entities in human head and neck carcinoma cell lines. RSC Med. Chem. 2020, 11, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Kong, X.; Zhang, S. Micromelalopha Troglodyta Sex Pheromone Attractant Composition, Lure and Preparation Method and Application Thereof. Patent CN 110959613, 17 April 2020. [Google Scholar]
- Alunda, J.M.; Cueto-Díaz, E.J.; Dardonville, C.; Gamarro, F.; Herraiz, T.; Manzano, J.I.; Olías-Molero, A.I.; Perea, A.; Torrado, J.J. Discovery and pharmacological studies of 4-hydroxyphenyl-derived phosphonium salts active in a mouse model of visceral leishmaniasis. J. Med. Chem. 2019, 62, 10664–10675. [Google Scholar]
- Mannu, A.; Di Pietro, M.E.; Priola, E.; Baldino, S.; Sacchetti, A.; Mele, A. Unconventional reactivity of epichlorohydrin in the presence of triphenylphosphine: Isolation of ((1,4-dioxane-2,5-diyl)-bis-(methylene))-bis-(triphenylphosphonium) chloride. Res. Chem. Intermed. 2021, 47, 1663–1674. [Google Scholar] [CrossRef]
- Wada, M.; Tsuboi, A. Reactions of tris(2,6-dimethoxyphenyl)phosphine with epoxides. J. Chem. Soc. Perkin Trans. 1 1987, 151–154. [Google Scholar] [CrossRef]
- Yamamoto, S.; Okuma, K.; Ohta, H. General synthesis of 2-, 3-, and 4-hydroxyalkylphosphonium salts by the reaction of triphenylphosphine with cyclic ethers in the presence of strong acids. Bull. Chem. Soc. Jpn. 1988, 61, 4476–4478. [Google Scholar] [CrossRef]
- Yamamoto, S.-I.; Takeuchi, H.; Tanaka, Y.; Okuma, K.; Ohta, H. Synthesis and reaction of optically active 2-and 3-hydroxyalkylphosphonium salts. Chem. Lett. 1991, 20, 113–116. [Google Scholar] [CrossRef]
- Plénat, F.; Grelet, D.; Ozon, V.; Cristau, H.-J. Synthesis of new functionalized hydroxyalkyl phosphonium salts. Synlett 1994, 4, 269–270. [Google Scholar] [CrossRef]
- Mironov, V.F.; Karaseva, A.N.; Nizamov, I.S.; Kedrov, I.S.; Konovalov, A.I. Triphenylphosphonium Trifluoromethanesulfonate in Reactions with Epoxy Derivatives. Russ. J. Org. Chem. 2004, 40, 910–911. [Google Scholar] [CrossRef]
- Christol, H.; Grelet, D.; Darvich, M.R.; Fallouh, F.; Plenat, F.; Cristau, H.-J. Réaction des oxiranes avec la triphénylphosphine en milieu phénolique. Bull. Soc. Chim. Fr. 1989, 477–483. [Google Scholar]
- Tolstikova, L.L.; Bel’skikh, A.V.; Shainyan, B.A. Protonation and alkylation of organophosphorus compounds with trifluoromethanesulfonic acid derivatives. Russ. J. Gen. Chem. 2011, 81, 474–480. [Google Scholar] [CrossRef]
- Parker, R.E.; Isaacs, N.S. Mechanisms of epoxide reactions. Chem. Rev. 1959, 59, 737–799. [Google Scholar] [CrossRef]
- Rosowsky, A. Ethylene Oxides. In Hеtеrocуclic Compounds with Three- and Four-Membered Rings; Weissberger, A., Ed.; Interscie: New York, NY, USA; London, UK; Sуdneу, Australia, 1964; Part I, pp. 1–523. [Google Scholar]
- Wohl, R.A. Mechanism of acid-catalyzed ring-opening of epoxides-reinterpretative review. Chimia 1974, 28, 1–5. [Google Scholar]
- Gray, G.A. Carbon-13 nuclear magnetic resonance of organophosphorus compounds. VIII. Triphenylphosphoranes and triphenylphosphonium salts. J. Am. Chem. Soc. 1973, 95, 7736–7742. [Google Scholar] [CrossRef]
- Flores-Gallardo, H.; Pollard, C.B. Epoxy ethers and ether amino alcohols. J. Org. Chem. 1947, 12, 831–833. [Google Scholar] [CrossRef]
- Giodini, L.; Lo Re, F.; Campagnol, D.; Marangon, E.; Posocco, B.; Dreussi, E.; Toffoli, G. Nanocarriers in cancer clinical practice: A pharmacokinetic issue. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 583–599. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Forssen, E.A. The design and development of DaunoXome® for solid tumor targeting in vivo. Adv. Drug Deliv. Rev. 1997, 24, 133–150. [Google Scholar] [CrossRef]
- Leonard, R.C.F.; Williams, S.; Tulpule, A.; Levine, A.M.; Oliveros, S. The design and development of DaunoXome® for solid tumor targeting in vivo. Breast 2009, 18, 218–224. [Google Scholar] [CrossRef]
- Bourquin, J.; Milosevic, A.; Hauser, D.; Lehner, R.; Blank, F.; Petri-Fink, A.; Rothen-Rutishauser, B. Biodistribution, clearance, and long-term fate of clinically relevant nanomaterials. Adv. Mater. 2018, 30, 1704307. [Google Scholar] [CrossRef]
- Tomsen-Melero, J.; Passemard, S.; Garcia-Aranda, N.; Diaz-Riascos, Z.V.; Gonzalez-Rioja, R.; Pedersen, J.N.; Lyngsø, J.; Merlo-Mas, J.; Cristobal-Lecina, E.; Corchero, J.L.; et al. Impact of chemical composition on the nanostructure and biological activity of α-galactosidase-loaded nanovesicles for fabry disease treatment. ACS Appl. Mater. Interfaces 2021, 13, 7825–7838. [Google Scholar] [CrossRef]
- Pashirova, T.N.; Sapunova, A.S.; Lukashenko, S.S.; Burilova, E.A.; Lubina, A.P.; Shaihutdinova, Z.M.; Gerasimova, T.P.; Kovalenko, V.I.; Voloshina, A.D.; Souto, E.B.; et al. Synthesis, structure-activity relationship and biological evaluation of tetracationic gemini Dabco-surfactants for transdermal liposomal formulations. Int. J. Pharm. 2020, 575, 118953. [Google Scholar] [CrossRef]
- Sęk, A.; Perczyk, P.; Wydro, P.; Gruszecki, W.I.; Szczes, A. Effect of trace amounts of ionic surfactants on the zeta potential of DPPC liposomes. Chem. Phys. Lipids 2021, 235, 105059. [Google Scholar] [CrossRef]
- Pashirova, T.N.; Burilova, E.A.; Tagasheva, R.G.; Zueva, I.V.; Gibadullina, E.M.; Nizameev, I.R.; Sudakov, I.A.; Vyshtakalyuk, A.B.; Voloshina, A.D.; Kadirov, M.K.; et al. Delivery nanosystems based on sterically hindered phenol derivatives containing a quaternary ammonium moiety: Synthesis, cholinesterase inhibition and antioxidant activity. Chem. Biol. Interact. 2019, 310, 108753. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.; SADABS. Program for Empirical X-ray Absorption Correction; Bruker-Nonius: Gottingen, Germany, 2004. [Google Scholar]
- APEX2 (Version 2.1), SAINTPlus. Data Reduction and Correction Program (Version 7.31A); BrukerAXS Inc.: Madison, WI, USA, 2006. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Straver, L.H.; Schierbeek, A.J. MOLEN. Structure Determination System. Program Description; Nonius, B.V.: Delft, The Netherlands, 1994; Volume 1, p. 180. [Google Scholar]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–459. [Google Scholar] [CrossRef]
- Al-Lal, A.M.; Garcнa-González, J.E.; Llamas, A.; Monjas, A.; Canoira, L.L. A new route to synthesize tert-butyl ethers of bioglycerol. Fuel 2012, 93, 632–637. [Google Scholar] [CrossRef]
- Solov’ev, D.V.; Kolomenskaya, L.V.; Rodin, A.A.; Zenkevich, I.G.; Lavrent’ev, A.N. Fluorine-containing 2, 3-epoxypropyl ethers. synthesis and spectral characteristics. J. Gen. Chem. USSR 1991, 61, 611–615. [Google Scholar]
- Il’in, A.A.; Il’in, A.N.; Bakhmutov, Y.L.; Furin, G.G.; Pokrovskii, L.M. Promising prospects for using partially fluorinated alcohols as O-nucleophilic reagents in organofluoric synthesis. Russ. J. Appl. Chem. 2007, 80, 405–418. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Bauer, K. Synthese des Bis-ß-chromanons. Chem. Ber. 1947, 80, 7–19. [Google Scholar] [CrossRef]
- Reyhanoglu, Y.; Sahmetlioglu, E.; Gokturk, E. Alternative approach for synthesizing polyglycolic acid copolymers from C1 feedstocks and fatty ester epoxides. ACS Sustain. Chem. Eng. 2019, 7, 5103–5110. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
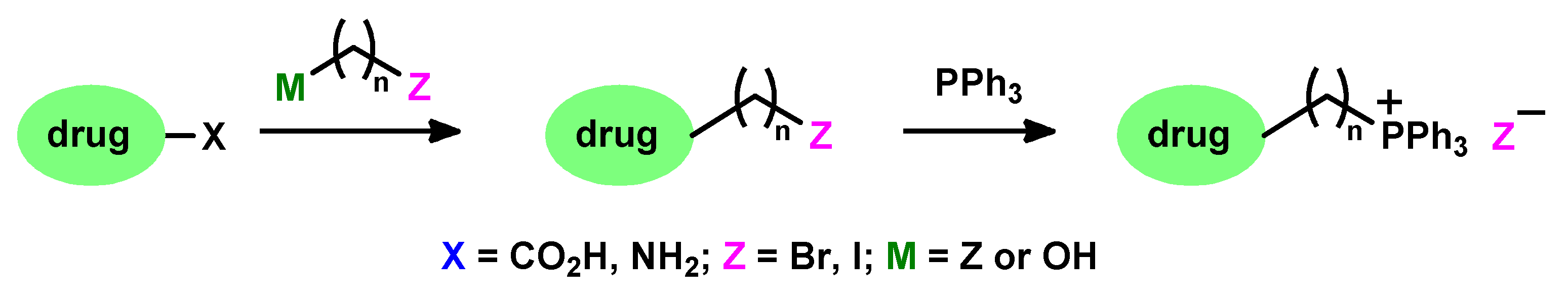{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
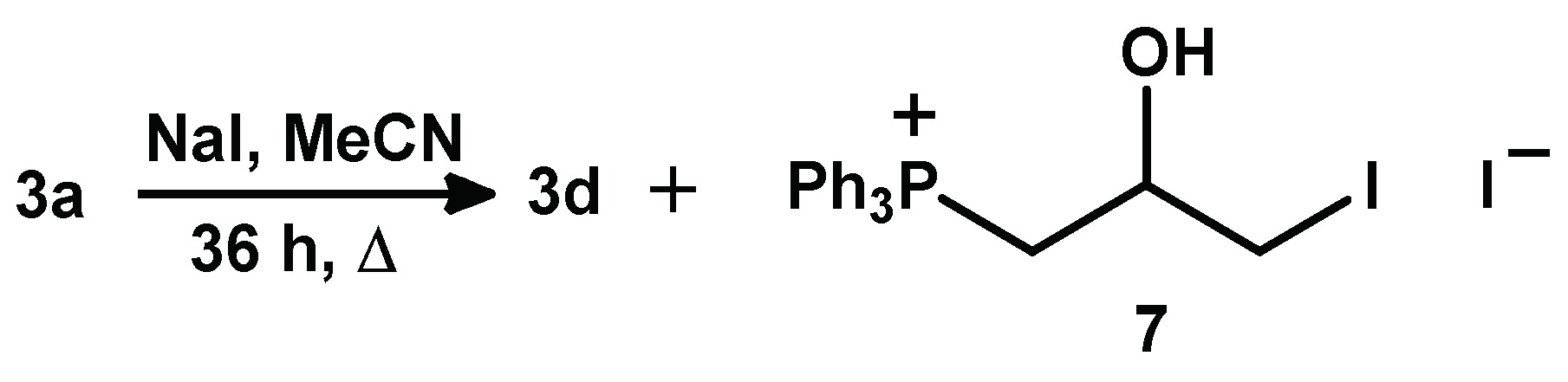{kind=link}
| Composition | Ratio, w/w, % | Concentration of Rhodamine B, (%, w/w) | Zaverage (nm) | PdI | Z (mV) | EE, (%) | LC, (%) |
|---|---|---|---|---|---|---|---|
| PC * | 100 | – | 119 ± 2 | 0.12 ± 0.02 | −7.0 ± 2 | ||
| PC/3k | 98/2 | – | 129 ± 1 | 0.14 ± 0.01 | +23.0 ± 2 | – | – |
| PC/3k | 95/5 | – | 125 ± 1 | 0.16 ± 0.02 | +39.0 ± 3 | – | – |
| PC/3k | 90/10 | – | 128 ± 1 | 0.16 ± 0.01 | +46.0 ± 3 | – | – |
| PC/3l | 98/2 | – | 127 ± 0.5 | 0.11 ± 0.01 | +6.1 ± 0.1 | – | – |
| PC/3l | 98/2 | – | 128 ± 0.5 | 0.12 ± 0.01 | +6.1 ± 0.1 | – | – |
| PC/3l | 95/5 | – | 116 ± 0.2 | 0.07 ± 0.01 | +4.4 ± 0.1 | – | – |
| PC/3l ** | 95/5 | – | 117 ± 0.2 | 0.09 ± 0.01 | +4.4 ± 0.1 | – | – |
| PC/3l | 90/10 | – | 111 ± 0.5 | 0.08 ± 0.01 | +7.8 ± 0.2 | – | – |
| PC/3l ** | 90/10 | – | 112 ± 0.5 | 0.09 ± 0.01 | +7.2 ± 0.2 | – | – |
| PC | 100 | 0.1 | 121 ± 1 | 0.14 ± 0.01 | +20.5 ± 2 | 87 ± 1 | 1.74 |
| PC ** | 100 | 0.1 | 122 ± 1 | 0.14 ± 0.01 | +12.0 ± 0.2 | – | – |
| PC/3l | 98/2 | 0.1 | 113 ± 1 | 0.13 ± 0.01 | +21.0 ± 1 | 80 ± 1 | 1.6 |
| PC/3l ** | 98/2 | 0.1 | 113 ± 1 | 0.08 ± 0.01 | +21.0 ± 0.1 | – | – |
| PC/3l | 95/5 | 0.1 | 109 ± 1 | 0.16 ± 0.01 | +22.0 ± 1 | 93 ± 1 | 1.86 |
| PC/3l ** | 95/5 | 0.1 | 109 ± 1 | 0.13 ± 0.01 | +22.0 ± 0.5 | – | – |
| PC/3l | 90/10 | 0.1 | 110 ± 1 | 0.17 ± 0.01 | +26.0 ± 1 | 93 ± 1 | 1.86 |
| PC/3l ** | 90/10 | 0.1 | 550 ± 50 | 0.4 ± 0.05 | +4.0 ± 0.5 | – | – |
| Test Compound | Cancer Cell Lines | Normal Cell Lines | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HuTu 80 a | PC3 b | M-HeLa c | MCF-7 d | A549 e | T98G f | WI38 g IC50 | |||||||
| IC50 | SI | IC50 | SI | IC50 | SI | IC50 | SI | IC50 | SI | IC50 | SI | ||
| 3a | 10.1 ± 0.8 | 4 | 45.0 ± 3.6 | ns | 66.7 ± 5.5 | ns | 56.1 ± 4.5 | ns | 64.4 ± 4.9 | ns | 64.3 ± 5.3 | ns | 37.5 ± 3.1 |
| 3b | 3.0 ± 0.1 | 5 | 23.3 ± 1.9 | ns | 30.1 ± 2.4 | ns | 34.0 ± 2.6 | ns | >100 | ns | 56.0 ± 4.5 | ns | 13.8 ± 1.1 |
| 3c | 19.1 ± 1.6 | 2.5 | 53.5 ± 4.2 | ns | 90.6 ± 8.6 | ns | 56.5 ± 4.5 | ns | >100 | ns | 52.1 ± 4.1 | ns | 47.3 ± 3.8 |
| 3d | 9.4 ± 0.7 | 4 | 39.1 ± 3.1 | ns | 50.0 ± 4.3 | ns | 88.6 ± 7.1 | ns | 74.2 ± 5.6 | ns | 64.0 ± 4.8 | ns | 35.2 ± 2.3 |
| 3f | 23.6 ± 1.8 | 1.5 | 55.2 ± 4.5 | ns | 56.2 ± 4.7 | ns | 60.7 ± 4.9 | ns | 39.0 ± 3.0 | ns | 60.4 ± 4.4 | ns | 35.6 ± 2.6 |
| 3g | 18.4 ± 1.5 | 1.6 | 48.9 ± 3.9 | ns | 63.7 ± 5.2 | ns | 66.8 ± 5.3 | ns | 42.2 ± 3.2 | ns | 62.9 ± 4.6 | ns | 30.1 ± 2.3 |
| 3h | 28.9 ± 2.1 | ns | 48.7 ± 3.7 | ns | 100 ± 8.6 | ns | 64.4 ± 5.0 | ns | 34.3 ± 2.6 | ns | 63.4 ± 4.8 | ns | 43.7 ± 3.5 |
| 3i | 7.1 ± 0.06 | 5 | 71.8 ± 6.6 | ns | 60.8 ± 5.2 | ns | 70.5 ± 5.6 | ns | 41.0 ± 3.3 | ns | 50.4 ± 4.0 | ns | 34.2 ± 2.7 |
| 3j | 0.5 ± 0.03 | 6.2 | 0.6 ± 0.05 | 5.2 | 1.1 ± 0.09 | 3 | 2.9 ± 0.2 | 1.1 | 3.7 ± 0.3 | ns | 1.7 ± 0.1 | 2 | 3.1 ± 0.2 |
| 3k | 3.5 ± 0.3 | 5 | 25.0 ± 2.0 | ns | 40.3 ± 3.5 | ns | 30.5 ± 2.5 | ns | 54.7 ± 4.4 | ns | 55.8 ± 4.7 | ns | 16.2 ± 1.3 |
| PC/3k | 3.8 ± 0.4 | 7.5 | 27.0 ± 2.2 | 1.1 | 48.9 ± 38 | ns | 35.8 ± 2.8 | ns | 88.7 ± 7.3 | ns | >100 | ns | 28.4 ± 2.4 |
| 3l | 45.0 ± 3.5 | ns | 53.8 ± 4.4 | ns | 59.2 ± 5.1 | ns | 52.8 ± 4.2 | ns | 75.1 ± 5.7 | ns | 55.9 ± 5.0 | ns | 50.0 ± 3.9 |
| PC/3l | 38.6 ± 3.1 | ns | 56.2 ± 4.5 | ns | 21.3 ± 1.7 | ns | 78.1 ± 6.2 | ns | >100 | ns | 64.7 ± 4.9 | ns | 26.8 ± 2.2 |
| 4a | 26.7 ± 2.1 | 1.4 | 50.3 ± 4.0 | ns | 57.3 ± 4.7 | ns | 75.7 ± 5.9 | ns | 36.0 ± 2.8 | ns | 55.3 ± 4.4 | ns | 36.5 ± 2.8 |
| 4b | 3.4 ± 0.3 | 7 | 42.4 ± 3.3 | ns | 68.4 ± 5.9 | ns | 60.3 ± 4.7 | ns | 39.1 ± 3.1 | ns | 49.7 ± 4.0 | ns | 22.9 ± 1.9 |
| 4c | 0.3 ± 0.02 | 12.3 | 0.7 ± 0.06 | 5.3 | 1.1 ± 0.09 | 3.4 | 2.7 ± 0.2 | 1.4 | 2.6 ± 0.2 | 1.4 | 2.0 ± 0.1 | 1.8 | 3.7 ± 0.3 |
| 4i | 5.0 ± 0.4 | 7 | 44.5 ± 3.5 | ns | 87.2 ± 7.9 | ns | 68.0 ± 5.5 | ns | 61.8 ± 5.0 | ns | >100 | ns | 34.2 |
| Dox | 0.2 ± 0.01 | 4 | 1.4 ± 0.1 | ns | 2.1 ± 0.1 | ns | 0.4 ± 0.03 | 2 | 0.7 ± 0.5 | 1.1 | 2.0 ± 0.1 | ns | 0.8 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mironov, V.F.; Nemtarev, A.V.; Tsepaeva, O.V.; Dimukhametov, M.N.; Litvinov, I.A.; Voloshina, A.D.; Pashirova, T.N.; Titov, E.A.; Lyubina, A.P.; Amerhanova, S.K.; et al. Rational Design 2-Hydroxypropylphosphonium Salts as Cancer Cell Mitochondria-Targeted Vectors: Synthesis, Structure, and Biological Properties. Molecules 2021, 26, 6350. https://doi.org/10.3390/molecules26216350
Mironov VF, Nemtarev AV, Tsepaeva OV, Dimukhametov MN, Litvinov IA, Voloshina AD, Pashirova TN, Titov EA, Lyubina AP, Amerhanova SK, et al. Rational Design 2-Hydroxypropylphosphonium Salts as Cancer Cell Mitochondria-Targeted Vectors: Synthesis, Structure, and Biological Properties. Molecules. 2021; 26(21):6350. https://doi.org/10.3390/molecules26216350
Chicago/Turabian StyleMironov, Vladimir F., Andrey V. Nemtarev, Olga V. Tsepaeva, Mudaris N. Dimukhametov, Igor A. Litvinov, Alexandra D. Voloshina, Tatiana N. Pashirova, Eugenii A. Titov, Anna P. Lyubina, Syumbelya K. Amerhanova, and et al. 2021. "Rational Design 2-Hydroxypropylphosphonium Salts as Cancer Cell Mitochondria-Targeted Vectors: Synthesis, Structure, and Biological Properties" Molecules 26, no. 21: 6350. https://doi.org/10.3390/molecules26216350
APA StyleMironov, V. F., Nemtarev, A. V., Tsepaeva, O. V., Dimukhametov, M. N., Litvinov, I. A., Voloshina, A. D., Pashirova, T. N., Titov, E. A., Lyubina, A. P., Amerhanova, S. K., Gubaidullin, A. T., & Islamov, D. R. (2021). Rational Design 2-Hydroxypropylphosphonium Salts as Cancer Cell Mitochondria-Targeted Vectors: Synthesis, Structure, and Biological Properties. Molecules, 26(21), 6350. https://doi.org/10.3390/molecules26216350