
Specificity of Molecular Fragments Binding to S100B versus S100A1 as Identified by NMR and Site Identification by Ligand Competitive Saturation (SILCS)

 , , and
, , and
Abstract
1. Introduction
2. Results
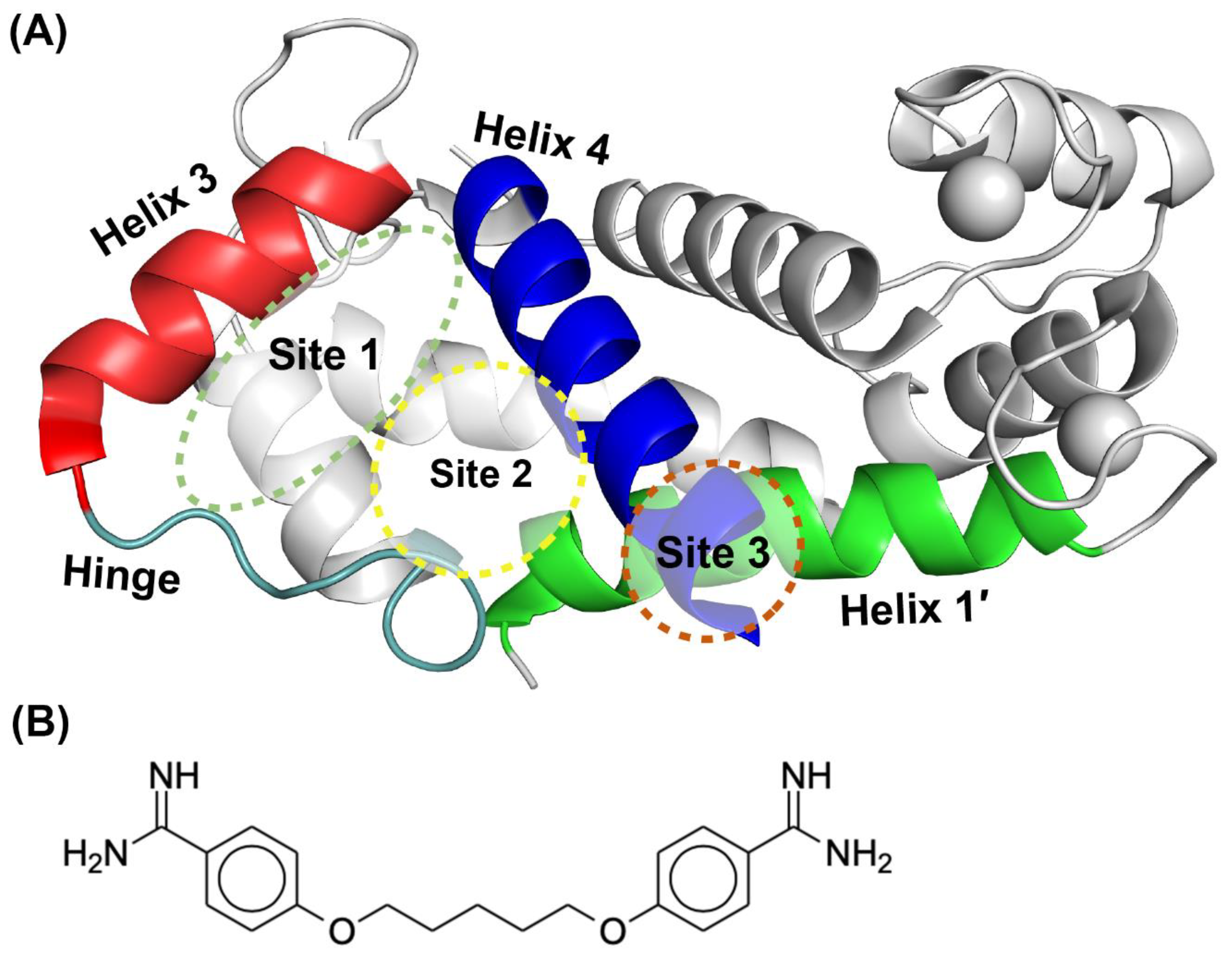
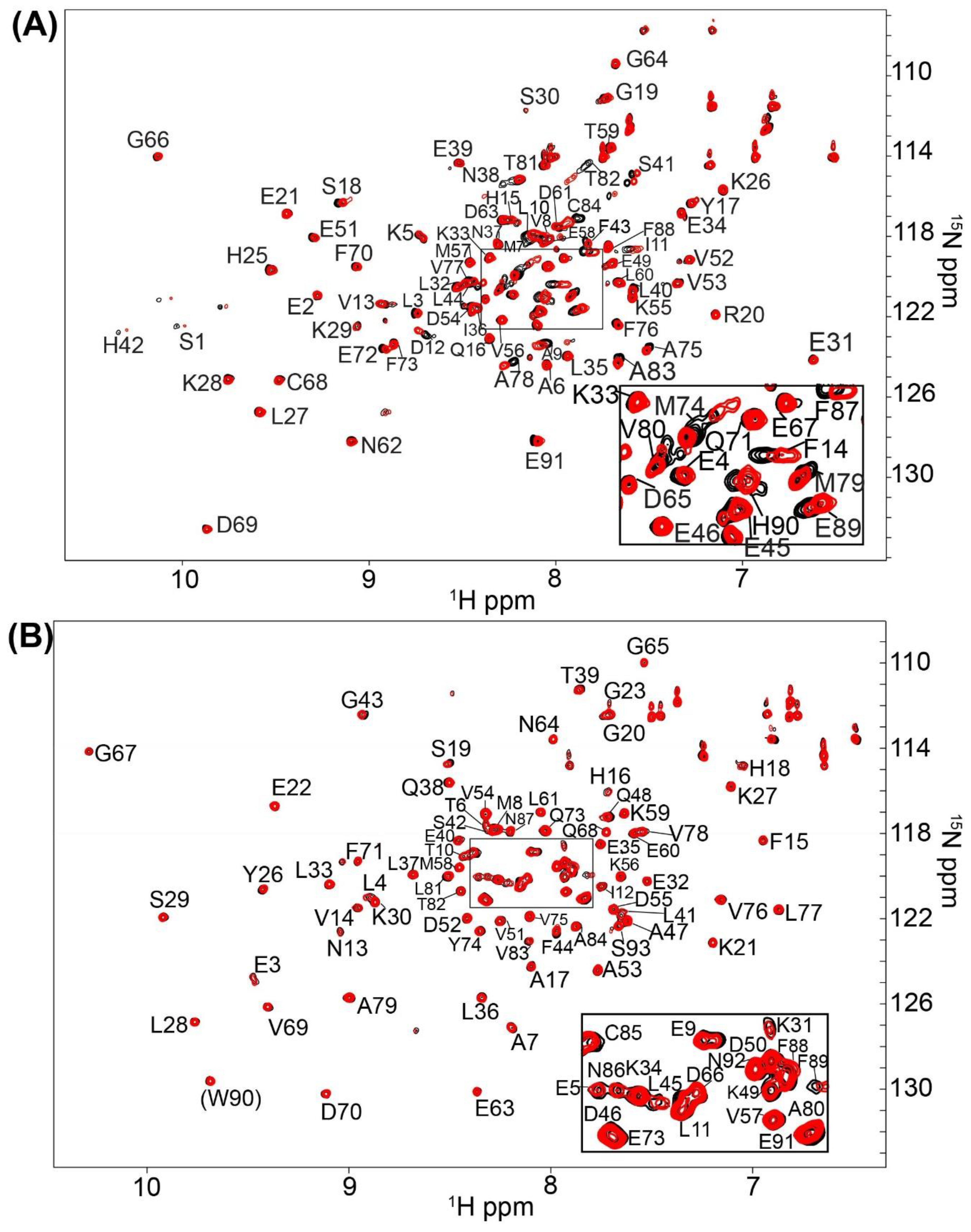
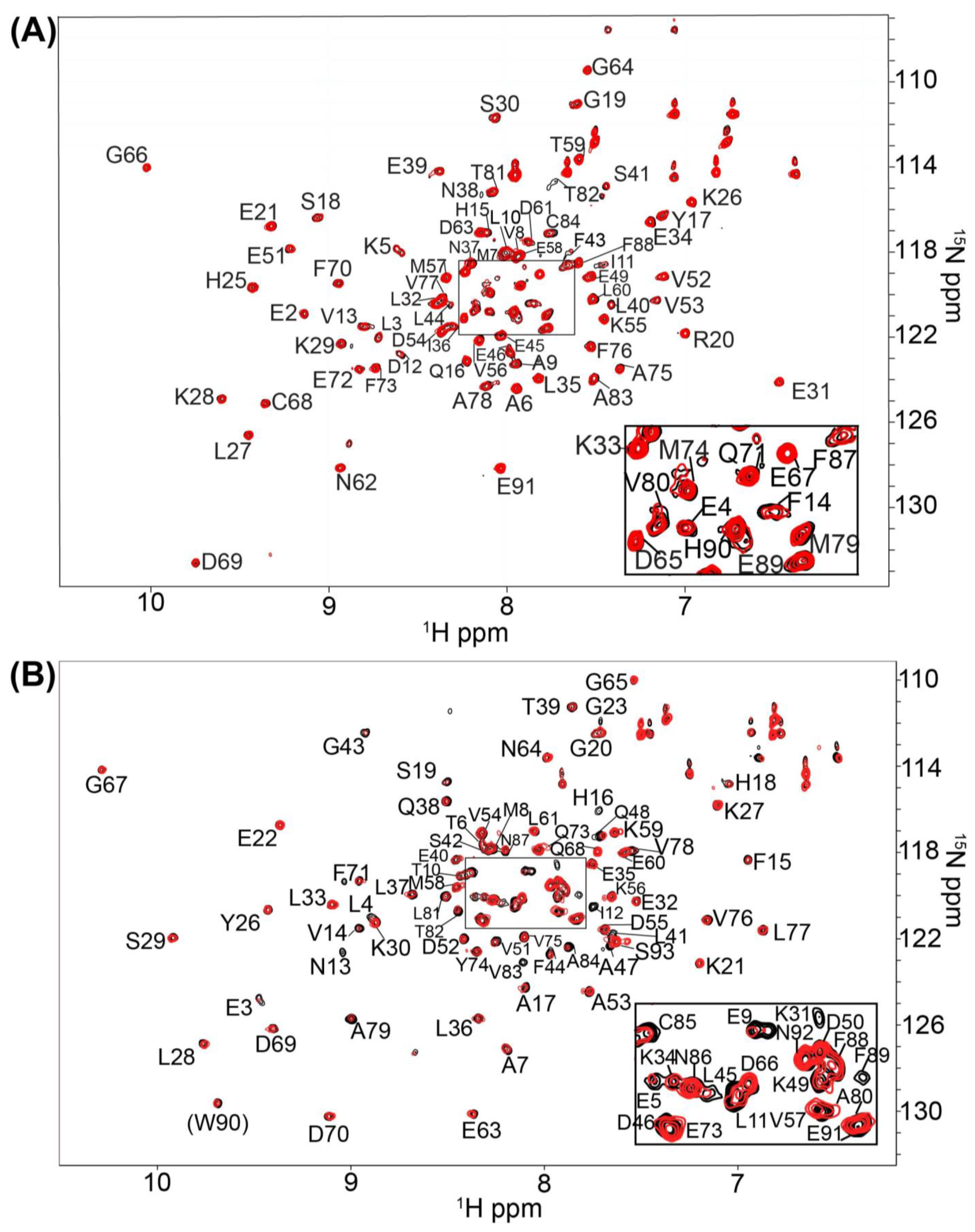
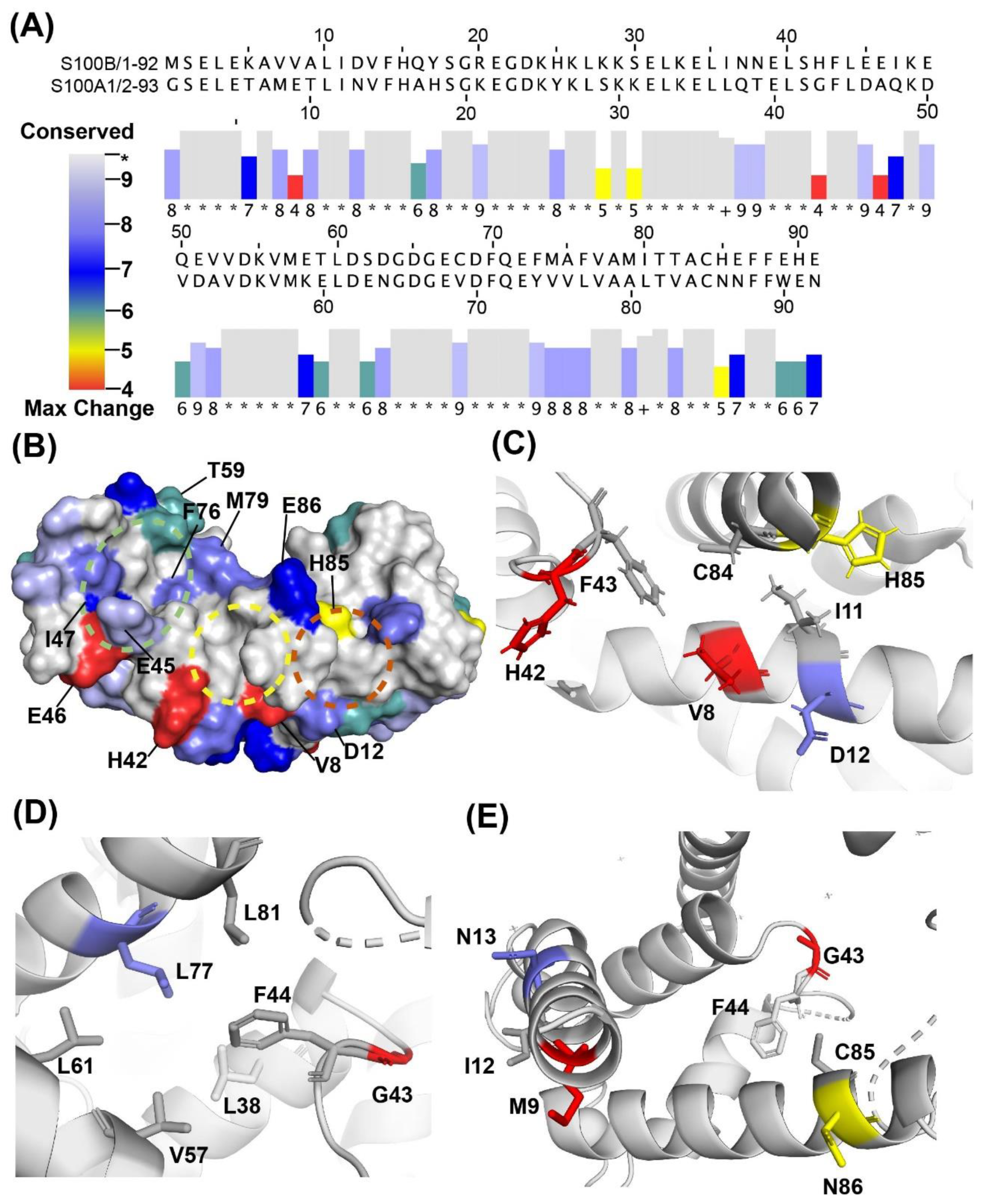
2.1. Fragment Compounds Specific for the Ca2+-Binding Proteins S100B or S100A1 were Identified Using NMR
2.2. Site Identification by Ligand Competitve Saturation (SILCS) Studies of S100A1 and S100B in the Ca2+-Bound States
2.3. Binding Sites on Ca2+-Bound S100A1 and S100B Predicted by SILCS-Hotspots
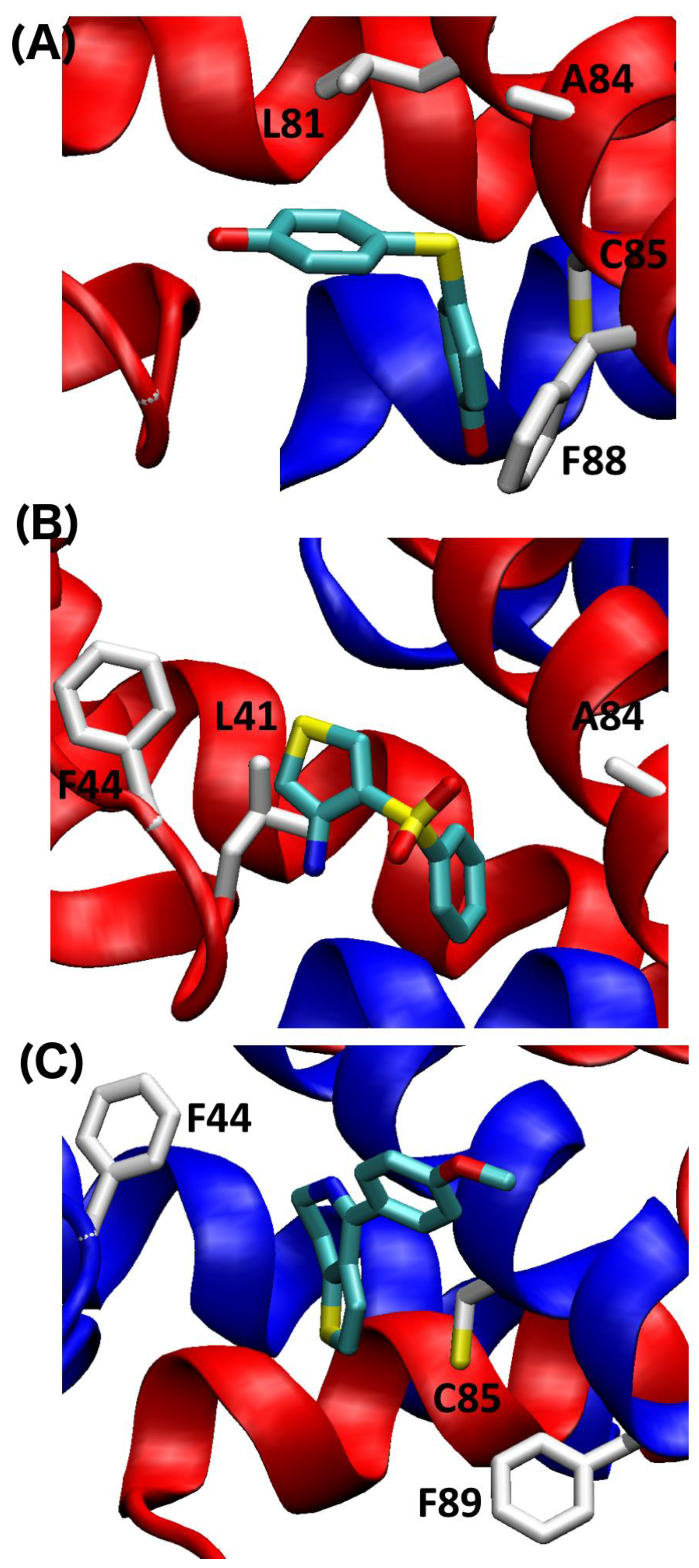
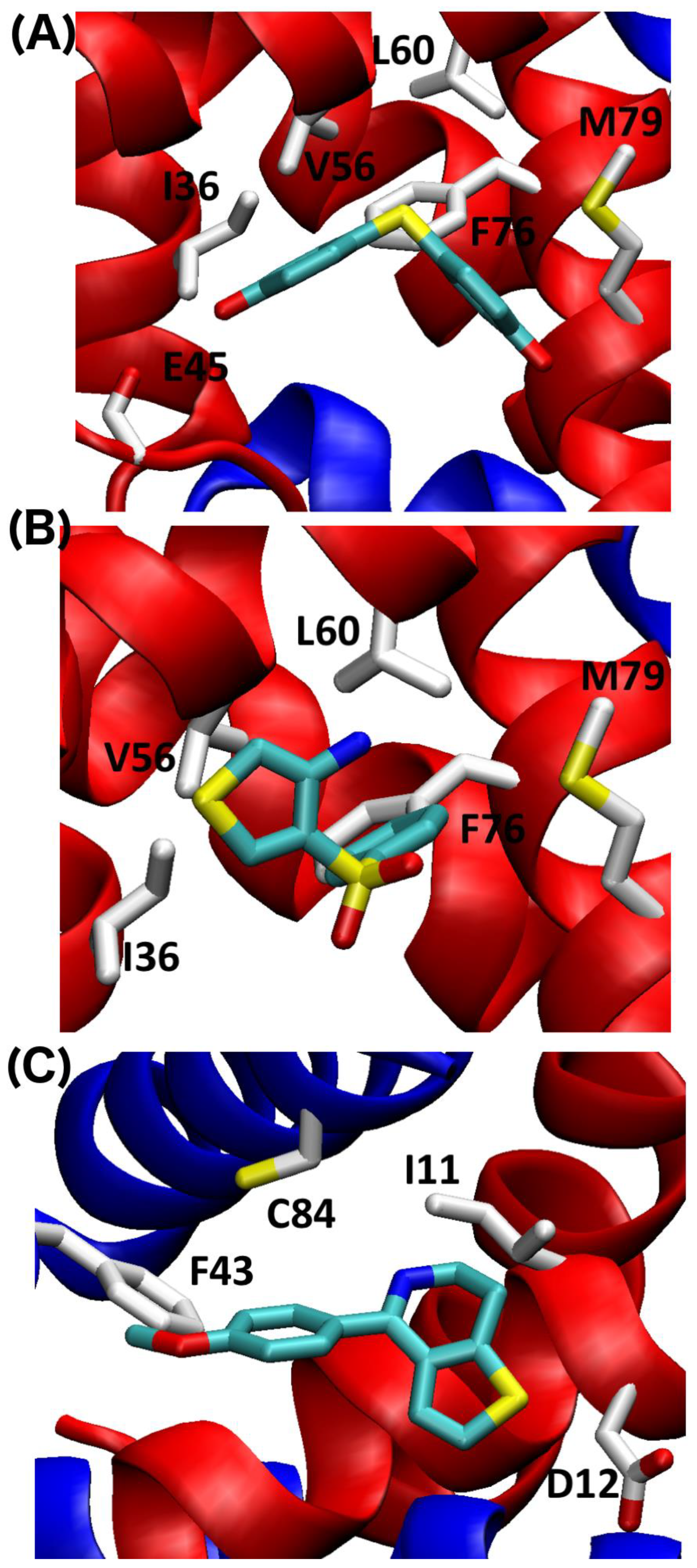
2.4. SILCS-MC Docking Poses and Lgfes Provide Insight into Fragment Specificity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.3. Screening and NMR Data Collection
4.4. Structure Models for S100B and S100A1
4.5. SILCS Simulation
4.6. SILCS-Hotspots Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Harpio, R.; Einarsson, R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin. Biochem. 2004, 37, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Engel, G.; Brenner, W.; Gläser, R.; Mönig, H.; Henze, E.; Christophers, E. S100B protein detection in serum is a significant prognostic factor in metastatic melanoma. Oncology 1999, 56, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.T.; Cannon, B.R.; Zimmer, D.B.; Weber, D.J. S100A1: Structure, Function, and Therapeutic Potential. Curr. Chem. Biol. 2009, 3, 138–145. [Google Scholar] [CrossRef]
- Wright, N.T.; Cannon, B.R.; Wilder, P.T.; Morgan, M.T.; Varney, K.M.; Zimmer, D.B.; Weber, D.J. Solution structure of S100A1 bound to the CapZ peptide (TRTK12). J. Mol. Biol. 2009, 386, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Inman, K.G.; Yang, R.; Rustandi, R.R.; Miller, K.E.; Baldisseri, D.M.; Weber, D.J. Solution NMR structure of S100B bound to the high-affinity target peptide TRTK-12. J. Mol. Biol. 2002, 324, 1003–1014. [Google Scholar] [CrossRef]
- Melville, Z.; Hernandez-Ochoa, E.O.; Pratt, S.J.P.; Liu, Y.; Pierce, A.D.; Wilder, P.T.; Adipietro, K.A.; Breysse, D.H.; Varney, K.M.; Schneider, M.F.; et al. The Activation of Protein Kinase A by the Calcium-Binding Protein S100A1 Is Independent of Cyclic AMP. Biochemistry 2017, 56, 2328–2337. [Google Scholar] [CrossRef]
- Prosser, B.L.; Wright, N.T.; Hernãndez-Ochoa, E.O.; Varney, K.M.; Liu, Y.; Olojo, R.O.; Zimmer, D.B.; Weber, D.J.; Schneider, M.F. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J. Biol. Chem. 2008, 283, 5046–5057. [Google Scholar] [CrossRef]
- Zimmer, D.B.; Chaplin, J.; Baldwin, A.; Rast, M. S100-mediated signal transduction in the nervous system and neurological diseases. Cell Mol. Biol. 2005, 51, 201–214. [Google Scholar]
- Zimmer, D.B.; Chessher, J.; Wilson, G.L.; Zimmer, W.E. S100A1 and S100B expression and target proteins in type I diabetes. Endocrinology 1997, 138, 5176–5183. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Barthelemy, A.; Feng, G.; Gentil-Perret, A.; Peoc’h, M.; Genin, C.; Tostain, J. S100A1: A powerful marker to differentiate chromophobe renal cell carcinoma from renal oncocytoma. Histopathology 2007, 50, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Pelc, P.; Vanmuylder, N.; Lefranc, F.; Heizmann, C.W.; Hassid, S.; Salmon, I.; Kiss, R.; Louryan, S.; Decaestecker, C. Differential expression of S100 calcium-binding proteins in epidermoid cysts, branchial cysts, craniopharyngiomas and cholesteatomas. Histopathology 2003, 42, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef] [PubMed]
- Drohat, A.C.; Amburgey, J.C.; Abildgaard, F.; Starich, M.R.; Baldisseri, D.; Weber, D.J. Solution structure of rat apo-S100B (beta beta) as determined by NMR spectroscopy. Biochemistry 1996, 35, 11577–11588. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, M.C.; Pierce, A.D.; Wilder, P.T.; Alasady, M.J.; Hartman, K.G.; Neau, D.B.; Foley, T.L.; Jadhav, A.; Maloney, D.J.; Simeonov, A.; et al. Covalent small molecule inhibitors of Ca(2+)-bound S100B. Biochemistry 2014, 53, 6628–6640. [Google Scholar] [CrossRef] [PubMed]
- Rustandi, R.R.; Baldisseri, D.M.; Weber, D.J. Structure of the negative regulatory domain of p53 bound to S100B (betabeta). Nat. Struct. Biol. 2000, 7, 570–574. [Google Scholar] [CrossRef]
- Rustandi, R.R.; Baldisseri, D.M.; Drohat, A.C.; Weber, D.J. Structural changes in the C-terminus of Ca2+-bound rat S100B (beta beta) upon binding to a peptide derived from the C-terminal regulatory domain of p53. Protein Sci. 1999, 8, 1743–1751. [Google Scholar] [CrossRef]
- Charpentier, T.H.; Wilder, P.T.; Liriano, M.A.; Varney, K.M.; Pozharski, E.; MacKerell, A.D.; Coop, A.; Toth, E.A.; Weber, D.J. Divalent metal ion complexes of S100B in the absence and presence of pentamidine. J. Mol. Biol. 2008, 382, 56–73. [Google Scholar] [CrossRef]
- Cavalier, M.C.; Melville, Z.; Aligholizadeh, E.; Raman, E.P.; Yu, W.; Fang, L.; Alasady, M.; Pierce, A.D.; Wilder, P.T.; MacKerell, A.D.; et al. Novel protein-inhibitor interactions in site 3 of Ca(2+)-bound S100B as discovered by X-ray crystallography. Acta Crystallogr. D Struct. Biol. 2016, 72, 753–760. [Google Scholar] [CrossRef]
- Hartman, K.G.; McKnight, L.E.; Liriano, M.A.; Weber, D.J. The evolution of S100B inhibitors for the treatment of malignant melanoma. Future Med. Chem. 2013, 5, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.; Chen, I.; Gitti, R.; Baldisseri, D.M.; Pan, Y.; Udan, R.; Carrier, F.; MacKerell, A.D.; Weber, D.J. Identification and characterization of small molecule inhibitors of the calcium-dependent S100B-p53 tumor suppressor interaction. J. Med. Chem. 2004, 47, 5085–5093. [Google Scholar] [CrossRef] [PubMed]
- Di Sante, G.; Amadio, S.; Sampaolese, B.; Clementi, M.E.; Valentini, M.; Volonté, C.; Casalbore, P.; Ria, F.; Michetti, F. The S100B Inhibitor Pentamidine Ameliorates Clinical Score and Neuropathology of Relapsing-Remitting Multiple Sclerosis Mouse Model. Cells 2020, 9, 748. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. BioMed Res. Int. 2015, 2015, 508342. [Google Scholar] [CrossRef]
- Seguella, L.; Rinaldi, F.; Marianecci, C.; Capuano, R.; Pesce, M.; Annunziata, G.; Casano, F.; Bassotti, G.; Sidoni, A.; Milone, M.; et al. Pentamidine niosomes thwart S100B effects in human colon carcinoma biopsies favouring wtp53 rescue. J. Cell Mol. Med. 2020, 24, 3053–3063. [Google Scholar] [CrossRef]
- Edwards, K.J.; Jenkins, T.C.; Neidle, S. Crystal structure of a pentamidine-oligonucleotide complex: Implications for DNA-binding properties. Biochemistry 1992, 31, 7104–7109. [Google Scholar] [CrossRef]
- Pathak, M.K.; Dhawan, D.; Lindner, D.J.; Borden, E.C.; Farver, C.; Yi, T. Pentamidine is an inhibitor of PRL phosphatases with anticancer activity. Mol. Cancer Ther. 2002, 1, 1255–1264. [Google Scholar]
- Kitamura, Y.; Arima, T.; Imaizumi, R.; Sato, T.; Nomura, Y. Inhibition of constitutive nitric oxide synthase in the brain by pentamidine, a calmodulin antagonist. Eur. J. Pharmacol. 1995, 289, 299–304. [Google Scholar] [CrossRef]
- Markowitz, J.; Mackerell, A.D.; Carrier, F.; Charpentier, T.H.; Weber, D.J. Design of Inhibitors for S100B. Curr. Top. Med. Chem. 2005, 5, 1093–1108. [Google Scholar] [CrossRef]
- Markowitz, J.; MacKerell, A.D.; Weber, D.J. A search for inhibitors of S100B, a member of the S100 family of calcium-binding proteins. Mini Rev. Med. Chem. 2007, 7, 609–616. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Bovey, F.; Mirau, P.; Gutowsky, H.S. Nuclear Magnetic Resonance Spectroscopy; Academic Press: Cambridge, MA, USA, 1988. [Google Scholar]
- Inman, K.G.; Baldisseri, D.M.; Miller, K.E.; Weber, D.J. Backbone dynamics of the calcium-signaling protein apo-S100B as determined by 15N NMR relaxation. Biochemistry 2001, 40, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Melville, Z.; Aligholizadeh, E.; McKnight, L.E.; Weber, D.J.; Pozharski, E. X-ray crystal structure of human calcium-bound S100A1. Acta Crystallogr. F Struct. Biol. Commun. 2017, 73, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, M.C.; Ansari, M.I.; Pierce, A.D.; Wilder, P.T.; McKnight, L.E.; Raman, E.P.; Neau, D.B.; Bezawada, P.; Alasady, M.J.; Charpentier, T.H.; et al. Small Molecule Inhibitors of Ca(2+)-S100B Reveal Two Protein Conformations. J. Med. Chem. 2016, 59, 592–608. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; MacKerell, A.D. Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput. Biol. 2009, 5, e1000435. [Google Scholar] [CrossRef]
- Lakkaraju, S.K.; Raman, E.P.; Yu, W.; MacKerell, A.D. Sampling of Organic Solutes in Aqueous and Heterogeneous Environments Using Oscillating Excess Chemical Potentials in Grand Canonical-like Monte Carlo-Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2281–2290. [Google Scholar] [CrossRef]
- Ustach, V.D.; Lakkaraju, S.K.; Jo, S.; Yu, W.; Jiang, W.; MacKerell, A.D. Optimization and Evaluation of Site-Identification by Ligand Competitive Saturation (SILCS) as a Tool for Target-Based Ligand Optimization. J. Chem. Inf. Model. 2019, 59, 3018–3035. [Google Scholar] [CrossRef]
- Levitt, M.; Lifson, S. Refinement of protein conformations using a macromolecular energy minimization procedure. J. Mol. Biol. 1969, 46, 269–279. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Physical. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef]
- Durell, S.R.; Brooks, B.R.; Ben-Naim, A. Solvent-Induced Forces between Two Hydrophilic Groups. J. Phys. Chem. 1994, 98, 2198–2202. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Raman, E.P.; Yu, W.; Lakkaraju, S.K.; MacKerell, A.D. Inclusion of multiple fragment types in the site identification by ligand competitive saturation (SILCS) approach. J. Chem. Inf. Model. 2013, 53, 3384–3398. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Jo, S.; Lakkaraju, S.K.; Lind, C.; Yu, W. Identification and characterization of fragment binding sites for allosteric ligand design using the site identification by ligand competitive saturation hotspots approach (SILCS-Hotspots). Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129519. [Google Scholar] [CrossRef]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 1993, 9, 745–756. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Specificity | S100A1 LGFE | S100B LGFE |
|---|---|---|---|
| BTB10184 | Both | −6.52 | −7.58 |
| KM01765 | S100A1 | −6.14 | −5.43 |
| SEW01483 | S100B | −5.97 | −6.13 |
| Compound | 2D Structure | Specificity | H-Bond Donor | H-Bond Acceptor | Polar Surface Area (Å2) A |
|---|---|---|---|---|---|
| SPB05355 |  | S100B | 2 | 2 | 45.8 |
| RJC03578 |  | S100B | 1 | 3 | 66.9 |
| SEW01483 |  | S100B | 0 | 3 | 49.8 |
| SEW01484 |  | S100B | 1 | 3 | 49.5 |
| GK00671 |  | S100A1 | 1 | 4 | 80.6 |
| KM01765 |  | S100A1 | 1 | 4 | 96.8 |
| BTB10184 |  | S100B/S100A1 | 2 | 3 | 65.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, B.D.; Yu, W.; Rodríguez, D.J.V.; Varney, K.M.; MacKerell, A.D., Jr.; Weber, D.J. Specificity of Molecular Fragments Binding to S100B versus S100A1 as Identified by NMR and Site Identification by Ligand Competitive Saturation (SILCS). Molecules 2021, 26, 381. https://doi.org/10.3390/molecules26020381
Young BD, Yu W, Rodríguez DJV, Varney KM, MacKerell AD Jr., Weber DJ. Specificity of Molecular Fragments Binding to S100B versus S100A1 as Identified by NMR and Site Identification by Ligand Competitive Saturation (SILCS). Molecules. 2021; 26(2):381. https://doi.org/10.3390/molecules26020381
Chicago/Turabian StyleYoung, Brianna D., Wenbo Yu, Darex J. Vera Rodríguez, Kristen M. Varney, Alexander D. MacKerell, Jr., and David J. Weber. 2021. "Specificity of Molecular Fragments Binding to S100B versus S100A1 as Identified by NMR and Site Identification by Ligand Competitive Saturation (SILCS)" Molecules 26, no. 2: 381. https://doi.org/10.3390/molecules26020381
APA StyleYoung, B. D., Yu, W., Rodríguez, D. J. V., Varney, K. M., MacKerell, A. D., Jr., & Weber, D. J. (2021). Specificity of Molecular Fragments Binding to S100B versus S100A1 as Identified by NMR and Site Identification by Ligand Competitive Saturation (SILCS). Molecules, 26(2), 381. https://doi.org/10.3390/molecules26020381