
Resources and Methods for Engineering “Designer” Glycan-Binding Proteins



Abstract
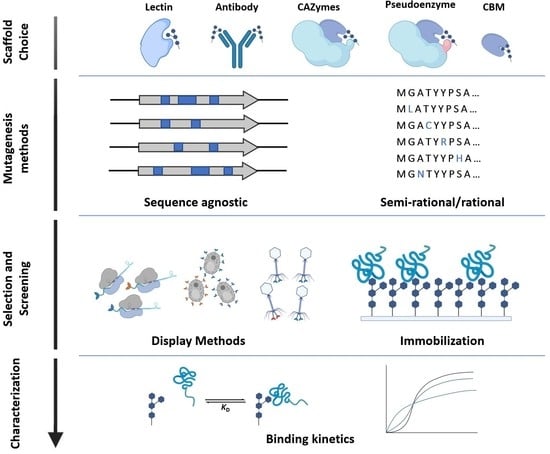
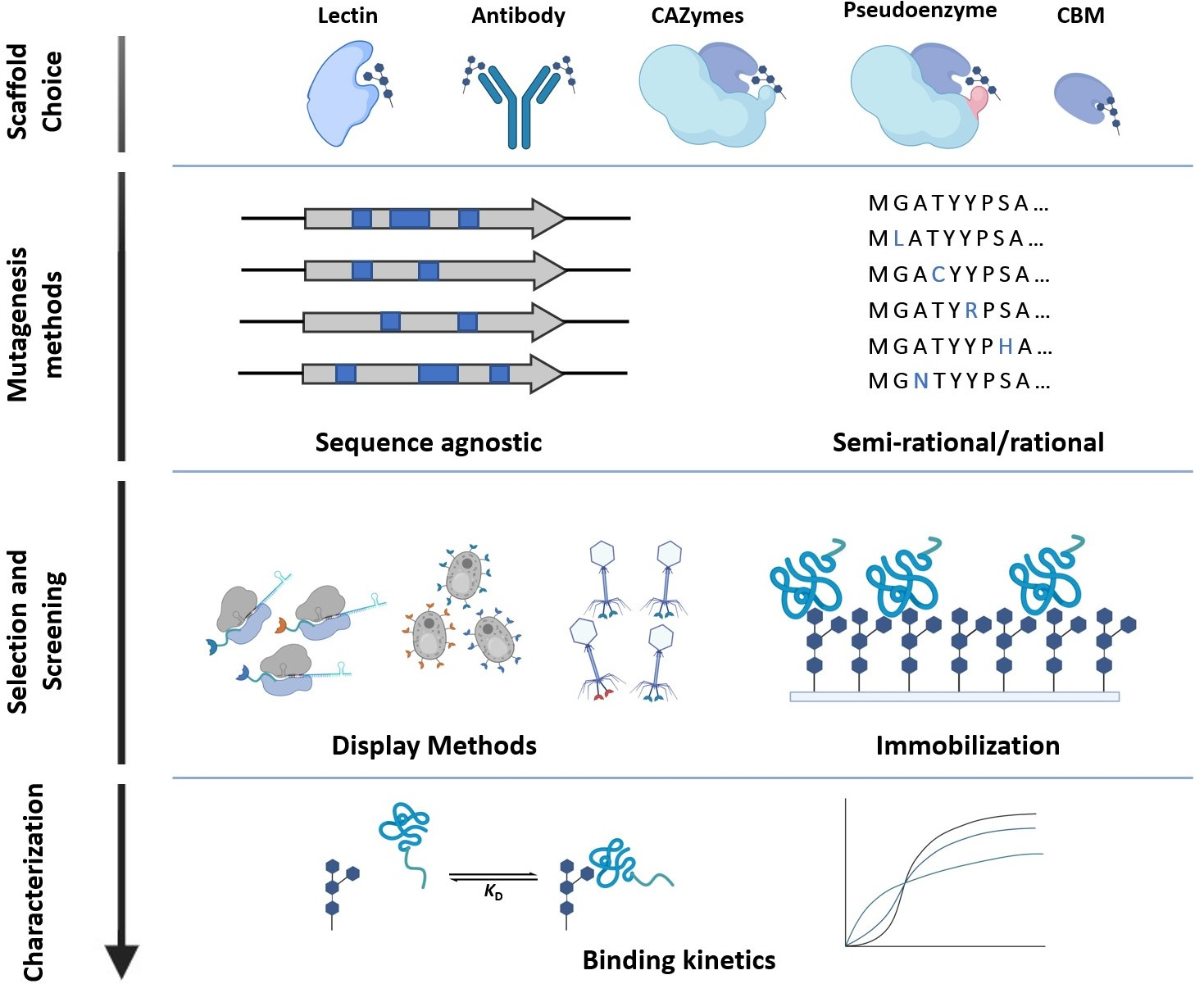
1. Introduction
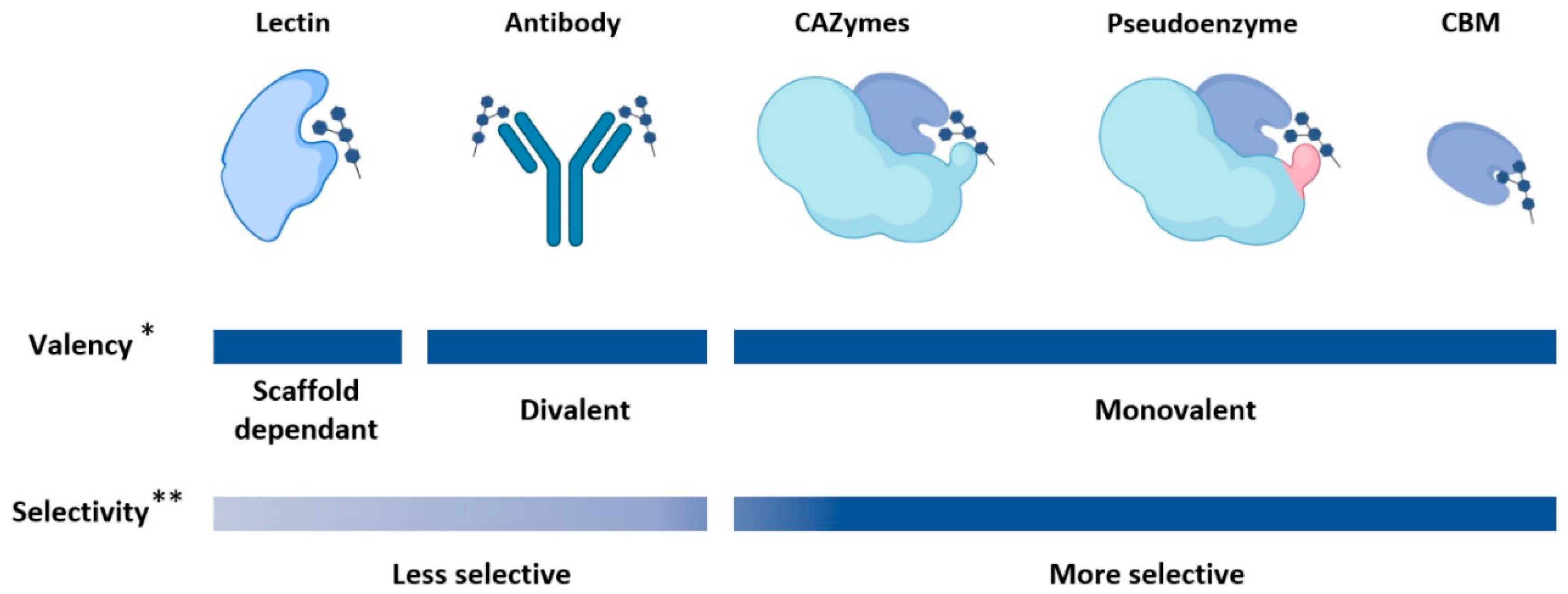
2. Glycan-Binding Protein Scaffolds
2.1. Lectins
2.2. Carbohydrate Binding Modules
2.3. Pseudoenzymes
2.4. Carbohydrate-Active Enzymes (CAZymes)
2.5. Antibody-Based Scaffolds
2.6. Summary on Available GBP Scaffolds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold Category | Scaffold Sub-Category | Description | Origin | Example Protein (PE) | PE Length | PE Ligand | PE Oligomeric State | PE Multivalency |
|---|---|---|---|---|---|---|---|---|
| Lectins | P-type | Lectin that binds to mannose 6-phosphate | Animal | Bovine CD-MPR binding domain [56] | 154 aa | Mannose 6-Phosphate | Dimer | Monovalent |
| I-type | Protein that is homologous to the immunoglobulin superfamily (IgSF) | Vertebrata | hCD22 domains 1-3 [57] | 324 aa | Sialoglycans | Monomer | Monovalent | |
| L-type | Proteins that are structurally similar to lectins found in the seeds of leguminous plants | All domains of life and viruses | Concanavalin A [58,59] | 237 aa | Trimannoside containing-oligosaccharides [59] | Oligomer | Divalent | |
| R-type | Proteins that are structurally similar to the carbohydrate recognition domain (CRD) in ricin | All domains of life and viruses | Ricin [60] | 267 aa | β1,4 galactose, N-acetylgalactosamine | Dimer | Divalent | |
| C-type | Ca2+ dependant proteins that share a primary and secondary homology in their CRDs | Animal | C-type domain of murine DCIR2 [61] | 129 aa | N-glycans | Monomer | Monovalent | |
| Galectin | Globular proteins that share primary structural homology in their CRDs | Animal | hGalectin-3 [62] | 146 aa | N-acetyllactosamine | Monomer | Monovalent | |
| Carbohydrate Binding Modules (CBMs) | Type A | Protein domain that binds to crystalline surfaces of cellulose and chitin | All domains of life and viruses | CBM from Cel7A [63] | 36 aa | Cellulose | Monomer | Monovalent |
| Type B | Protein domain that binds endo-glycan chains | All domains of life and viruses | CBM4-2 from xylanase [33] | 150 aa | Xylans, β-glucans | Monomer | Monovalent | |
| Type C | Protein domain that binds exo-type glycan chains | All domains of life and viruses | Cp-CBM 32 of hexosaminidase [64] | 150 aa | N-acetyllactosamine | Monomer | Monovalent | |
| Pseudoenzymes | Pseudoglycosidase | Carbohydrate binding proteins that evolved from glycosidases but are no longer catalytically active | Possibly all domains of life* | hYKL-39 [36] | 365 aa | Chitooligo-saccharides | Monomer | Monovalent |
| Pseudoesterase | Carbohydrate binding proteins that evolved from carbohydrate esterases but are no longer catalytically active | Possibly all domains of life * | C-terminal domain of PgaB [42] | 367 aa | Poly-1,6-N-acetylgluco-samine | Monomer | Monovalent | |
| Carbohydrate- Active Enzymes (CAZymes) | Glycoside hydrolase | Enzymes that cleave glycosidic linkages | All domains of life and viruses | Endo-NF (GH58) [44] | 811 aa | Polysialic acid | Trimer | Multivalent |
| Carbohydrate esterase | Enzymes that hydrolyze ester linkages of acyl groups attached to carbohydrates | All domains of life and viruses | CtCE2 [47] | 333 aa | Cellooligo-saccharides | Monomer | Monovalent | |
| Other CAZymes (glycosyltransferase, polysaccharide lyase, auxiliary activities) | Enzymes involved in the assembly, break-down, and modification of carbohydrates | All domains of life and viruses | − | − | − | − | − | |
| Antibodies | N/A | Naturally or synthetically produced proteins with an immunoglobulin, or derived from an immunoglobulin-like structure | Vertebrata | hu3S193 [65] | LC: 219 aa HC: 222 aa | LewisY | Dimeric | Divalent |
3. Mutagenesis Methods for Library Generation
3.1. Sequence Agnostic Random Mutagenesis
3.2. Rational and Semi-Rational Mutagenesis
| Method | Definition | Pros | Cons | References |
|---|---|---|---|---|
| PCR site-directed mutagenesis | Primers containing the desired mutation(s) are used to alter the original gene | Not limited by the availability of nearby restriction enzyme cut sites | Primer design can be complicated when introducing multiple mutations | [43,44,45,46,75,76,77,82] |
| Site-saturation mutagenesis | A set of codons is substituted with every amino acid using degenerate codons | Allows for the screening of ideal amino acids at different positions | Mutation bias from the degenerate codon | [78,80] |
| Motif- and domain-swapping | Uses a “cassette” DNA fragment containing the mutations, which replaces the unmutated segment in the original gene | High mutation efficiency | Is limited by the domains/motifs that are used. | [81,83] |
3.3. Computational Tools for Rational and Semi-Rational Mutagenesis
| Computational Approach | Definition | Examples | Utility in Protein Engineering | References |
|---|---|---|---|---|
| Homology model | Constructs an atomic resolution model of a protein based on available structural data of related homologous proteins. | Phyre2, ROBETTA, SWISS-MODEL | Predicted structures can be used in other computational methods, such as docking and molecular dynamics simulations. | [84] |
| Molecular Dynamics | Predicts the conformational energy landscape available to a protein based on the structure. | YASARA, Enlighten2, MOE, GROMACS | Dynamics can indicate how certain mutations can affect the behavior of protein such as folding, stability, ligand interaction and enzymatic activity. | [76,91,92] |
| Deep learning | Uses known protein sequences and properties to predict the properties of uncharacterized or novel proteins. | CPred, UniRep | Allows for the generation of more efficiency mutant libraries, by focusing on the most promising candidates. | [88,89,93] |
4. Library Selection and Screening Methods for GBPs
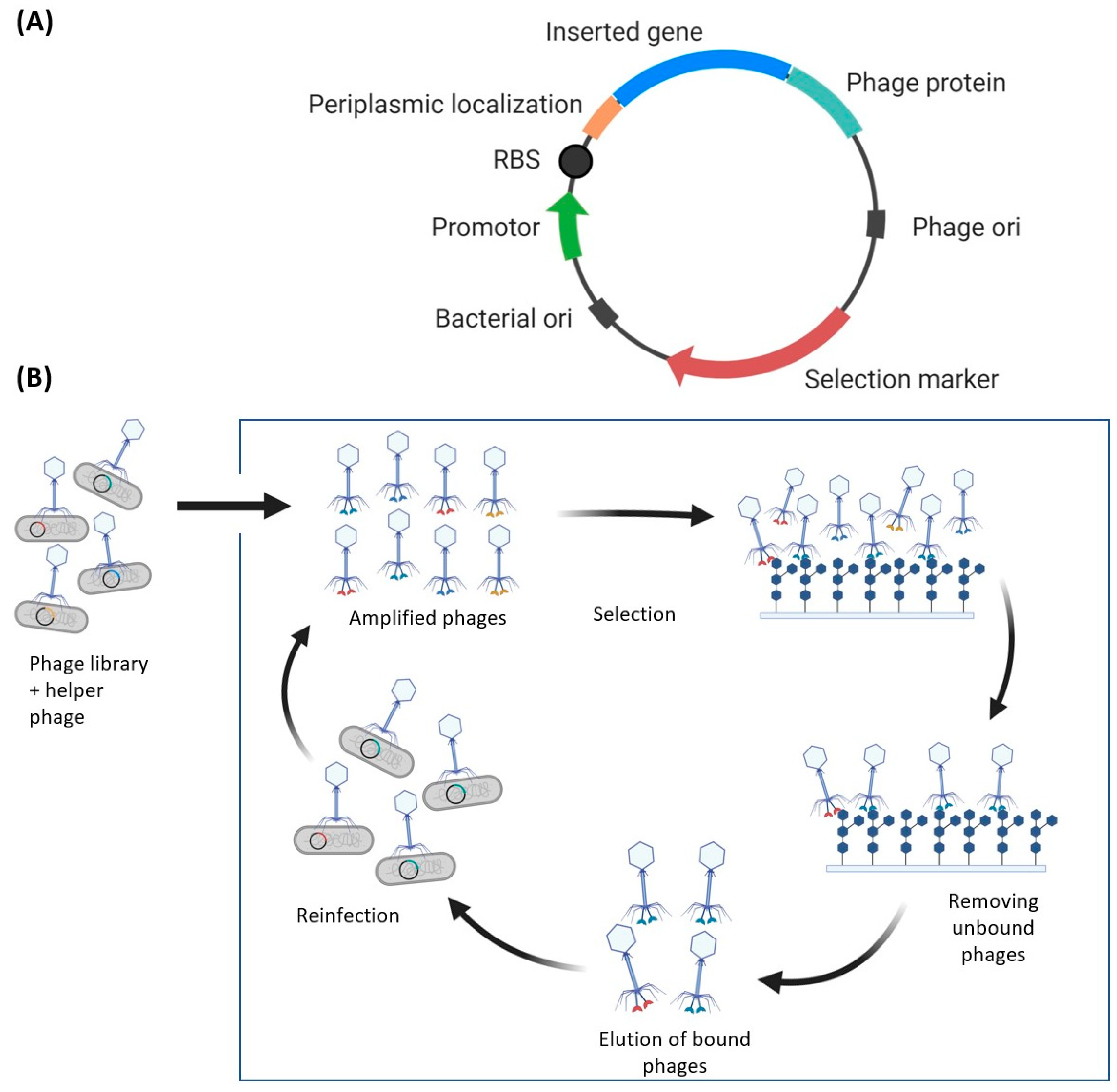
4.1. Phage Display
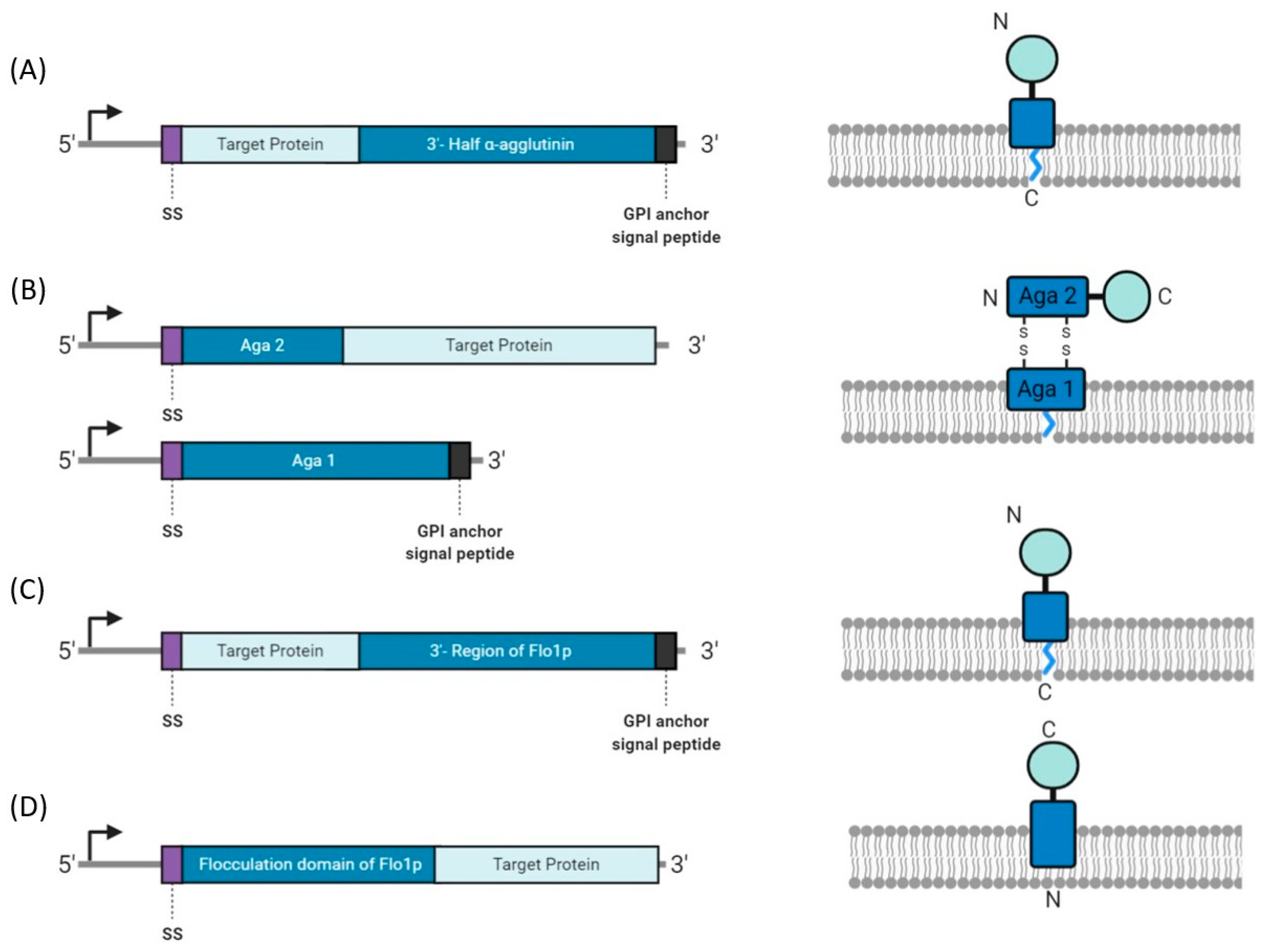
4.2. Yeast Display
4.3. Ribosome Display
4.4. mRNA Display
4.5. Glycan Immobilization Strategies for GBP Selection
5. Binding Characterization of GBPs
5.1. Frontal Affinity Chromatography
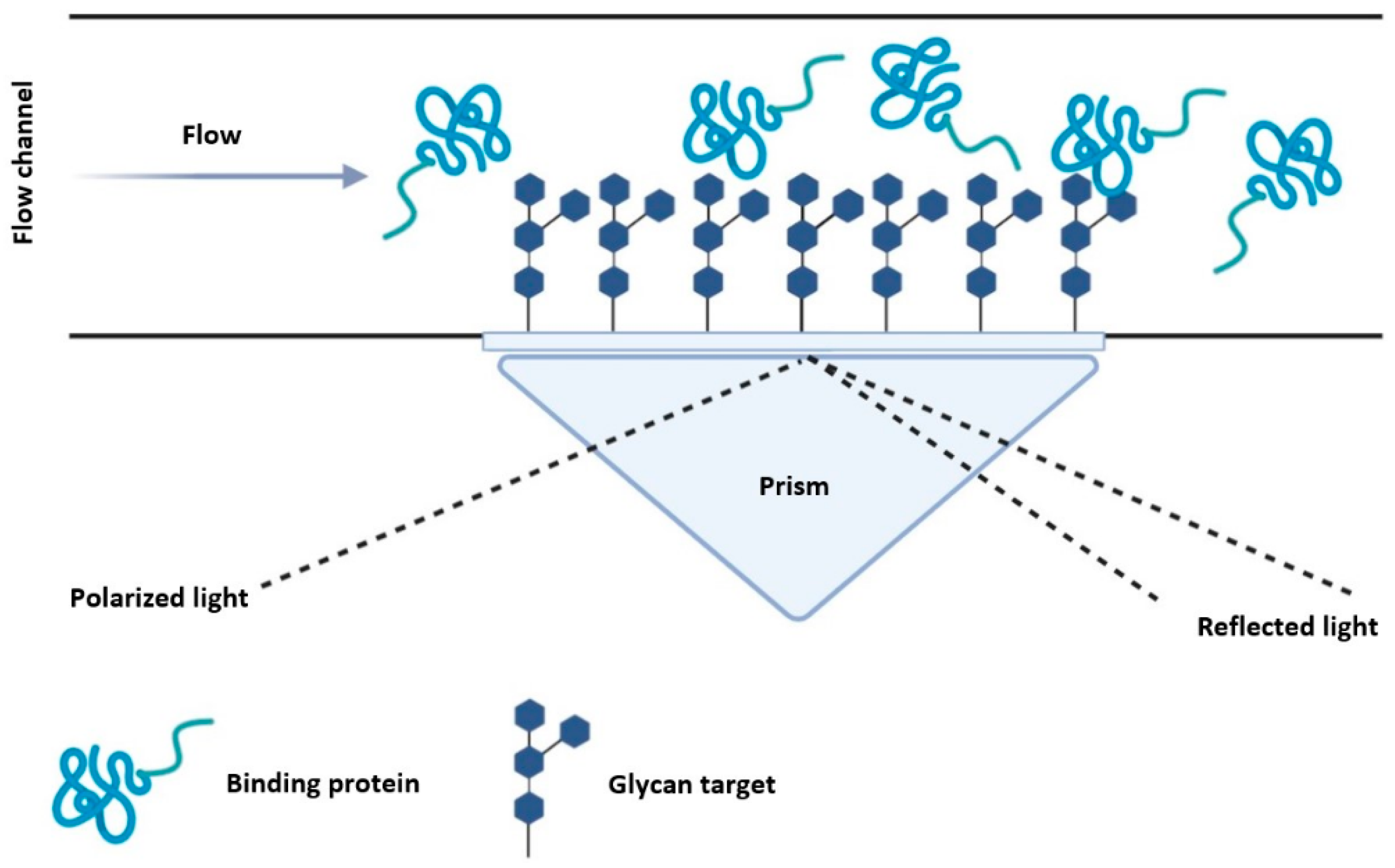
5.2. Surface Plasmon Resonance
5.3. Titration Calorimetry
6. Current Limitations in Lectin Engineering
6.1. Glycan Availability
6.2. Glycan Binding Protein Scaffolds
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Heimburg-Molinaro, J.; Lum, M.; Vijay, G.; Jain, M.; Almogren, A.; Rittenhouse-Olson, K. Cancer vaccines and carbohydrate epitopes. Vaccine 2011, 29, 8802–8826. [Google Scholar] [CrossRef] [PubMed]
- Polonskaya, Z.; Deng, S.; Sarkar, A.; Kain, L.; Comellas-Aragones, M.; McKay, C.S.; Kaczanowska, K.; Holt, M.; McBride, R.; Palomo, V.; et al. T cells control the generation of nanomolar-affinity anti-glycan antibodies. J. Clin. Investig. 2017, 127, 1491–1504. [Google Scholar] [CrossRef]
- Power, B.E.; Caine, J.M.; Burns, J.E.; Shapira, D.R.; Hattarki, M.K.; Tahtis, K.; Lee, F.-T.; Smyth, F.E.; Scott, A.M.; Kortt, A.A.; et al. Construction, expression and characterisation of a single-chain diabody derived from a humanised anti-LewisY cancer targeting antibody using a heat-inducible bacterial secretion vector. Cancer Immunol. Immunother. 2001, 50, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J. Targeting the glycans of gp120: A novel approach aimed at the Achilles heel of HIV. Lancet Infect. Dis. 2005, 5, 726–731. [Google Scholar] [CrossRef]
- Akkouh, O.; Ming, I.D.T.; Singh, S.S.; Yin, C.M.; Dan, X.; Chan, Y.S.; Pan, W.; Cheung, R.C.F. Lectins with Anti-HIV Activity: A Review. Molecules 2015, 20, 648–668. [Google Scholar] [CrossRef]
- Jandú, J.J.B.; Neto, R.N.M.; Zagmignan, A.; De Sousa, E.M.; Brelaz-De-Castro, M.C.A.; Correia, M.T.D.S.; Da Silva, L.C.N. Targeting the Immune System with Plant Lectins to Combat Microbial Infections. Front. Pharmacol. 2017, 8, 8. [Google Scholar] [CrossRef]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-Specific GlcNAcylation of Human Erythrocyte Proteins: Potential Biomarker(s) for Diabetes. Diabetes 2008, 58, 309–317. [Google Scholar] [CrossRef]
- Ercan, A.; Cui, J.; Hazen, M.M.; Batliwalla, F.; Royle, L.; Rudd, P.M.; Coblyn, J.S.; Shadick, N.A.; Weinblatt, M.; Gregersen, P.K.; et al. Hypogalactosylation of serum N-glycans fails to predict clinical response to methotrexate and TNF inhibition in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R43. [Google Scholar] [CrossRef]
- Kuno, A.; Miyoshi, E.; Nakayama, J.; Ohyama, C.; Togayachi, A. Glycan Biomarkers for Cancer and Various Disease. In Glycoscience: Basic Science to Applications: Insights from the Japan Consortium for Glycobiology and Glycotechnology (JCGG); Taniguchi, N., Endo, T., Hirabayashi, J., Nishihara, S., Kadomatsu, K., Akiyoshi, K., Aoki-Kinoshita, K.F., Eds.; Springer: Singapore, 2019; pp. 297–309. ISBN 9789811358562. [Google Scholar]
- Pearce, O.M. Cancer glycan epitopes: Biosynthesis, structure and function. Glycobiology 2018, 28, 670–696. [Google Scholar] [CrossRef] [PubMed]
- Velkov, V.V.; Medvinsky, A.B.; Sokolov, M.S.; Marchenko, A.I. Will transgenic plants adversely affect the environment? J. Biosci. 2005, 30, 515–548. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.C.M.; Carvalho, V.; Domingues, L.; Gama, M. Recombinant CBM-fusion technology—Applications overview. Biotechnol. Adv. 2015, 33, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Kalum, L.; Andersen, B.G. Enzymatic Treatment of Denim. U.S. Patent 6,146,428, 14 November 2000. Available online: https://patents.google.com/patent/US6146428A/en (accessed on 3 November 2020).
- Von der Osten, C.; Bjornvad, M.E.; Vind, J.; Rasmussen, M.D. Process and Composition for Desizing Cellulosic Fabric with an Enzyme Hybrid. U.S. Patent 6,017,751, 25 January 2000. [Google Scholar]
- Dang, K.; Zhang, W.; Jiang, S.; Lin, X.; Qian, A. Application of Lectin Microarrays for Biomarker Discovery. ChemistryOpen 2020, 9, 285–300. [Google Scholar] [CrossRef]
- Barondes, S.H. Bifunctional properties of lectins: Lectins redefined. Trends Biochem. Sci. 1988, 13, 480–482. [Google Scholar] [CrossRef]
- Kocourek, J.; Horejsi, V. Defining a lectin. Nat. Cell Biol. 1981, 290, 188. [Google Scholar] [CrossRef]
- Feng, Y.; Guo, Y.; Li, Y.; Tao, J.; Ding, L.; Wu, J.; Ju, H. Lectin-mediated in situ rolling circle amplification on exosomes for probing cancer-related glycan pattern. Anal. Chim. Acta 2018, 1039, 108–115. [Google Scholar] [CrossRef]
- Hashim, O.H.; Jayapalan, J.J.; Lee, C.-S. Lectins: An effective tool for screening of potential cancer biomarkers. PeerJ 2017, 5, e3784. [Google Scholar] [CrossRef]
- Bertozzi, C.R. Chemical Glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef]
- Liener, I.E.; Sharon, N.; Goldstein, I.J. The Lectins: Properties, Functions, and Applications in Biology and Medicine; Academic Press: Cambridge, MA, USA, 1986. [Google Scholar]
- Hu, D.; Tateno, H.; Hirabayashi, J. Lectin Engineering, a Molecular Evolutionary Approach to Expanding the Lectin Utilities. Molecules 2015, 20, 7637–7656. [Google Scholar] [CrossRef]
- Bonnardel, F.; Mariethoz, J.; Salentin, S.; Robin, X.; Schroeder, M.; Perez, S.; Lisacek, F.; Imberty, A. UniLectin3D, a database of carbohydrate binding proteins with curated information on 3D structures and interacting ligands. Nucleic Acids Res. 2019, 47, D1236–D1244. [Google Scholar] [CrossRef] [PubMed]
- Armenta, S.; Moreno-Mendieta, S.; Sánchez-Cuapio, Z.; Sanchez, S.; Rodríguez-Sanoja, R. Advances in molecular engineering of carbohydrate-binding modules. Proteins Struct. Funct. Bioinform. 2017, 85, 1602–1617. [Google Scholar] [CrossRef] [PubMed]
- Guillén, D.; Sanchez, S.; Rodríguez-Sanoja, R. Carbohydrate-binding domains: Multiplicity of biological roles. Appl. Microbiol. Biotechnol. 2009, 85, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Simpson, H.D.; Barras, F. Functional Analysis of the Carbohydrate-Binding Domains of Erwinia chrysanthemi Cel5 (Endoglucanase Z) and an Escherichia coli Putative Chitinase. J. Bacteriol. 1999, 181, 4611–4616. [Google Scholar] [CrossRef]
- Simpson, P.J.; Jamieson, S.J.; Hachem, M.A.; Karlsson, E.N.; Gilbert, H.J.; Holst, O.; Williamson, M.P. The Solution Structure of the CBM4-2 Carbohydrate Binding Module from a Thermostable Rhodothermus marinus Xylanase. Biochemistry 2002, 41, 5712–5719. [Google Scholar] [CrossRef]
- Boraston, A.B.; Creagh, A.L.; Alam, M.; Kormos, J.M.; Tomme, P.; Haynes, C.A.; Warren, R.A.J.; Kilburn, U.G. Binding specificity and thermodynamics of a family 9 carbohydrate-binding module from Thermotoga maritima xylanase 10A. Biochemistry 2001, 40, 6240–6247. [Google Scholar] [CrossRef]
- Furtado, G.P.; Lourenzoni, M.R.; Fuzo, C.A.; Fonseca-Maldonado, R.; Guazzaroni, M.-E.; Ribeiro, L.F.; Ward, R.J. Engineering the affinity of a family 11 carbohydrate binding module to improve binding of branched over unbranched polysaccharides. Int. J. Biol. Macromol. 2018, 120, 2509–2516. [Google Scholar] [CrossRef]
- Gunnarsson, L.C.; Karlsson, E.N.; Albrekt, A.; Andersson, M.; Holst, O.; Ohlin, M. A carbohydrate binding module as a diversity-carrying scaffold. Protein Eng. Des. Sel. 2004, 17, 213–221. [Google Scholar] [CrossRef]
- Sakata, T.; Takakura, J.; Miyakubo, H.; Osada, Y.; Wada, R.; Takahashi, H.; Yatsunami, R.; Fukui, T.; Nakamura, S. Improvement of binding activity of xylan-binding domain by amino acid substitution. Nucleic Acids Symp. Ser. 2006, 50, 253–254. [Google Scholar] [CrossRef]
- Eyers, P.A.; Murphy, J.M. The evolving world of pseudoenzymes: Proteins, prejudice and zombies. BMC Biol. 2016, 14, 1–6. [Google Scholar] [CrossRef]
- Schimpl, M.; Rush, C.L.; Betou, M.; Eggleston, I.M.; Recklies, A.D.; Van Aalten, D.M. Human YKL-39 is a pseudo-chitinase with retained chitooligosaccharide-binding properties. Biochem. J. 2012, 446, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Da Silva, C.A.; Cruz, C.S.D.; Ahangari, F.; Ma, B.; Kang, M.-J.; He, C.-H.; Takyar, S.; Elias, J.A. Role of Chitin and Chitinase/Chitinase-Like Proteins in Inflammation, Tissue Remodeling, and Injury. Annu. Rev. Physiol. 2011, 73, 479–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, J.; Mohanty, J.; Sousa, L.P.; Tome, F.; Pardon, E.; Steyaert, J.; Lemmon, M.A.; Lax, I.; Schlessinger, J. Structures of β-klotho reveal a ‘zip code’-like mechanism for endocrine FGF signalling. Nat. Cell Biol. 2018, 553, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.; Wong, W.; Thompson, J.K.; Healer, J.; Goddard-Borger, E.D.; Lawrence, M.C.; Cowman, A.F. Structural basis for inhibition of erythrocyte invasion by antibodies to Plasmodium falciparum protein CyRPA. eLife 2017, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Favuzza, P.; Guffart, E.; Tamborrini, M.; Scherer, B.; Dreyer, A.M.; Rufer, A.C.; Erny, J.; Hoernschemeyer, J.; Thoma, R.; Schmid, G.; et al. Structure of the malaria vaccine candidate antigen CyRPA and its complex with a parasite invasion inhibitory antibody. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Huang, R.; Menant, S.; Hong, C.; Sandow, J.J.; Birkinshaw, R.W.; Healer, J.; Hodder, A.N.; Kanjee, U.; Tonkin, C.J.; et al. Structure of Plasmodium falciparum Rh5–CyRPA–Ripr invasion complex. Nat. Cell Biol. 2018, 565, 118–121. [Google Scholar] [CrossRef]
- Little, D.J.; Li, G.; Ing, C.; DiFrancesco, B.R.; Bamford, N.C.; Robinson, H.; Nitz, M.; Pomès, R.; Howell, P.L. Modification and periplasmic translocation of the biofilm exopolysaccharide poly-1,6-N-acetyl-D-glucosamine. Proc. Natl. Acad. Sci. USA 2014, 111, 11013–11018. [Google Scholar] [CrossRef]
- Stummeyer, K.; Dickmanns, A.; Mühlenhoff, M.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of the polysialic acid–degrading endosialidase of bacteriophage K1F. Nat. Struct. Mol. Biol. 2004, 12, 90–96. [Google Scholar] [CrossRef]
- Jakobsson, E.; Jokilammi, A.; Aalto, J.; Ollikka, P.; Lehtonen, J.V.; Hirvonen, H.; Finne, J. Identification of amino acid residues at the active site of endosialidase that dissociate the polysialic acid binding and cleaving activities in Escherichia coli K1 bacteriophages. Biochem. J. 2007, 405, 465–472. [Google Scholar] [CrossRef]
- Yu, C.-C.; Hill, T.; Kwan, D.; Chen, H.-M.; Lin, C.-C.; Wakarchuk, W.; Withers, S.G. A plate-based high-throughput activity assay for polysialyltransferase from Neisseria meningitidis. Anal. Biochem. 2014, 444, 67–74. [Google Scholar] [CrossRef]
- Yu, C.-C.; Huang, L.-D.; Kwan, D.; Wakarchuk, W.; Withers, S.G.; Lin, C.-C. A glyco-gold nanoparticle based assay for α-2,8-polysialyltransferase from Neisseria meningitidis. Chem. Commun. 2013, 49, 10166–10168. [Google Scholar] [CrossRef] [PubMed]
- Montanier, C.; Money, V.A.; Pires, V.M.R.; Flint, J.E.; Pinheiro, B.A.; Goyal, A.; Prates, J.A.M.; Izumi, A.; Stålbrand, H.; Morland, C.; et al. The Active Site of a Carbohydrate Esterase Displays Divergent Catalytic and Noncatalytic Binding Functions. PLoS Biol. 2009, 7, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.J.; Yang, L. Glycan-Specific Analytical Tools. U.S. Patent 9,926,612, 27 March 2018. [Google Scholar]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L. Antibody fragments. mAbs 2010, 2, 77–83. [Google Scholar] [CrossRef]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. ScFv Antibody: Principles and Clinical Application. J. Immunol. Res. 2012. Available online: https://www.hindawi.com/journals/jir/2012/980250/ (accessed on 2 January 2021). [CrossRef]
- Holliger, P.; Prospero, T.; Winter, G. Diabodies: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef]
- Sterner, E.; Flanagan, N.; Gildersleeve, J.C. Perspectives on Anti-Glycan Antibodies Gleaned from Development of a Community Resource Database. ACS Chem. Biol. 2016, 11, 1773–1783. [Google Scholar] [CrossRef]
- Stewart, A.; Liu, Y.; Lai, J.R. A strategy for phage display selection of functional domain-exchanged immunoglobulin scaffolds with high affinity for glycan targets. J. Immunol. Methods 2012, 376, 150–155. [Google Scholar] [CrossRef]
- Olson, L.J.; Hindsgaul, O.; Dahms, N.M.; Kim, J.-J.P. Structural Insights into the Mechanism of pH-dependent Ligand Binding and Release by the Cation-dependent Mannose 6-Phosphate Receptor. J. Biol. Chem. 2008, 283, 10124–10134. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Sicard, T.; Cui, H.; Mazhab-Jafari, M.T.; Benlekbir, S.; Guarné, A.; Rubinstein, J.L.; Julien, J.-P. Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Parkin, S.; Rupp, B.; Hope, H. Atomic Resolution Structure of Concanavalin A at 120 K. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.K.; Kishore, N.; Brewer, C.F. Thermodynamics of Lectin-Carbohydrate Interactions. Titration Microcalorimetry Measurements of the Binding of N-Linked Carbohydrates and Ovalbumin to Concanavalin A. Biochemistry 1994, 33, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Rutenber, E.; Katzin, B.J.; Ernst, S.; Collins, E.J.; Mlsna, D.; Ready, M.P.; Robertus, J.D. Crystallographic refinement of ricin to 2.5 Å. Proteins Struct. Funct. Bioinform. 1991, 10, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Nagae, M.; Yamanaka, K.; Hanashima, S.; Ikeda, A.; Morita-Matsumoto, K.; Satoh, T.; Matsumoto, N.; Yamamoto, K.; Yamaguchi, Y. Recognition of Bisecting N-Acetylglucosamine. J. Biol. Chem. 2013, 288, 33598–33610. [Google Scholar] [CrossRef]
- Sörme, P.; Arnoux, P.; Kahl-Knutsson, B.; Leffler, H.; Rini, J.M.; Nilsson, U.J. Structural and Thermodynamic Studies on Cation−Π Interactions in Lectin−Ligand Complexes: High-Affinity Galectin-3 Inhibitors through Fine-Tuning of an Arginine−Arene Interaction. J. Am. Chem. Soc. 2005, 127, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Werneérus, H.; Lehtiö, J.; Teeri, T.; Nygren, P.-Å.; Ståahl, S. Generation of Metal-Binding Staphylococci through Surface Display of Combinatorially Engineered Cellulose-Binding Domains. Appl. Environ. Microbiol. 2001, 67, 4678–4684. [Google Scholar] [CrossRef]
- Ficko-Blean, E.; Boraston, A.B. The Interaction of a Carbohydrate-binding Module from aClostridium perfringens N-Acetyl-β-hexosaminidase with Its Carbohydrate Receptor. J. Biol. Chem. 2006, 281, 37748–37757. [Google Scholar] [CrossRef]
- Ramsland, P.A.; Farrugia, W.; Bradford, T.M.; Hogarth, P.M.; Scott, A.M. Structural Convergence of Antibody Binding of Carbohydrate Determinants in Lewis Y Tumor Antigens. J. Mol. Biol. 2004, 340, 809–818. [Google Scholar] [CrossRef]
- McCullum, E.O.; Williams, B.A.R.; Zhang, J.; Chaput, J.C. Random Mutagenesis by Error-Prone PCR. In In Vitro Mutagenesis Protocols, 3rd ed.; Braman, J., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 103–109. ISBN 978-1-60761-652-8. [Google Scholar]
- Coco, W.M.; Levinson, W.E.; Crist, M.J.; Hektor, H.J.; Darzins, A.; Pienkos, P.T.; Squires, C.H.; Monticello, D.J. DNA shuffling method for generating highly recombined genes and evolved enzymes. Nat. Biotechnol. 2001, 19, 354–359. [Google Scholar] [CrossRef]
- Muteeb, G.; Sen, R. Random Mutagenesis Using a Mutator Strain. In In Vitro Mutagenesis Protocols, 3rd ed.; Braman, J., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 411–419. ISBN 978-1-60761-652-8. [Google Scholar]
- Poluri, K.M.; Gulati, K. Protein Engineering Techniques: Gateways to Synthetic Protein Universe; Springer: Singapore, 2017; ISBN 978-981-10-2731-4. [Google Scholar]
- Yabe, R.; Suzuki, R.; Kuno, A.; Fujimoto, Z.; Jigami, Y.; Hirabayashi, J. Tailoring a Novel Sialic Acid-Binding Lectin from a Ricin-B Chain-like Galactose-Binding Protein by Natural Evolution-Mimicry. J. Biochem. 2006, 141, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, W.P.C. Rapid evolution of a protein in vitro by DNA shuffling. Nat. Cell Biol. 1994, 370, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Melzer, S.; Sonnendecker, C.; Föllner, C.; Zimmermann, W. Stepwise error-prone PCR and DNA shuffling changed the pH activity range and product specificity of the cyclodextrin glucanotransferase from an alkaliphilicBacillussp. FEBS Open Bio 2015, 5, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Ihssen, J.; Haas, J.; Kowarik, M.; Wiesli, L.; Wacker, M.; Schwede, T.; Thöny-Meyer, L. Increased efficiency of Campylobacter jejuni N -oligosaccharyltransferase PglB by structure-guided engineering. Open Biol. 2015, 5, 140227. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, L.M.; Marana, S.R. Single mutations outside the active site affect the substrate specificity in a β-glycosidase. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Pokorná, M.; Sabin, C.; Mitchell, E.; Imberty, A.; Wimmerová, M. Engineering of PA-IIL lectin from Pseudomonas aeruginosa—Unravelling the role of the specificity loop for sugar preference. BMC Struct. Biol. 2007, 7, 36. [Google Scholar] [CrossRef]
- Kunstmann, S.; Engström, O.; Wehle, M.; Widmalm, G.; Santer, M.; Barbirz, S. Increasing the Affinity of an O-Antigen Polysaccharide Binding Site in Shigella flexneri Bacteriophage Sf6 Tailspike Protein. Chem. A Eur. J. 2020, 26, 7263–7273. [Google Scholar] [CrossRef]
- Allhorn, M.; Olin, A.I.; Nimmerjahn, F.; Collin, M. Human IgG/FcγR Interactions Are Modulated by Streptococcal IgG Glycan Hydrolysis. PLoS ONE 2008, 3, e1413. [Google Scholar] [CrossRef]
- O’Donohue, M.J.; Kneale, G.G. Site-Directed and Site-Saturation Mutagenesis Using Oligonucleotide Primers. In DNA-Protein Interactions: Principles and Protocols; Kneale, G.G., Ed.; Humana Press: Totowa, NJ, USA, 1994; pp. 211–225. ISBN 978-1-59259-517-4. [Google Scholar]
- Hutchison, C.A.; Phillips, S.; Edgell, M.H.; Gillam, S.; Jahnke, P.; Smith, M. Mutagenesis at a specific position in a DNA sequence. J. Biol. Chem. 1978, 253, 6551–6560. [Google Scholar] [CrossRef]
- Imamura, K.; Takeuchi, H.; Yabe, R.; Tateno, H.; Hirabayashi, J. Engineering of the glycan-binding specificity of Agrocybe cylindracea galectin towards α(2,3)-linked sialic acid by saturation mutagenesis. J. Biochem. 2011, 150, 545–552. [Google Scholar] [CrossRef]
- Yamamoto, K.; Konami, Y.; Osawa, T. A chimeric lectin formed from Bauhinia purpurea lectin and Lens culinaris lectin recognizes a unique carbohydrate structure. J. Biochem. 2000, 127, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nandwani, N.; Surana, P.; Negi, H.; Mascarenhas, N.M.; Udgaonkar, J.B.; Das, R.; Gosavi, S. A five-residue motif for the design of domain swapping in proteins. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Lienemann, M.; Boer, H.; Paananen, A.; Cottaz, S.; Koivula, A. Toward understanding of carbohydrate binding and substrate specificity of a glycosyl hydrolase 18 family (GH-18) chitinase from Trichoderma harzianum. Glycobiology 2009, 19, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Pleiss, J. Identification of selectivity-determining residues in cytochrome P450 monooxygenases: A systematic analysis of the substrate recognition site 5. Proteins Struct. Funct. Bioinform. 2009, 74, 1028–1035. [Google Scholar] [CrossRef]
- Cheriyan, M.; Toone, E.J.; Fierke, C.A. Mutagenesis of the phosphate-binding pocket of KDPG aldolase enhances selectivity for hydrophobic substrates. Protein Sci. 2007, 16, 2368–2377. [Google Scholar] [CrossRef]
- Schneider, S.; Gutiérrez, M.; Sandalova, T.; Schneider, G.; Clapés, P.; Sprenger, G.A.; Samland, A.K. Redesigning the Active Site of Transaldolase TalB from Escherichia coli: New Variants with Improved Affinity towards Nonphosphorylated Substrates. ChemBioChem 2010, 11, 681–690. [Google Scholar] [CrossRef]
- Lo, W.-C.; Wang, L.-F.; Liu, Y.-Y.; Dai, T.; Hwang, J.-K.; Lyu, P.-C. CPred: A web server for predicting viable circular permutations in proteins. Nucleic Acids Res. 2012, 40, W232–W237. [Google Scholar] [CrossRef]
- Stephen, P.; Tseng, K.-L.; Liu, Y.-N.; Lyu, P.-C. Circular permutation of the starch-binding domain: Inversion of ligand selectivity with increased affinity. Chem. Commun. 2012, 48, 2612–2614. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Zinovjev, K.; Van der Kamp, M.W. Enlighten2: Molecular Dynamics Simulations of Protein-Ligand Systems Made Accessible. chemRxiv 2020. [Google Scholar] [CrossRef]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [PubMed]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.; Lai, J. Protein and Antibody Engineering by Phage Display. Methods Enzym. 2016, 580, 45–87. [Google Scholar] [CrossRef]
- Peltomaa, R.; Benito-Peña, E.; Barderas, R.; Bondi, M.C.C.M. Phage Display in the Quest for New Selective Recognition Elements for Biosensors. ACS Omega 2019, 4, 11569–11580. [Google Scholar] [CrossRef]
- Chasteen, L.; Ayriss, J.; Pavlik, P.; Bradbury, A. Eliminating helper phage from phage display. Nucleic Acids Res. 2006, 34, e145. [Google Scholar] [CrossRef]
- Yuasa, N.; Koyama, T.; Fujita-Yamaguchi, Y. Purification and refolding of anti-T-antigen single chain antibodies (scFvs) expressed in Escherichia coli as inclusion bodies. Biosci. Trends 2014, 8, 24–31. [Google Scholar] [CrossRef]
- Ng, S.; Tjhung, K.F.; Paschal, B.M.; Noren, C.J.; Derda, R. Chemical Posttranslational Modification of Phage-Displayed Peptides. In Peptide Libraries: Methods and Protocols; Derda, R., Ed.; Springer: New York, NY, USA, 2015; pp. 155–172. ISBN 978-1-4939-2020-4. [Google Scholar]
- Boder, E.T.; Wittrup, K.D. Yeast Surface Display for Directed Evolution of Protein Expression, Affinity, and Stability. Methods Enzymol. 2000, 328, 430–444. [Google Scholar] [CrossRef]
- Hong, X.; Ma, M.Z.; Gildersleeve, J.C.; Chowdhury, S.; Barchi, J.J.; Mariuzza, R.A.; Murphy, M.B.; Mao, L.; Pancer, Z. Sugar-Binding Proteins from Fish: Selection of High Affinity Lambodies That Recognize Biomedically Relevant Glycans. ACS Chem. Biol. 2012, 8, 152–160. [Google Scholar] [CrossRef]
- Lu, Z.; Kamat, K.; Johnson, B.P.; Yin, C.C.; Scholler, N.; Abbott, K.L. Generation of a Fully Human scFv that binds Tumor-Specific Glycoforms. Sci. Rep. 2019, 9, 5101. [Google Scholar] [CrossRef]
- Hanes, J.; Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef]
- Kurz, M.; Gu, K.; Lohse, P.A. Psoralen Photo-Crosslinked MRNA–Puromycin Conjugates: A Novel Template for the Rapid and Facile Preparation of MRNA–Protein Fusions. Nucleic Acids Res. 2000, 28, e83. [Google Scholar] [CrossRef]
- Nemoto, N.; Miyamoto-Sato, E.; Husimi, Y.; Yanagawa, H. In vitro virus: Bonding of mRNA bearing puromycin at the 3′-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997, 414, 405–408. [Google Scholar] [CrossRef]
- Newton, M.S.; Cabezas-Perusse, Y.; Tong, C.L.; Seelig, B. In Vitro Selection of Peptides and Proteins—Advantages of mRNA Display. ACS Synth. Biol. 2019, 9, 181–190. [Google Scholar] [CrossRef]
- Niwa, T.; Kanamori, T.; Ueda, T.; Taguchi, H. Global analysis of chaperone effects using a reconstituted cell-free translation system. Proc. Natl. Acad. Sci. USA 2012, 109, 8937–8942. [Google Scholar] [CrossRef]
- Lieberoth, A.; Splittstoesser, F.; Katagihallimath, N.; Jakovcevski, I.; Loers, G.; Ranscht, B.; Karagogeos, D.; Schachner, M.; Kleene, R. Lewisx and 2,3-Sialyl Glycans and Their Receptors TAG-1, Contactin, and L1 Mediate CD24-Dependent Neurite Outgrowth. J. Neurosci. 2009, 29, 6677–6690. [Google Scholar] [CrossRef]
- Sprung, R.; Nandi, A.; Chen, Y.; Kim, S.C.; Barma, D.; Falck, J.R.; Zhao, Y. Tagging-via-Substrate Strategy for Probing O-GlcNAc Modified Proteins. J. Proteome Res. 2005, 4, 950–957. [Google Scholar] [CrossRef]
- Mahal, L.K.; Yarema, K.J.; Bertozzi, C.R. Engineering Chemical Reactivity on Cell Surfaces Through Oligosaccharide Biosynthesis. Science 1997, 276, 1125–1128. [Google Scholar] [CrossRef]
- Luchansky, S.J.; Argade, S.; Hayes, B.K.; Bertozzi, C.R. Metabolic Functionalization of Recombinant Glycoproteins. Biochemistry 2004, 43, 12358–12366. [Google Scholar] [CrossRef]
- Bryan, M.C.; Lee, L.V.; Wong, C.-H. High-throughput identification of fucosyltransferase inhibitors using carbohydrate microarrays. Bioorg. Med. Chem. Lett. 2004, 14, 3185–3188. [Google Scholar] [CrossRef]
- Winzor, D.J. Determination of binding constants by affinity chromatography. J. Chromatogr. A 2004, 1037, 351–367. [Google Scholar] [CrossRef]
- Kasai, K. Frontal Affinity Chromatography: A Unique Research Tool for Biospecific Interaction That Promotes Glycobiology. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Schasfoort, R.B.M. Chapter 1: Introduction to Surface Plasmon Resonance. In Handbook of Surface Plasmon Resonance; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–26. [Google Scholar]
- Gutiérrez-Gallego, R.; Haseley, S.R.; Van Miegem, V.F.L.; Vliegenthart, J.F.; Kamerling, J.P. Identification of carbohydrates binding to lectins by using surface plasmon resonance in combination with HPLC profiling. Glycobiology 2004, 14, 373–386. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bellapadrona, G.; Tesler, A.B.; Grünstein, D.; Hossain, L.H.; Kikkeri, R.; Gilmore, K.; Vaskevich, A.; Rubinstein, I. Optimization of Localized Surface Plasmon Resonance Transducers for Studying Carbohydrate–Protein Interactions. Anal. Chem. 2011, 84, 232–240. [Google Scholar] [CrossRef]
- Huang, C.-F.; Yao, G.-H.; Liang, R.-P.; Qiu, J.-D. Graphene oxide and dextran capped gold nanoparticles based surface plasmon resonance sensor for sensitive detection of concanavalin A. Biosens. Bioelectron. 2013, 50, 305–310. [Google Scholar] [CrossRef]
- Liu, L.; Prudden, A.R.; Capicciotti, C.J.; Bosman, G.P.; Yang, J.-Y.; Chapla, D.G.; Moremen, K.W.; Boons, G.-J. Streamlining the chemoenzymatic synthesis of complex N-glycans by a stop and go strategy. Nat. Chem. 2019, 11, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Palcic, M.M. Glycosyltransferases as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 226–233. [Google Scholar] [CrossRef]
- Shilova, O.N.; Deyev, S.M. DARPins: Promising Scaffolds for Theranostics. Acta Nat. 2019, 11, 42–53. [Google Scholar] [CrossRef]
- Hofmeister, D.L.; Thoden, J.B.; Holden, H.M. Investigation of a sugar N-formyltransferase from the plant pathogen Pantoea ananatis. Protein Sci. 2019, 28, 707–716. [Google Scholar] [CrossRef]






| Method | Definition | Pros | Cons | References |
|---|---|---|---|---|
| Error prone PCR (epPCR) | epPCR relies on increased error rate of the polymerase | Efficient amplification of mutants | Library size is limited by cloning efficiency | [66] |
| DNA shuffling | DNA shuffling randomly recombines point mutations during PCR | Can be followed up with epPCR | Mutation efficiency is highly dependant on the shuffled library | [67] |
| In vivo mutagenesis | Mutations are introduced in bacteria using chemical or physical means, chemical mutagens or mutators strains | Wider variety of mutations without bias | More labor intensive, mutator strains get progressively sick from mutations | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warkentin, R.; Kwan, D.H. Resources and Methods for Engineering “Designer” Glycan-Binding Proteins. Molecules 2021, 26, 380. https://doi.org/10.3390/molecules26020380
Warkentin R, Kwan DH. Resources and Methods for Engineering “Designer” Glycan-Binding Proteins. Molecules. 2021; 26(2):380. https://doi.org/10.3390/molecules26020380
Chicago/Turabian StyleWarkentin, Ruben, and David H. Kwan. 2021. "Resources and Methods for Engineering “Designer” Glycan-Binding Proteins" Molecules 26, no. 2: 380. https://doi.org/10.3390/molecules26020380
APA StyleWarkentin, R., & Kwan, D. H. (2021). Resources and Methods for Engineering “Designer” Glycan-Binding Proteins. Molecules, 26(2), 380. https://doi.org/10.3390/molecules26020380