
Melatonin Induces Autophagy via Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Pathway in Colorectal Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
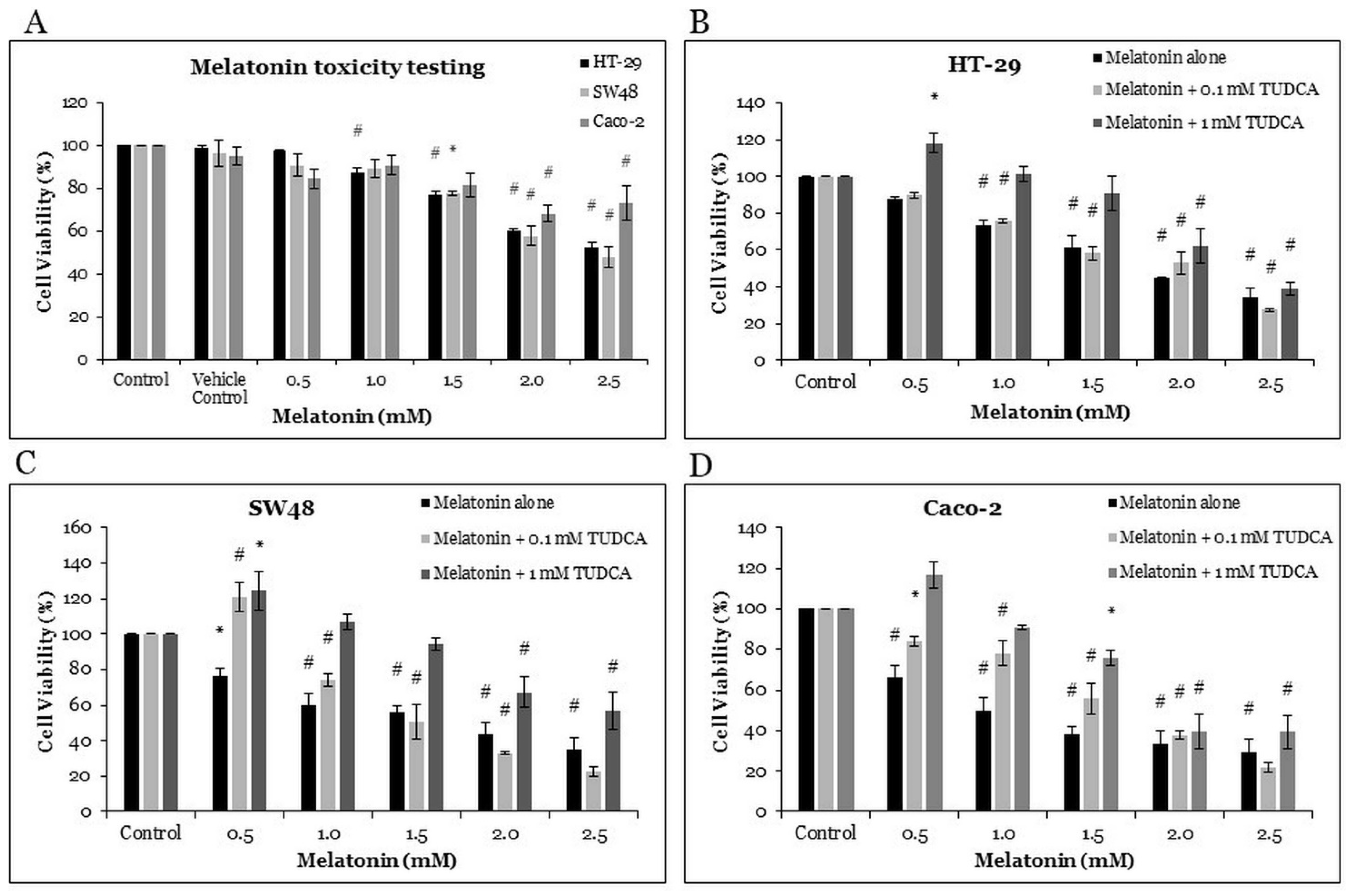
2.1. The Effects of Melatonin and Tauroursodeoxycholic Acid (TUDCA) on Cell Viability of CRC Cells
2.2. Melatonin Induces Oxidative Stress in HT-29 Cells
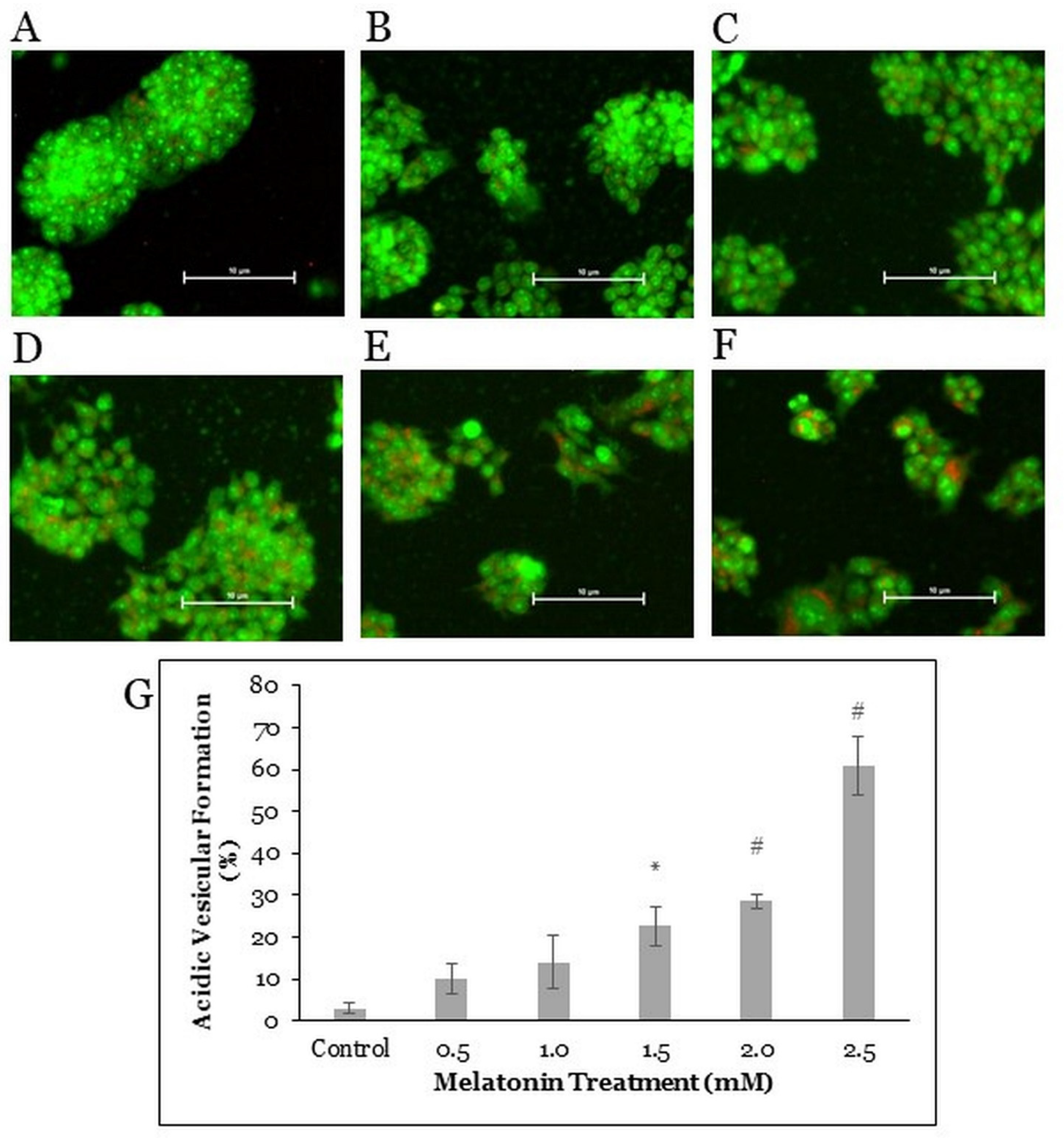
2.3. Melatonin Treatment Induces Autophagic Vacuoles Formation in HT-29 Cells
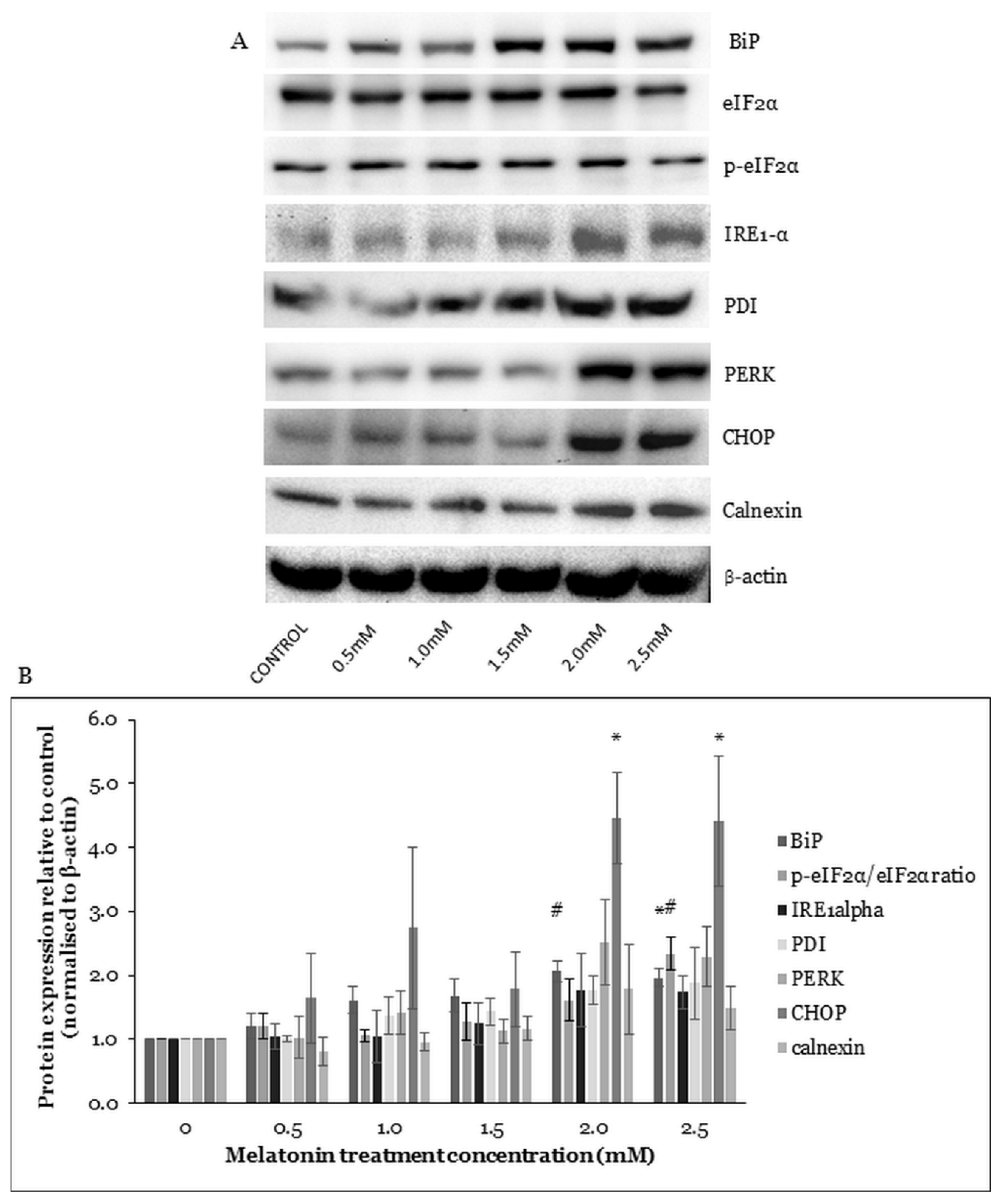
2.4. Melatonin Alters ER Stress-Related Protein Expressions in HT-29 Cells
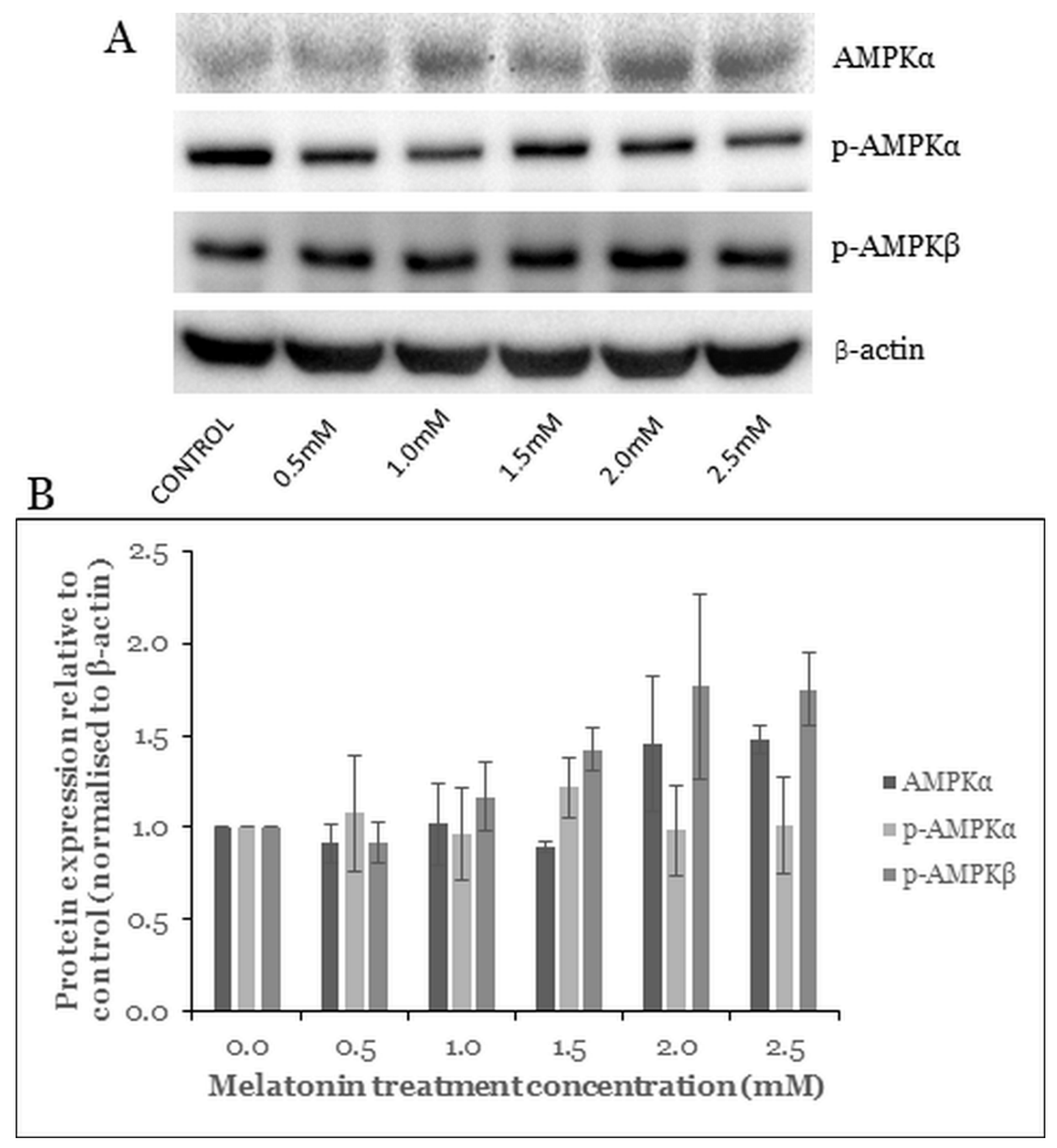
2.5. Melatonin Induces Changes in the AMPK Pathway
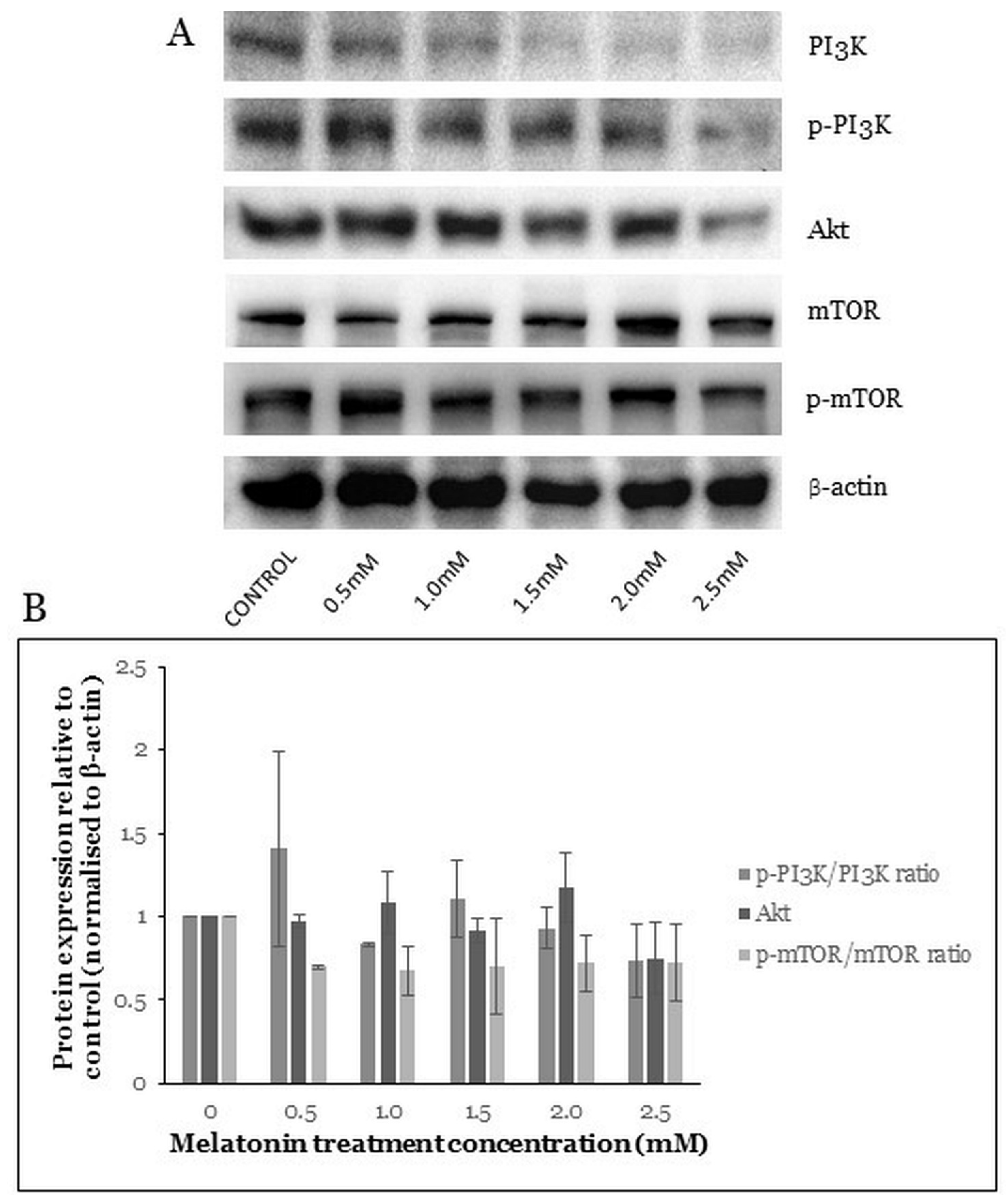
2.6. Melatonin Induces Changes in the PI3K/Akt/mTOR Pathway
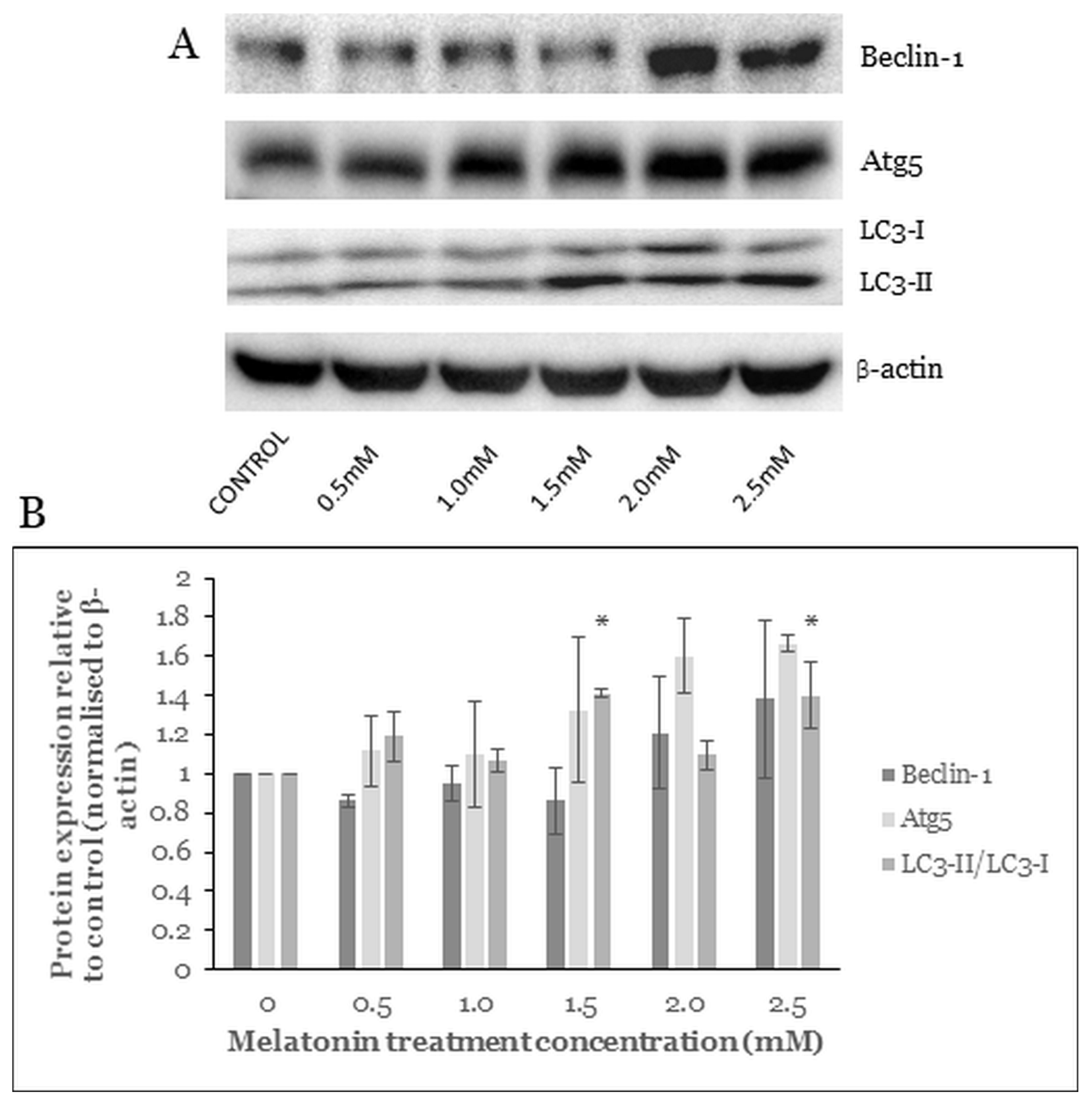
2.7. Melatonin Alters the Level of Autophagy Proteins
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. MTT Cytotoxicity Assay
4.4. Trypan Blue Cell Viability Assay
4.5. ROS Level Analysis
4.6. Fluorescence Staining
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Araghi, M.; Soerjomataram, I.; Jenkins, M.; Brierley, J.; Morris, E.; Bray, F.; Arnold, M. Global trends in colorectal cancer mortality: Projections to the year 2035. Int. J. Cancer 2019, 144, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D.; ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii1–iii9. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Lincz, L.; Adler, K.; Scorgie, F.; Ackland, S.P.; Sakoff, J.A. Predicting 5-fluorouracil toxicity in colorectal cancer patients from peripheral blood cell telomere length: A multivariate analysis. Br. J. Cancer 2012, 107, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, A.; Haas, M.; Viney, R.; Pearson, S.-A.; Haywood, P.; Brown, C.; Ward, R. Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study. PLoS ONE 2017, 12, e0184360. [Google Scholar] [CrossRef]
- Plaimee, P.; Weerapreeyakul, N.; Barusrux, S.; Johns, N.P. Melatonin potentiates cisplatin-induced apoptosis and cell cycle arrest in human lung adenocarcinoma cells. Cell Prolif. 2015, 48, 67–77. [Google Scholar] [CrossRef]
- Gao, Y.; Xiao, X.; Zhang, C.; Yu, W.; Guo, W.; Zhang, Z.; Li, Z.; Feng, X.; Hao, J.; Zhang, K.; et al. Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-κB/iNOS signaling pathways. J. Pineal Res. 2016, 62, e12380. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-X.; Chen, D.-L.; Wang, D.-S.; Chen, L.-Z.; Mo, H.-Y.; Sheng, H.; Bai, L.; Wu, Q.-N.; Yu, H.-E.; Xie, D.; et al. Melatonin enhances sensitivity to fluorouracil in oesophageal squamous cell carcinoma through inhibition of Erk and Akt pathway. Cell Death Dis. 2016, 7, e2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xiao, X.; Zhang, Y.; Shi, D.; Chen, W.; Fu, L.; Liu, L.; Xie, F.; Kang, T.; Huang, W.; et al. Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J. Pineal Res. 2012, 53, 77–90. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Ordoñez, R.; Benet, M.; Jover, R.; Palomo, A.G.; Mauriz, J.L.; González-Gallego, J. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 2013, 109, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Akbar, M.A.; Movassaghpour, A.A.; Ghanbari, H.; Kheirandish, M.; Maroufi, N.F.; Rahbarghazi, R.; Nouri, M.; Samadi, N. The potential therapeutic effect of melatonin on human ovarian cancer by inhibition of invasion and migration of cancer stem cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Kasi, R.; Yeo, P.L.; Yen, N.K.; Koh, R.Y.; Ponnudurai, G.; Tiong, Y.L.; Chye, S.M.; Ng, K.Y. Melatonin Induces Apoptosis and Inhibits the Proliferation of Cancer Cells via Reactive Oxygen Species-mediated MAPK and mTOR Pathways. Clin. Cancer Drugs 2020, 7, 44–56. [Google Scholar] [CrossRef]
- Tiong, Y.L.; Ng, K.Y.; Koh, R.Y.; Ponnudurai, G.; Chye, S.M. Melatonin Prevents Oxidative Stress-Induced Mitochondrial Dysfunction and Apoptosis in High Glucose-Treated Schwann Cells via Upregulation of Bcl2, NF-κB, mTOR, Wnt Signalling Pathways. Antioxidants 2019, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Fu, A.; Hoffman, A.E.; Zheng, T.; Zhu, Y. Melatonin enhances DNA repair capacity possibly by affecting genes involved in DNA damage responsive pathways. BMC Cell Biol. 2013, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Kontek, R.; Jakubczak, M.; Matlawska-Wasowska, K. The antioxidants, vitamin A and E but not vitamin C and melatonin enhance the proapoptotic effects of irinotecan in cancer cells in vitro. Toxicol. In Vitro 2014, 28, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef]
- Bejarano, I.; Espino, J.; Barriga, C.; Reiter, R.J.; Pariente, J.A.; Rodríguez, A.B. Pro-Oxidant Effect of Melatonin in Tumour Leucocytes: Relation with its Cytotoxic and Pro-Apoptotic Effects. Basic Clin. Pharmacol. Toxicol. 2010, 108, 14–20. [Google Scholar] [CrossRef]
- Burada, F. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Hou, Y.-F.; Melan, M.; Chen, X.; Stolz, D.B.; Shao, Z.-M.; Yin, X.-M. Differential Effects of Endoplasmic Reticulum Stress-induced Autophagy on Cell Survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Lepine, S.; Allegood, J.C.; Park, M.; Dent, P.W.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2010, 18, 350–361. [Google Scholar] [CrossRef]
- Basile, V.; Belluti, S.; Ferrari, E.; Gozzoli, C.; Ganassi, S.; Quaglino, D.; Saladini, M.; Imbriano, C. bis-Dehydroxy-Curcumin Triggers Mitochondrial-Associated Cell Death in Human Colon Cancer Cells through ER-Stress Induced Autophagy. PLoS ONE 2013, 8, e53664. [Google Scholar] [CrossRef]
- Feng, Q.; Wang, H.; Pang, J.; Ji, L.; Han, J.; Wang, Y.; Qi, X.; Liu, Z.; Lu, L. Prevention of Wogonin on Colorectal Cancer Tumorigenesis by Regulating p53 Nuclear Translocation. Front. Pharmacol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Dutta, D.; Chakraborty, B.; Sarkar, A.; Chowdhury, C.; Das, P. A potent betulinic acid analogue ascertains an antagonistic mechanism between autophagy and proteasomal degradation pathway in HT-29 cells. BMC Cancer 2016, 16, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelkerke, A.; Sweep, F.C.; Geurts-Moespot, A.; Bussink, J.; Span, P.N. Therapeutic targeting of autophagy in cancer. Part I: Molecular pathways controlling autophagy. Semin. Cancer Biol. 2015, 31, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.; Ordõñez, R.; Reiter, R.J.; González-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Rela-tion to autophagy and apoptosis. J. Pineal Res. 2015, 59, 292–307. [Google Scholar] [CrossRef]
- Kranz, P.; Neumann, F.; Wolf, A.; Classen, F.; Pompsch, M.; Ocklenburg, T.; Baumann, J.; Janke, K.; Baumann, M.; Goepelt, K.; et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. 2017, 8, e2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, J.J.; Brenner, M.B.; Thomas, D.; Williams, D.B. Calnexin: A membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 1994, 19, 124–128. [Google Scholar] [CrossRef]
- Carling, D. The AMP-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.-S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Song, N.; Huang, Y.; Chen, Y. Irisin inhibits pancreatic cancer cell growth via the AMPK-mTOR pathway. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Xie, Q.; Liu, Y.; Li, X. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl. Oncol. 2020, 13, 100871. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Gurlo, T.; Ravier, M.; Wojtusciszyn, A.; Mathieu, J.; Brown, M.R.; Broca, C.; Bertrand, G.; Butler, P.C.; Matveyenko, A.V.; et al. Proteasomal degradation of the histone acetyl transferase p300 contributes to beta-cell injury in a diabetes environment. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Jiang, G.; Liu, H.; Li, Z.; Pei, Y.; Wang, H.; Pan, H.; Cui, H.; Long, J.; Wang, J.; et al. Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone Res. 2020, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, L.; Qin, X.; Turdi, S.; Sun, D.; Culver, B.; Reiter, R.J.; Wang, X.; Zhou, H.; Ren, J. ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, S.; Son, M.; Cheon, J.H.; Park, Y.S. Melatonin controls microbiota in colitis by goblet cell differentiation and antimicrobial peptide production through Toll-like receptor 4 signalling. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Talib, W.H. Melatonin and Cancer Hallmarks. Molecules 2018, 23, 518. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Lei, H.; He, M.; Gong, R.; Wang, Y.; He, X.; Li, G.; Pang, P.; Li, X.; Yu, S.; et al. Melatonin triggers autophagic cell death by regulating RORC in Hodgkin lymphoma. Biomed. Pharmacother. 2020, 123, 109811. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Cheng, L.; Fan, L.; Wang, F.; Yu, H.; Liu, Q.; Li, Y.; Bu, L.; Li, X.; et al. Melatonin increases the anti-tumor ef-fects of sorafenib on human hepatoma cell lines via down-regulating autophagy. Int. J. Clin. Exp. Med. 2017, 10, 14109–14120. [Google Scholar]
- Polo, R.-A.G.; Boya, P.; Pauleau, A.-L.; Jalil, A.; Larochette, N.; Souquère, S.; Eskelinen, E.-L.; Pierron, G.; Saftig, P.; Kroemer, G. The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell Sci. 2005, 118, 3091–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yuan, K.; Tang, M.; Yue, J.; Bao, L.; Wu, S.; Zhang, Y.; Li, Y.; Wang, Y.; Ou, X.; et al. Melatonin inhibiting the survival of human gastric cancer cells under ER stress involving autophagy and Ras-Raf-MAPK signalling. J. Cell. Mol. Med. 2021, 25, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zhou, W.; Li, X.; Du, J.; Li, X.; Hao, H. Melatonin inhibits proliferation and viability and promotes apoptosis in colorectal cancer cells via upregulation of the microRNA-34a/449a cluster. Mol. Med. Rep. 2021, 23, 1. [Google Scholar] [CrossRef]
- Farriol, M.; Venereo, Y.; Orta, X.; Castellanos, J.M.; Segovia-Silvestre, T. In vitro effects of melatonin on cell prolifera-tion in a colon adenocarcinoma line. J. Appl. Toxicol. 2000, 20, 21–24. [Google Scholar] [CrossRef]
- Ghorbani-Anarkooli, M.; Dabirian, S.; Moladoust, H.; Zendedel, A.; Bahadori, M.H. Comparison of MTT, trypan blue, and clonogenic assay, to determine the viability in human anaplastic thyroid cancer cell line. Tehran Univ. Med. J. 2019, 77, 26–32. [Google Scholar]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.-S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.-J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2011, 441, 523–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.-J.; Chien, M.-H.; Chow, J.-M.; Chang, J.-L.; Wen, Y.-C.; Lin, Y.-W.; Cheng, C.-W.; Lai, G.-M.; Hsiao, M.; Lee, L.-M. Nonautophagic cytoplasmic vacuolation death induction in human PC-3M prostate cancer by curcumin through reactive oxygen species -mediated endoplasmic reticulum stress. Sci. Rep. 2015, 5, srep10420. [Google Scholar] [CrossRef]
- Yao, W.; Lin, Z.; Shi, P.; Chen, B.; Wang, G.; Huang, J.; Sui, Y.; Liu, Q.; Li, S.; Lin, X.; et al. Delicaflavone induces ROS-mediated apoptosis and inhibits PI3K/AKT/mTOR and Ras/MEK/Erk signaling pathways in colorectal cancer cells. Biochem. Pharmacol. 2020, 171, 113680. [Google Scholar] [CrossRef]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar]
- Thomé, M.P.; Chiela, E.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Cho, T.J.; Woo, B.H.; Choi, K.U.; Lee, C.H.; Ryu, M.H.; Park, H.R. Curcumin-induced autophagy contributes to the decreased survival of oral cancer cells. Arch. Oral Biol. 2012, 57, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, D.; Chowdhury, R.; Trudel, L.J.; Tee, A.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yun, C.W.; Han, Y.-S.; Kim, S.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.-J.; Lee, S.H. Melatonin and 5-fluorouracil co-suppress colon cancer stem cells by regulating cellular prion protein-Oct4 axis. J. Pineal Res. 2018, 65, e12519. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, T.-J.; Yoo, Y.-M. Melatonin Combined with Endoplasmic Reticulum Stress Induces Cell Death via the PI3K/Akt/mTOR Pathway in B16F10 Melanoma Cells. PLoS ONE 2014, 9, e92627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.-C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt Activates the Mammalian Target of Rapamycin by Regulating Cellular ATP Level and AMPK Activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2011, 13, 7–12. [Google Scholar] [CrossRef]
- Rodolfo, C.; Rocco, M.; Cattaneo, L.; Tartaglia, M.; Sassi, M.; Aducci, P.; Scaloni, A.; Camoni, L.; Marra, M. Ophiobolin A Induces Autophagy and Activates the Mitochondrial Pathway of Apoptosis in Human Melanoma Cells. PLoS ONE 2016, 11, e0167672. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chok, K.C.; Koh, R.Y.; Ng, M.G.; Ng, P.Y.; Chye, S.M. Melatonin Induces Autophagy via Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Pathway in Colorectal Cancer Cells. Molecules 2021, 26, 5038. https://doi.org/10.3390/molecules26165038
Chok KC, Koh RY, Ng MG, Ng PY, Chye SM. Melatonin Induces Autophagy via Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Pathway in Colorectal Cancer Cells. Molecules. 2021; 26(16):5038. https://doi.org/10.3390/molecules26165038
Chicago/Turabian StyleChok, Kian Chung, Rhun Yian Koh, Ming Guan Ng, Pei Ying Ng, and Soi Moi Chye. 2021. "Melatonin Induces Autophagy via Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Pathway in Colorectal Cancer Cells" Molecules 26, no. 16: 5038. https://doi.org/10.3390/molecules26165038
APA StyleChok, K. C., Koh, R. Y., Ng, M. G., Ng, P. Y., & Chye, S. M. (2021). Melatonin Induces Autophagy via Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Pathway in Colorectal Cancer Cells. Molecules, 26(16), 5038. https://doi.org/10.3390/molecules26165038