
Synthesis and Antiviral Evaluation of Nucleoside Analogues Bearing One Pyrimidine Moiety and Two D-Ribofuranosyl Residues

 , ,
, ,  , , and
, , and
Abstract

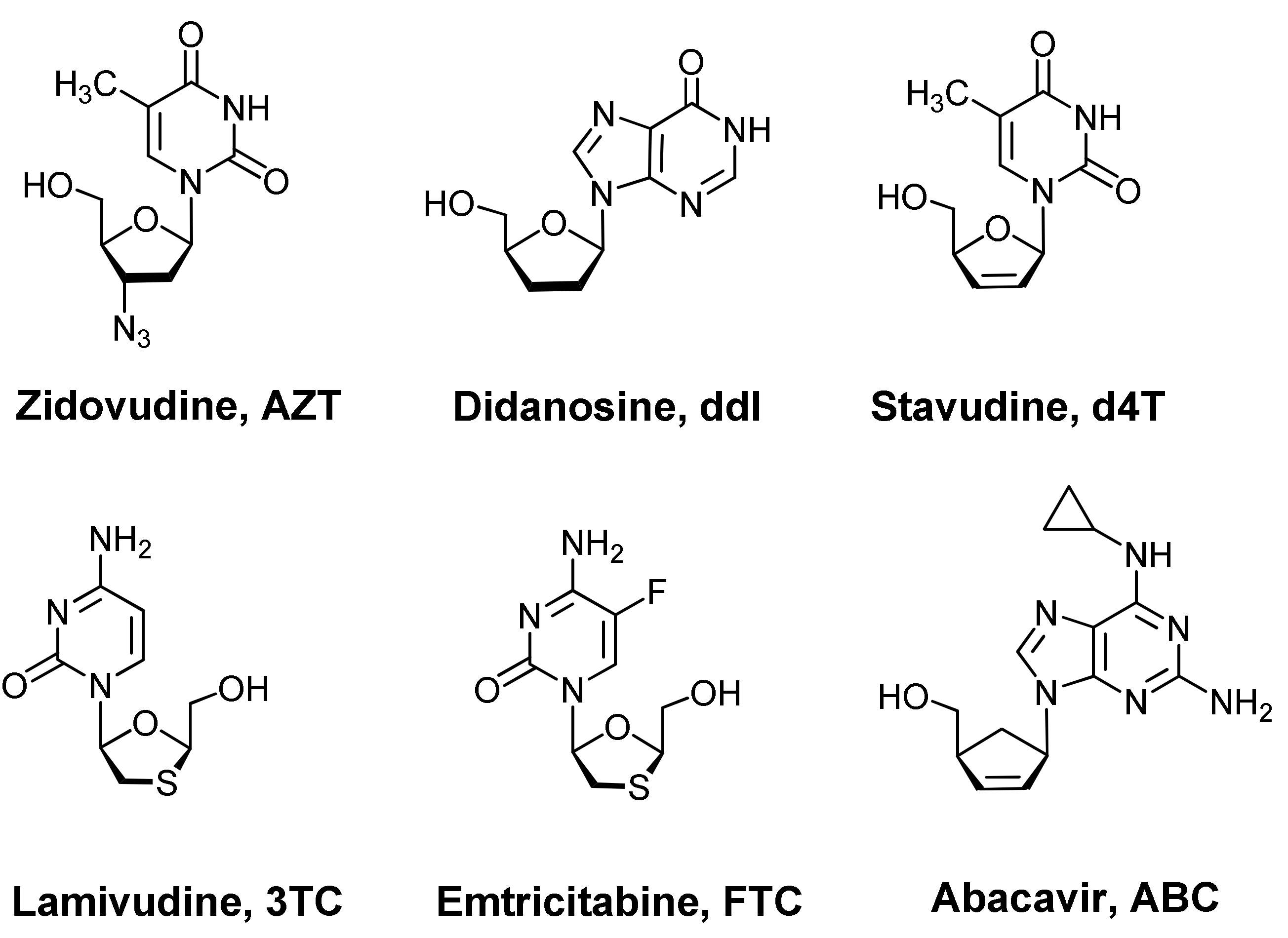
1. Introduction
2. Results and Discussion
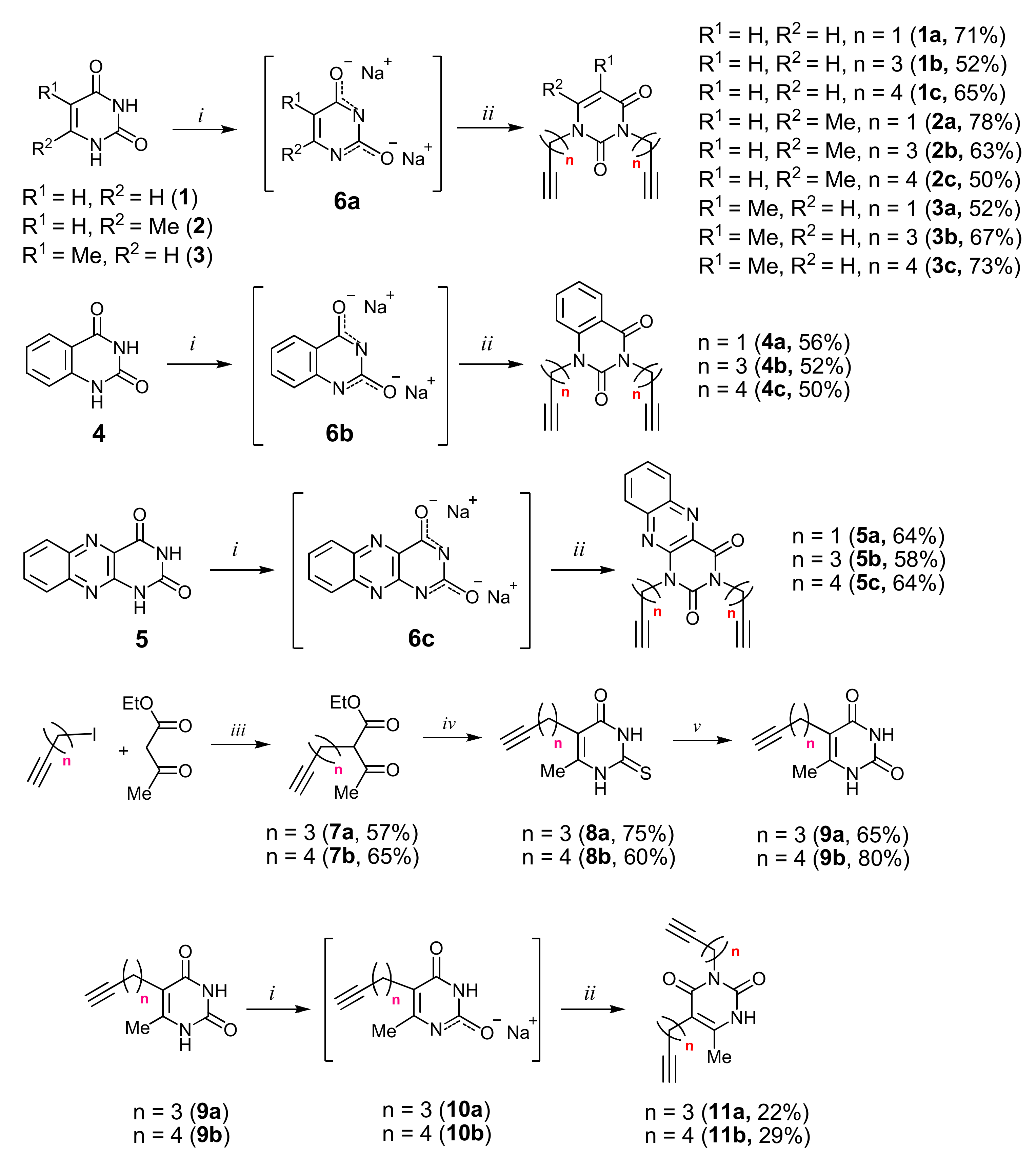
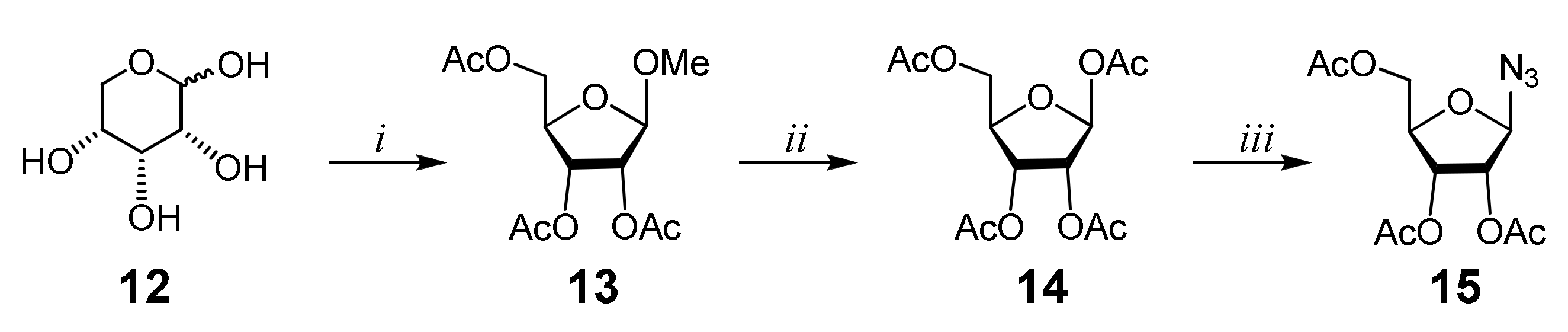
2.1. Chemistry
2.2. Antiviral Evaluation
2.3. Molecular Docking
2.3.1. Influenza Virus A H1N1
2.3.2. Coxsackievirus B3
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- De Clercq, E.; Lia, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, L.A.; Efremenkova, O.V.; Andronova, V.L.; Galegov, G.A.; Solyev, P.N.; Karpenko, I.L.; Kochetkov, S.N. 5-(4-alkyl-1,2,3-triazol-1-yl)methyl derivatives of 2′-deoxyuridine as inhibitors of viral and bacterial growth. Russ. J. Bioorganic Chem. 2016, 42, 677–684. [Google Scholar] [CrossRef]
- Zhurilo, N.I.; Chudinov, M.V.; Matveev, A.V.; Smirnova, O.S.; Konstantinova, I.D.; Miroshnikov, A.I.; Prutkov, A.N.; Grebenkina, L.E.; Pulkova, N.V.; Shvets, V.I. Isosteric ribavirin analogues: Synthesis and antiviral activities. Bioorganic Med. Chem. Lett. 2018, 28, 11–14. [Google Scholar] [CrossRef]
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Tirumalasetty, M.C.B.; Wudayagiri, R.; Donthabakthuni, S.; Maroju, R.; Baburao, K.; Parasa, L.S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396. [Google Scholar] [CrossRef]
- Kohgo, S.; Imoto, S.; Tokuda, R.; Takamatsu, Y.; Higashi-Kuwata, N.; Aoki, M.; Amano, M.; Kansui, H.; Onitsuka, K.; Maeda, K.; et al. Synthesis of 4′-substituted purine 2′-deoxynucleosides and their activity against human immunodeficiency virus type 1 and hepatitis B virus. ChemistrySelect 2018, 3, 3313–3317. [Google Scholar] [CrossRef]
- Wang, P.; Hong, J.H.; Cooperwood, J.S.; Chu, C.K. Recent advances in l-nucleosides: Chemistry and biology. Antivir. Res. 1998, 40, 19–44. [Google Scholar] [CrossRef]
- Kim, H.O.; Shanmuganathan, K.; Alves, A.J.; Jeong, L.S.; Beach, J.W.; Schinazi, R.F.; Chang, C.N.; Cheng, Y.C.; Chu, C.K. Potent anti-HIV and anti-HBV activities of (−)-L-β-dioxolane-C and (+)-L-β-dioxolane-T and their asymmetric syntheses. Tetrahedron Lett. 1992, 33, 6899–6902. [Google Scholar] [CrossRef]
- Roy, B.; Depaix, A.; Périgaud, C.; Peyrottes, S. Recent Trends in Nucleotide Synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2007, 28, 278–308. [Google Scholar] [CrossRef]
- Petrova, K.T.; Potewar, T.M.; Correia-Da-Silva, P.; Barros, M.T.; Calhelha, R.C.; Ciric, A.; Sokovic, M.; Ferreira, I.C.F.R. Antimicrobial and cytotoxic activities of 1,2,3-triazole-sucrose derivatives. Carbohydr. Res. 2015, 417, 66–71. [Google Scholar] [CrossRef]
- Park, S.M.; Yang, H.; Park, S.-K.; Kim, H.M.; Kim, B.H. Design, synthesis, and anticancer activities of novel perfluoroalkyltriazole-appended 2′-deoxyuridines. Russ. J. Bioorganic Chem. Med. Chem. Lett. 2010, 20, 5831–5834. [Google Scholar] [CrossRef]
- Montagu, A.; Roy, V.; Balzarini, J.; Snoeck, R.; Andrei, G.; Agrofoglio, L.A. Synthesis of new C5-(1-substituted-1,2,3-triazol-4 or 5-yl)-2′-deoxyuridines and their antiviral evaluation. Eur. J. Med. Chem. 2011, 46, 778–786. [Google Scholar] [CrossRef]
- Shmalenyuk, E.R.; Chernousova, L.N.; Karpenko, I.L.; Kochetkov, S.N.; Smirnova, T.G.; Andreevskaya, S.N.; Chizhov, A.O.; Efremenkova, O.; Alexandrova, L.A. Inhibition of Mycobacterium tuberculosis strains H37Rv and MDR MS-115 by a new set of C5 modified pyrimidine nucleosides. Bioorganic Med. Chem. 2013, 21, 4874–4884. [Google Scholar] [CrossRef]
- Kumar, J.M.; Idris, M.M.; Srinivas, G.; Kumar, P.V.; Meghah, V.; Kavitha, M.; Reddy, C.R.; Mainkar, P.S.; Pal, B.; Chandrasekar, S.; et al. Phenyl 1,2,3-triazole-thymidine ligands stabilize g-quadruplex DNA, inhibit DNA synthesis and potentially reduce tumor cell proliferation over 3′-azido deoxythymidine. PLoS ONE 2013, 8, e70798. [Google Scholar] [CrossRef]
- Chatzileontiadou, D.S.M.; Parmenopoulou, V.; Manta, S.; Kantsadi, A.L.; Kylindri, P.; Griniezaki, M.; Kontopoulou, F.; Telopoulou, A.; Prokova, H.; Panagopoulos, D.; et al. Triazole double-headed ribonucleosides as inhibitors of eosinophil derived neurotoxin. Bioorganic Chem. 2015, 63, 152–165. [Google Scholar] [CrossRef]
- Elayadi, H.; Mesnaoui, M.; Korba, B.E.; Smietana, M.; Vasseur, J.-J.; Secrist, J.A.; Lazrek, H.B. Preparation of 1,4-disubstituted-1,2,3-triazolo ribonucleosides by Na2CuP2O7 catalyzed azide-alkyne 1,3-dipolar cycloaddition. Arkivoc 2012, 2012, 76. [Google Scholar] [CrossRef]
- Amant, A.H.S.; Bean, L.A.; Guthrie, J.P.; Hudson, R.H.E. Click fleximers: A modular approach to purine base-expanded ribonucleoside analogues. Org. Biomol. Chem. 2012, 10, 6521. [Google Scholar] [CrossRef]
- Chatzileontiadou, D.S.M.; Tsika, A.C.; Diamantopoulou, Z.; Delbé, J.; Badet, J.; Courty, J.; Skamnaki, V.T.; Parmenopoulou, V.; Komiotis, D.; Hayes, J.M.; et al. Evidence for Novel Action at the Cell-Binding Site of Human Angiogenin Revealed by Heteronuclear NMR Spectroscopy, in silico and in vivo Studies. ChemMedChem 2018, 13, 259–269. [Google Scholar] [CrossRef]
- Andreeva, O.V.; Belenok, M.G.; Saifina, L.F.; Shulaeva, M.M.; Dobrynin, A.B.; Sharipova, R.R.; Voloshina, A.D.; Saifina, A.F.; Gubaidullin, A.T.; Khairutdinov, B.I.; et al. Synthesis of novel 1,2,3-triazolyl nucleoside analogues bearing uracil, 6-methyluracil, 3,6-dimethyluracil, thymine, and quinazoline-2,4-dione moieties. Tetrahedron Lett. 2019, 60, 151276. [Google Scholar] [CrossRef]
- Sharipova, R.R.; Saifina, L.F.; Belenok, M.G.; Semenov, V.E.; Kataev, V.E. First analog of pyrimidine nucleosides with two d-ribofuranose residues. Russ. J. Org. Chem. 2020, 56, 181–184. [Google Scholar] [CrossRef]
- Andreeva, O.V.; Garifullin, B.F.; Zarubaev, V.V.; Slita, A.V.; Yesaulkova, I.L.; ·Saifina, L.F.; Shulaeva, M.M.; Belenok, M.G.; Semenov, V.E.; Kataev, V.E. Synthesis of 1,2,3-triazolyl nucleoside analogues and their antiviral activity. Mol. Divers. 2020, 25, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Voloshina, A.D.; Sapunova, A.S.; Kulik, N.V.; Belenok, M.G.; Strobykina, I.Y.; Lyubina, A.P.; Gumerova, S.K.; Kataev, V.E. Antimicrobial and cytotoxic effects of ammonium derivatives of diterpenoids steviol and isosteviol. Bioorganic Med. Chem. 2021, 32, 115974. [Google Scholar] [CrossRef] [PubMed]
- Stevaert, A.; Naesens, L. The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design. Med. Res. Rev. 2016, 36, 1127–1173. [Google Scholar] [CrossRef] [PubMed]
- Jockusch, S.; Tao1, C.; Li1, X.; Anderson, T.K.; Chien, M.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. A library of nucleotide analogues terminate RNA synthesis catalyzed by polymerases of coronaviruses that cause SARS and COVID-19. Antivir. Res. 2020, 180, 104857. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Velthuis, A.J.T. The RNA-dependent RNA polymerase of the influenza A virus. Futur. Virol. 2014, 9, 863–876. [Google Scholar] [CrossRef]
- Pala, N.; Stevaert, A.; Dallocchio, R.; Dessì, A.; Rogolino, D.; Carcelli, M.; Sanna, V.; Sechi, M.; Naesens, L. Virtual Screening and Biological Validation of Novel Influenza Virus PA Endonuclease Inhibitors. ACS Med. Chem. Lett. 2015, 6, 866–871. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Fudo, S.; Yamamoto, N.; Nukaga, M.; Odagiri, T. Two Distinctive Binding Modes of Endonuclease Inhibitors to the N-Terminal Region of Influenza Virus Polymerase Acidic Subunit. Biochemistry 2016, 55, 2646–2660. [Google Scholar] [CrossRef]
- Muckelbauer, J.K.; Kremer, M.; Minor, I.; Tong, L.; Zlotnick, A.; Johnson, J.E.; Rossmann, M.G. Structure determination of coxsackievirus B3 to 3.5 Å resolution. Acta Cryst. 1995, D51, 871–887. [Google Scholar] [CrossRef]
- Bermsn, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- HyperChem Professional 8.0. Hypercube, Inc. 2007. Available online: https://www.chemits.com/en/software/molecular-modeling/hyperchem/ (accessed on 14 September 2020).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Das, K.; Aramini, J.M.; Ma, L.-C.; Krug, R.M.; Arnold, E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010, 17, 530–538. [Google Scholar] [CrossRef]
- Yan, V.C.; Muller, F.L. Advantages of the Parent Nucleoside GS-441524 over Remdesivir for Covid-19 Treatment. ACS Med. Chem. Lett. 2020, 11, 1361–1366. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide analogues as inhibitors of SARSCoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Wagner, C.R.; Iyer, V.V.; Mcintee, E.J. Pronucleotides: Toward thein vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. [Google Scholar] [CrossRef]
- Adams, M.J.; King, A.M.Q.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch. Virol. 2013, 158, 2023–2030. [Google Scholar] [CrossRef]
- Hogle, J.M.; Chow, M.; Filman, D.J. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 1985, 229, 1358–1365. [Google Scholar] [CrossRef]
- Liu, Y.; Sheng, J.; Fokine, A.; Meng, G.; Shin, W.-H.; Long, F.; Kuhn, R.J.; Kihara, D.; Rossmann, M.G. Structure and inhibition of EV-D68, a virus that causes respiratory illness in children. Science 2015, 347, 71–74. [Google Scholar] [CrossRef]
- Smyth, M.; Tate, J.; Hoey, E.; Lyons, C.; Martin, S.; Stuart, D. Implications for viral uncoating from the structure of bovine enterovirus. Nat. Struct. Mol. Biol. 1995, 2, 224–231. [Google Scholar] [CrossRef]
- Rossmann, M.G. Viral cell recognition and entry. Protein Sci. 1994, 3, 1712–1725. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | CC50 1 (μM) | IC50 2 (μM) | SI 3 |
|---|---|---|---|---|
| 2g |  | >543.0 | >543.0 | 1 |
| 2j |  | >885 | >885 | 1 |
| 2h |  | >492.9 | >492.9 | 1 |
| 2k |  | 254 ± 19 4 | 43 ± 5 4 | 6 4 |
| 2f |  | >337.5 | 96.3 ± 12.4 | 4 |
| 2i |  | >471.2 | 57.5 ± 7.1 | 8 |
| 2m |  | 311 ± 27 4 | 48 ± 6 4 | 6 4 |
| 4g |  | >509.8 | 180.1 ± 19.9 | 3 |
| 4j |  | 132 ± 9 | 42 ± 5 | 3 |
| 4h |  | >465.4 | 186.2 ± 21.6 | 3 |
| 4k |  | >744 4 | 212 ± 25 4 | 4 4 |
| 4f |  | >324.0 | 103.9 ± 15.1 | 3 |
| 4i |  | >446.0 | 297.3 ± 32.4 | 2 |
| 4m |  | >719 | 30 ± 4 | 24 |
| 5g |  | >468.3 | 187.3 ± 20.8 | 3 |
| 5h |  | >430.6 | 114.8 ± 15.2 | 4 |
| 5f |  | >307.7 | 91.6 ± 10.8 | 3 |
| 5i |  | >414.0 | 24.3 ± 3.2 | 17 |
| 11c |  | >349.0 | 29.2 ± 4.3 | 12 |
| 11g |  | 104 ± 8 4 | 67 ± 8 4 | 2 4 |
| 11e |  | >492.9 | 164.3 ± 19.2 | 3 |
| 11h |  | 79 ± 6 | 15 ± 3 | 5 |
| 11d |  | >338.0 | 119.3 ± 8.7 | 3 |
| 11i |  | >592 4 | >592 4 | 1 4 |
| 11f |  | >471.2 | >471.2 | 1 |
| 11j |  | >787 | >787 | 1 |
| Rimantadine | 340 ± 16 | 77 ± 8 | 4 | |
| Oseltamivir carboxylate | >200 | 0.3 ± 0.06 | >667 |
| Compound | Structure | CC50 1 (μM) | IC50 2 (μM) | SI 3 |
|---|---|---|---|---|
| 2g |  | >543.0 | >543.0 | 1 |
| 2j |  | >1180 4 | >1180 4 | 1 4 |
| 2h |  | >492.9 | >492.9 | 1 |
| 2k |  | >1090 4,3 | >1090 4,3 | 1 4,3 |
| 2f |  | 17.7 ± 0.6 | >12.4 | 1 |
| 2i |  | >471.2 | >471.2 | 1 |
| 2m |  | 656 ± 33 4,3 | 106 ± 11 4,3 | 6 4,3 |
| 4g |  | 380.6 ± 26.1 | >169.9 | 2 |
| 4j |  | 166 ± 12 4 | 133 ± 154 | 1 4 |
| 4h |  | >465.4 | >465.4 | 1 |
| 4k |  | 101 ± 8 4,3 | >82 4,3 | 1 4,3 |
| 4i |  | >446.0 | >446.0 | 1 |
| 4m |  | 101 ± 9 4,3 | >79 4,3 | 1 4,3 |
| 5g |  | >468.3 | >468.3 | 1 |
| 5h |  | >430.6 | >430.6 | 1 |
| 5f |  | 17.8 ± 1.1 | >11.3 | 2 |
| 5i |  | >414.0 | >414.0 | 1 |
| 11e |  | 394.3 ± 21.1 | >164.3 | 2 |
| 11g |  | 140 ± 10 4 | >34 4 | 4 4 |
| 11f |  | >471.2 | >471.2 | 1 |
| 11h |  | 525 ± 31 4 | >263 4 | 2 4 |
| Pleconaril | >1000 | 21.6 | 46 | |
| Compound | Structure | IC50 (µM) | |||
|---|---|---|---|---|---|
| Cancer Cell Lines | Normal Cell Line | ||||
| M-HeLa 2 | HuTu-80 3 | PC-3 4 | WI-38 5 | ||
| 2f |  | 88.1 ± 7.4 | >100 | >100 | >100 |
| 2i |  | 100 ± 8.5 | >100 | >100 | >100 |
| 5f |  | 71.4 ± 6.3 | >100 | 66.8 ± 5.3 | >100 |
| 5i |  | 71 ± 6.1 | >100 | >100 | >100 |
| 5-Fluorouracil | 62.0 ± 4.9 | 65.2 ± 5.5 | 10.3 ± 0.8 | 82.5 ± 6.6 | |
| Compound | Structure | IC50 (µM) | −Ebind (kkal/mol) | Ligand–Protein Interactions |
|---|---|---|---|---|
| 2i |  | 57.5 | 8.7 | H-bonding: Val122, Lys134, Lys137 |
| 2i-TP |  | − | 8.8 | H-bonding: Tyr24, Val122, Lys134, Lys137, Arg196 π-cation: Lys34 |
| 5i |  | 24.3 | 9.3 | H-bonding: Tyr24, Gly81, Leu106 π-cation: Mn2+, Lys134 |
| 5i-TP |  | − | 9.1 | H-bonding: Tyr24, Gly81, Arg84, Lys137 π-π: Tyr24 |
| 11c |  | 29.2 | 8.1 | H-bonding: Arg84, Leu106, Lys134, Lys137, Arg196 π-cation: Mn2+. |
| Compound | Structure | IC50 (µM) | −Ebind (kkal/mol) | Ligand–Protein Interactions |
|---|---|---|---|---|
| 2f |  | >12.4 | 8.3 | H-bonding: Ser209 (VP2) π-π: Tyr189 (VP1) |
| 5f |  | >11.3 | 9.2 | H-bonding: Tyr143 (VP1), Ser190 (VP1), Arg196 (VP2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreeva, O.V.; Garifullin, B.F.; Zarubaev, V.V.; Slita, A.V.; Yesaulkova, I.L.; Volobueva, A.S.; Belenok, M.G.; Man’kova, M.A.; Saifina, L.F.; Shulaeva, M.M.; et al. Synthesis and Antiviral Evaluation of Nucleoside Analogues Bearing One Pyrimidine Moiety and Two D-Ribofuranosyl Residues. Molecules 2021, 26, 3678. https://doi.org/10.3390/molecules26123678
Andreeva OV, Garifullin BF, Zarubaev VV, Slita AV, Yesaulkova IL, Volobueva AS, Belenok MG, Man’kova MA, Saifina LF, Shulaeva MM, et al. Synthesis and Antiviral Evaluation of Nucleoside Analogues Bearing One Pyrimidine Moiety and Two D-Ribofuranosyl Residues. Molecules. 2021; 26(12):3678. https://doi.org/10.3390/molecules26123678
Chicago/Turabian StyleAndreeva, Olga V., Bulat F. Garifullin, Vladimir V. Zarubaev, Alexander V. Slita, Iana L. Yesaulkova, Alexandrina S. Volobueva, Mayya G. Belenok, Maria A. Man’kova, Liliya F. Saifina, Marina M. Shulaeva, and et al. 2021. "Synthesis and Antiviral Evaluation of Nucleoside Analogues Bearing One Pyrimidine Moiety and Two D-Ribofuranosyl Residues" Molecules 26, no. 12: 3678. https://doi.org/10.3390/molecules26123678
APA StyleAndreeva, O. V., Garifullin, B. F., Zarubaev, V. V., Slita, A. V., Yesaulkova, I. L., Volobueva, A. S., Belenok, M. G., Man’kova, M. A., Saifina, L. F., Shulaeva, M. M., Voloshina, A. D., Lyubina, A. P., Semenov, V. E., & Kataev, V. E. (2021). Synthesis and Antiviral Evaluation of Nucleoside Analogues Bearing One Pyrimidine Moiety and Two D-Ribofuranosyl Residues. Molecules, 26(12), 3678. https://doi.org/10.3390/molecules26123678