
Effect of Solvents on Fe–Lignin Precursors for Production Graphene-Based Nanostructures


Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Fe–Lignin Mixture
2.3. Catalytic Graphitization
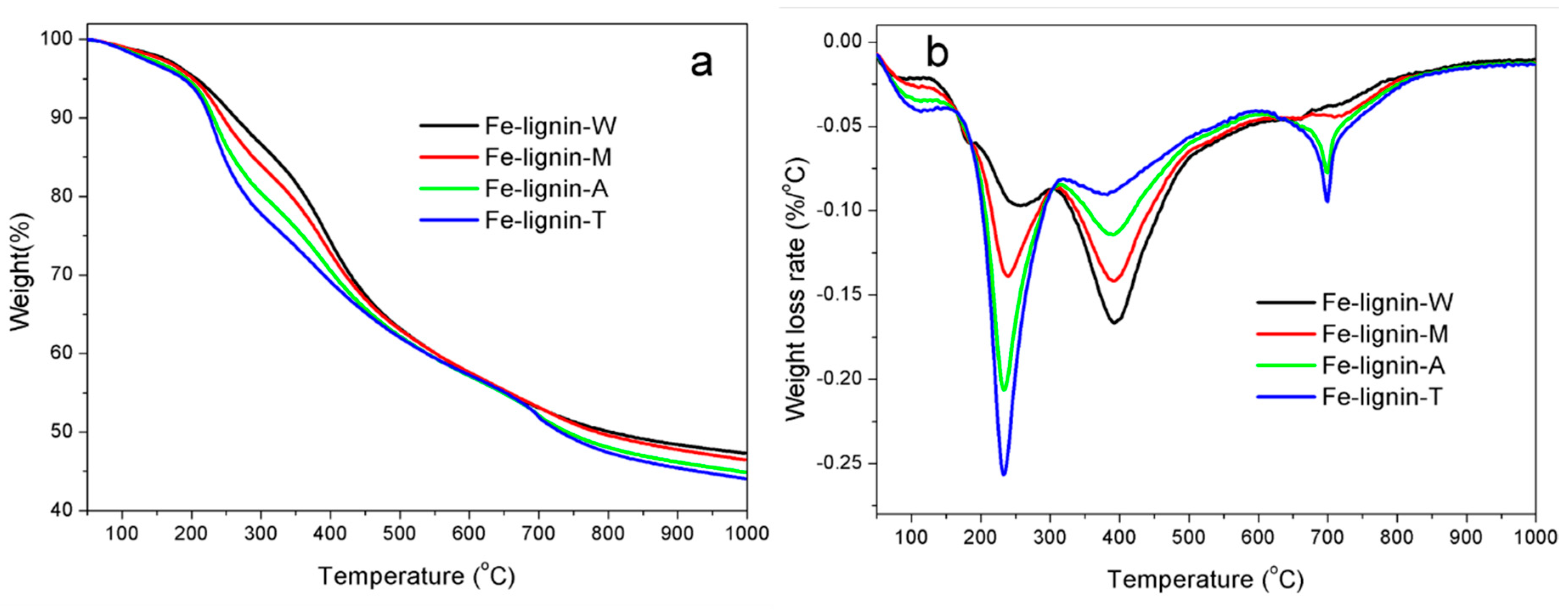
2.4. Thermogravimetric Analyses (TGA)
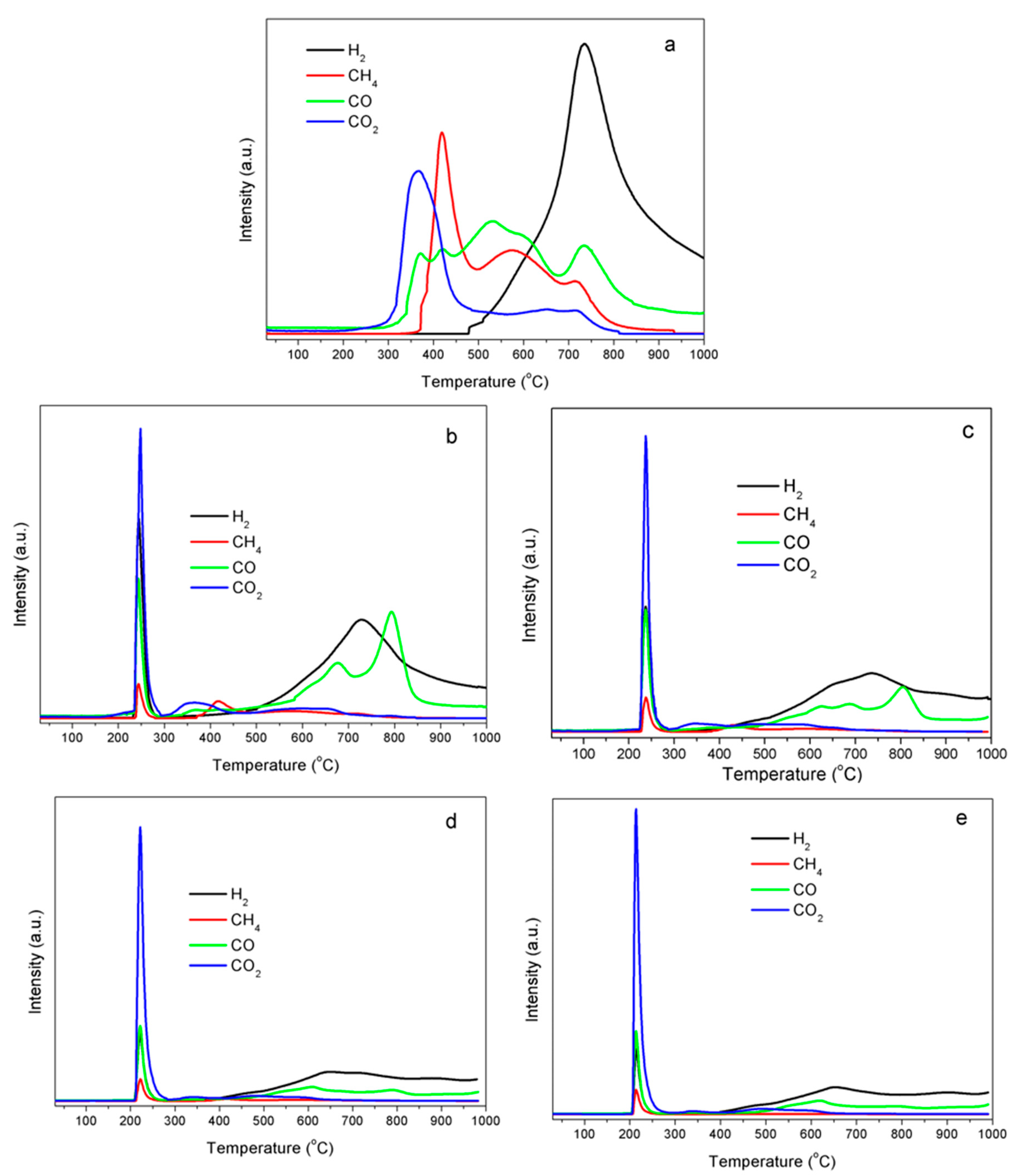
2.5. Formation of Gaseous Products by Temperature-programmed Decomposition (TPD)
2.6. Characterization
3. Results and Discussion
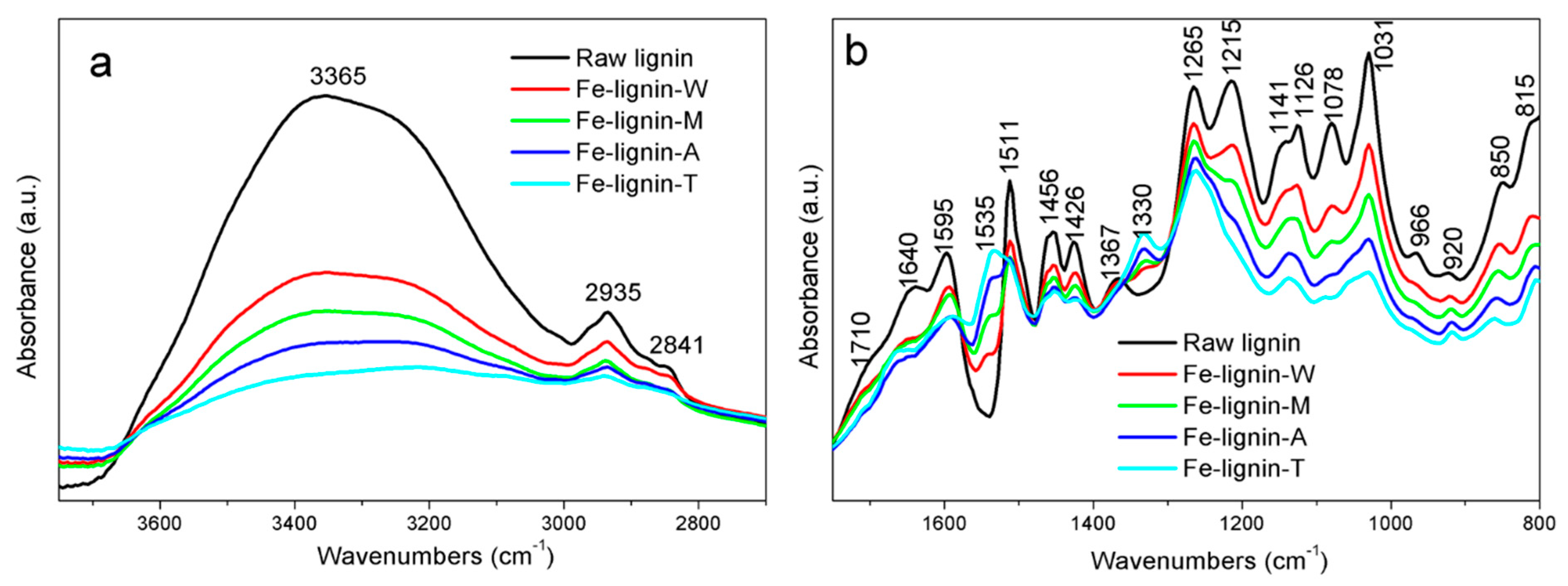
3.1. FTIR
3.2. Thermal Analyses of Fe–Lignin Precursors Prepared Using Different Solvents
3.3. Gaseous Product Analyses by Temperature-Programmed Decomposition (TPD)
3.4. Effects of Solvents on Fe–Lignin Performance—Product Distributions
3.5. Solid Product Characterization
3.5.1. Surface Area
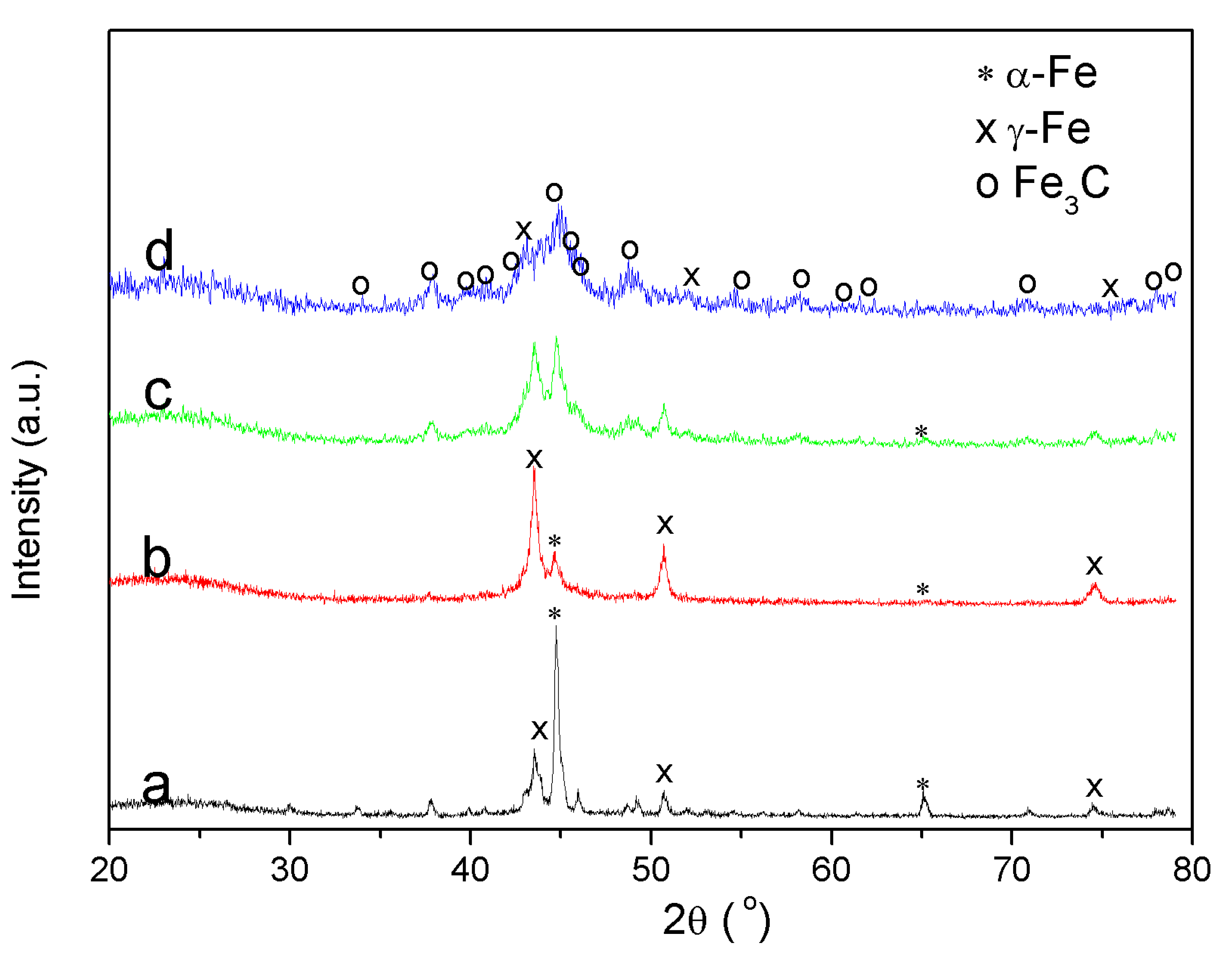
3.5.2. X-ray Diffraction (XRD)
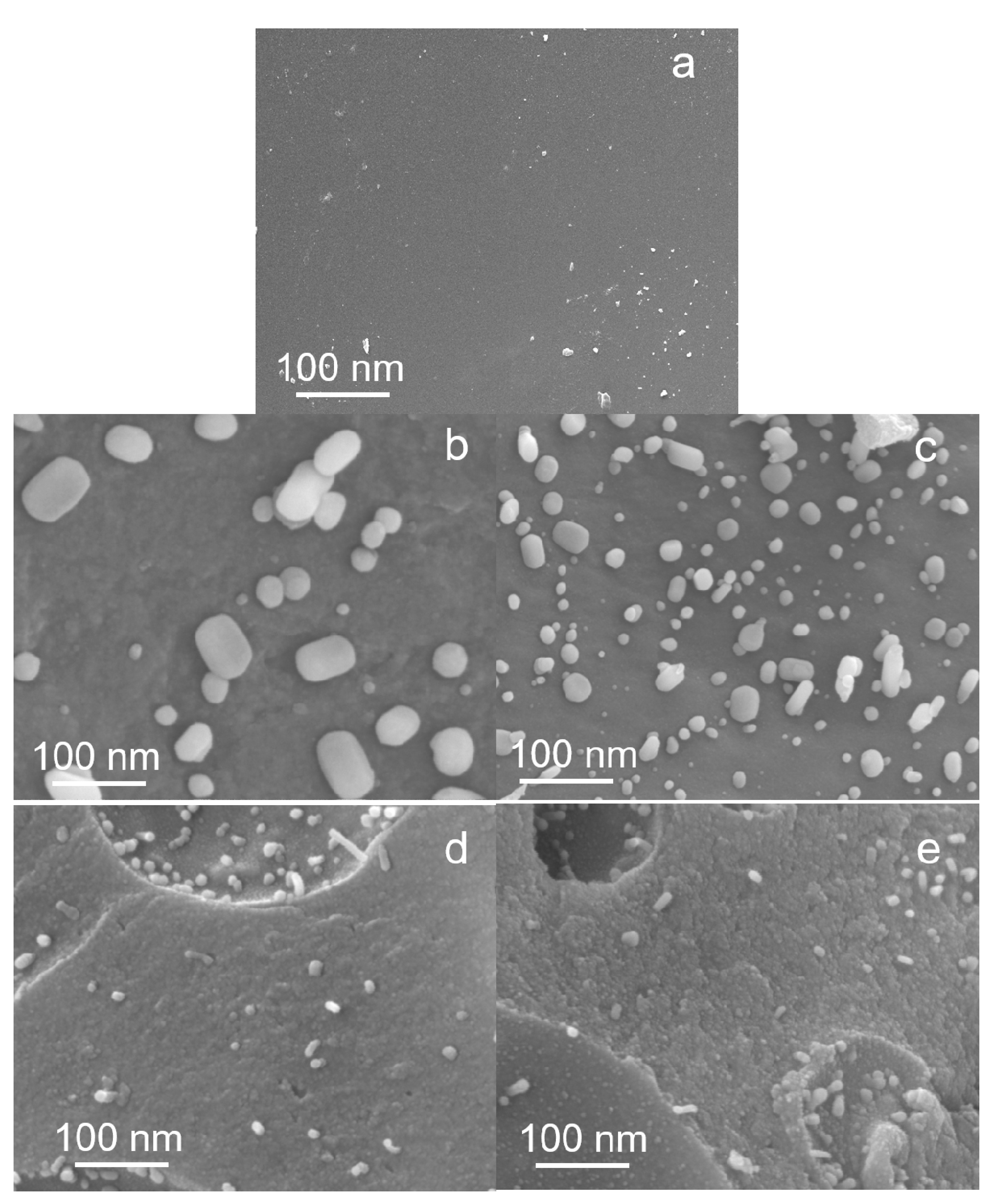
3.5.3. Scanning Electron Microscope (SEM)
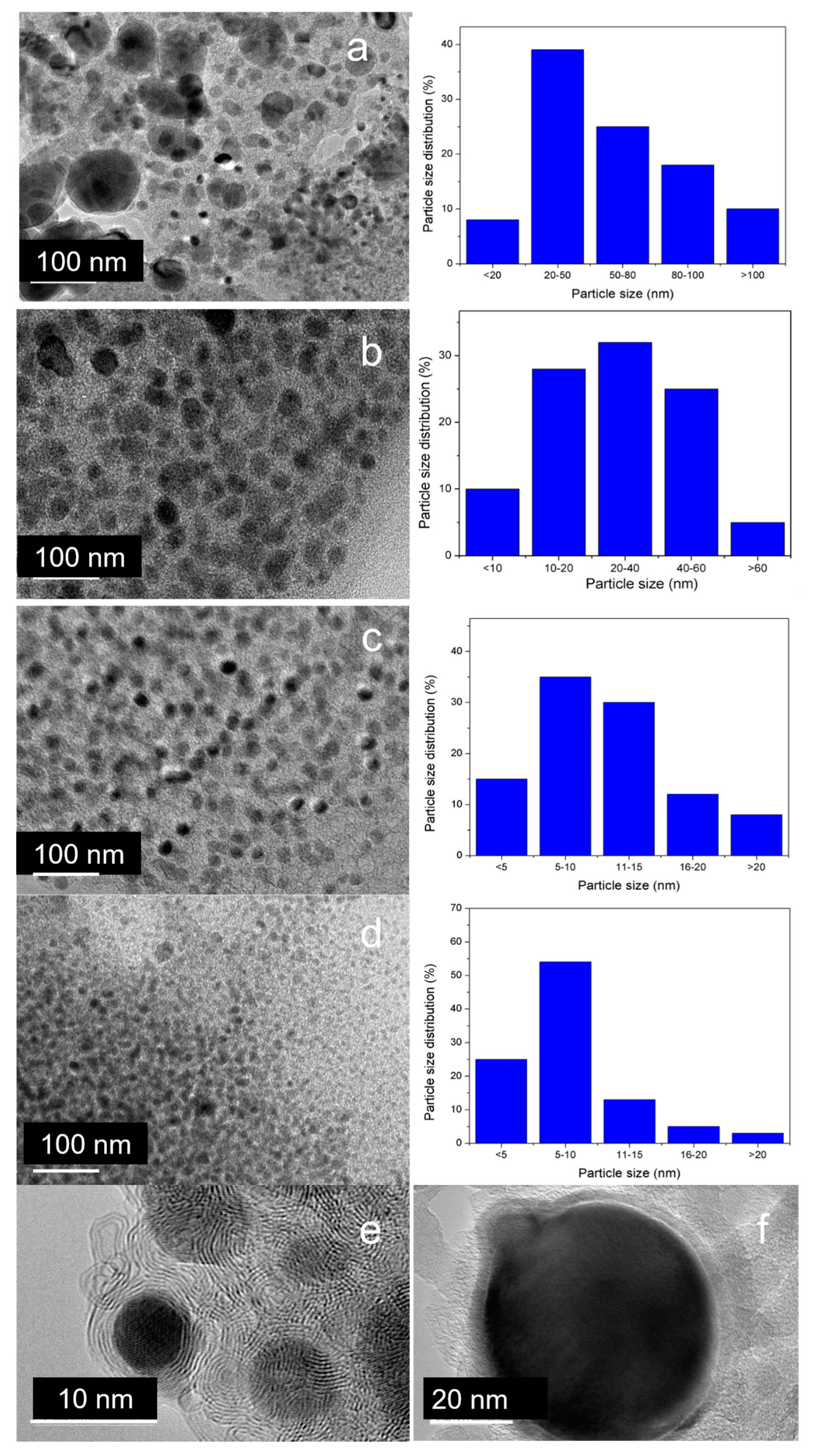
3.5.4. High-Resolution Transmission Electron Microscopy (HRTEM)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, Q.; Li, J.; Zhang, X.; Zhang, J.; Cai, Z. Mass production of graphene materials from solid carbon sources using a molecular cracking and welding method. J. Mater. Chem. A 2019, 7, 13978–13985. [Google Scholar] [CrossRef]
- Yan, Q. Catalytic Thermal Conversion of Kraft Lignin to Multi-Layer Graphene Materials. Ph.D. Thesis, Mississippi State University, Starkville, MS, USA, 2017. Available online: https://search.proquest.com/docview/1889555601?pq-origsite=gscholar (accessed on 15 March 2020).
- Cai, Z.; Yan, Q.; Li, J.; Zhang, J.; Marcoccia, B.; Freiberg, J. Methods for synthesizing graphene from encapsulated particles. US Patent Pub. No. 20180194630, 8 December 2018. [Google Scholar]
- Yan, Q.; Li, J.; Zhang, X.; Zhang, J.; Cai, Z. Synthetic Bio-Graphene Based Nanomaterials through Different Iron Catalysts. Nanomaterials 2018, 8, 840. [Google Scholar] [CrossRef]
- Demir, M.; Kahveci, Z.; Aksoy, B.; Palapati, N.K.R.; Subramanian, A.; Cullinan, H.T.; El-Kaderi, H.M.; Harris, C.T.; Gupta, R.B. Graphitic Biocarbon from Metal-Catalyzed Hydrothermal Carbonization of Lignin. Ind. Eng. Chem. Res. 2015, 54, 10731–10739. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wan, C.; Liu, J.; Gao, J.; Yu, F.; Zhang, J.; Cai, Z. Iron nanoparticles in situ encapsulated in biochar-based carbon as an effective catalyst for the conversion of biomass-derived syngas to liquid hydrocarbons. Green Chem. 2013, 15, 1631–1640. [Google Scholar] [CrossRef]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Salvado, J. Analytical methods for determining functional groups in various technical lignins. Ind. Crop. Prod. 2007, 26, 116–124. [Google Scholar] [CrossRef]
- Schuerch, C. The Solvent Properties of Liquids and Their Relation to the Solubility, Swelling, Isolation and Fractionation of Lignin. J. Am. Chem. Soc. 1952, 74, 5061–5067. [Google Scholar] [CrossRef]
- Saito, T.; Perkins, J.H.; Vautard, F.; Meyer, H.M.; Messman, J.M.; Tolnai, B.; Naskar, A.K. Methanol Fractionation of Softwood Kraft Lignin: Impact on the Lignin Properties. ChemSusChem 2014, 7, 221–228. [Google Scholar] [CrossRef]
- Jiang, X.; Savithri, D.; Du, X.; Pawar, S.; Jameel, H.; Chang, H.-M.; Zhou, X. Fractionation and characterization of kraft lignin by sequential precipitation with various organic solvent. ACS Sustain. Chem. Eng. 2017, 5, 835–842. [Google Scholar] [CrossRef]
- Shukry, N.; Fadel, S.M.; Agblevor, F.A.; El-Kalyoubi, S.F. Some physical properties of acetosolv lignins from bagasse. J. Appl. Polym. Sci. 2008, 109, 434–444. [Google Scholar] [CrossRef]
- Sameni, J.; Krigstin, S.; Sain, M. Solubility of lignin and acetylated lignin in organic solvents. BioResources 2017, 12, 1548–1565. [Google Scholar] [CrossRef]
- Olarte, M.V. Base-Catalyzed Depolymerization of Lignin and Hydrodeoxygenation of Lignin Model Compounds for Alternative Fuel Production. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2011. [Google Scholar]
- Melro, E.; Alves, L.; Antunes, F.; Medronho, B. A brief overview on lignin dissolution. J. Mol. Liq. 2018, 265, 578–584. [Google Scholar] [CrossRef]
- Sun, J.; Dutta, T.; Parthasarathi, R.; Kim, K.H.; Tolic, N.; Chu, R.K.; Isern, N.G.; Cort, J.R.; Simmons, B.A.; Singh, S. Rapid room temperature solubilization and depolymerization of polymeric lignin at high loadings. Green Chem. 2016, 18, 6012–6020. [Google Scholar] [CrossRef]
- Rashid, T.; Kait, C.F.; Regupathi, I.; Murugesan, T. Dissolution of kraft lignin using Protic Ionic Liquids and characterization. Ind. Crop. Prod. 2016, 84, 284–293. [Google Scholar] [CrossRef]
- Achinivu, E.C.; Howard, R.M.; Li, G.; Gracz, H.; Henderson, W.A. Lignin extraction from biomass with protic ionic liquids. Green Chem. 2014, 16, 1114–1119. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Fiţigău, F.I.; Gosselink, R.J.; Frissen, A.E.; Stoutjesdijk, J.; Peter, F. Fractionation of five technical lignins by selective extraction in green solvents and characterisation of isolated fractions. Ind. Crop. Prod. 2014, 62, 481–490. [Google Scholar] [CrossRef]
- ASTM D4442-07(2007). Standard Test Methods for Direct Moisture Content Measurement of Wood and Wood-Based Materials; ASTM Int.: West Conshohocken, PA, USA, 2007. [Google Scholar]
- Yan, Q.; Li, J.; Zhang, X.; Zhang, J.; Cai, Z. In situ formation of graphene-encapsulated iron nanoparticles in carbon frames through catalytic graphitization of kraft lignin. Nanomater. Nanotechnol. 2018, 8. [Google Scholar] [CrossRef]
- Cheng, F.; Cao, Q.; Guan, Y.; Cheng, H.; Wang, X.; Miller, J.D. FTIR analysis of water structure and its influence on the flotation of arcanite (K2SO4). Int. J. Miner. Process. 2013, 122, 36–42. [Google Scholar] [CrossRef]
- Awal, A.; Sain, M. Spectroscopic studies and evaluation of thermorheological properties of softwood and hardwood lignin. J. Appl. Polym. Sci. 2011, 122, 956–963. [Google Scholar] [CrossRef]
- KuboJohn, S.; Kadla, F. Hydrogen Bonding in Lignin: A Fourier Transform Infrared Model Compound Study. Biomacromolecules 2005, 6, 52815–52821. [Google Scholar]
- Ashkenazy, R.; Gottlieb, L.; Yannai, S. Characterization of acetone-washed yeast biomass functional groups involved in lead biosorption. Biotechnol. Bioeng. 1997, 55, 1–10. [Google Scholar] [CrossRef]
- Painter, P.; Starsinic, M.; Coleman, M. In Fourier Transform Infrared Spectroscopy; Academic Press: New York, NY, USA, 1985; Volume 4, pp. 169–241. [Google Scholar]
- Zawadzki, J. IR spectroscopic investigations of the mechanism of oxidation of carbonaceous films with HNO3 solution. Carbon 1980, 18, 281–285. [Google Scholar] [CrossRef]
- El-Hendawy, A.A. Influence of HNO3 oxidation on the structure and adsorptive properties of corncob-based activated carbon. Carbon 2003, 41, 713–722. [Google Scholar] [CrossRef]
- Bautista-Toledo, I.; Rivera-Utrilla, J.; Ferro-Garcia, M.A.; Moreno-Castilla, C. Influence of the oxygen surface complexes of activated carbons on the adsorption of chromium ions from aqueous solutions: Effect of sodium chloride and humic acid. Carbon 1994, 32, 93–100. [Google Scholar] [CrossRef]
- Bassilakis, R.; Carangelo, R.M.; Wójtowicz, M.A. TG-FTIR analysis of biomass pyrolysis. Fuel 2001, 80, 1765–1786. [Google Scholar] [CrossRef]
- Freudenberg, K.; Nash, A.C. Constitution and Biosynthesis of Lignin; Springer: Berlin, Germany, 1968. [Google Scholar]
- Yan, Q.; Li, J.; Zhang, J.; Cai, Z. Thermal decomposition of kraft lignin under of argon gas atmospheres, hydrogen, and carbon dioxide. Polymers 2018, 10, 729. [Google Scholar] [CrossRef]
- Wang, S.; Lin, H.; Ru, B.; Sun, W.; Wang, Y.; Luo, Z. Comparison of the pyrolysis behavior of pyrolytic lignin and milled wood lignin by using TG–FTIR analysis. J. Anal. Appl. Pyrolysis 2014, 108, 78–85. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zheng, Y.; Luo, Z.; Cen, K. Mechanism study of wood lignin pyrolysis by using TG–FTIR analysis. J. Anal. Appl. Pyrolysis 2008, 82, 170–177. [Google Scholar] [CrossRef]
- Zhao, J.; Xiuwen, W.; Hu, J.; Liu, Q.; Shen, D.; Xiao, R. Thermal degradation of softwood lignin and hardwood lignin by TG-FTIR and Py-GC/MS. Polym. Degrad. Stab. 2014, 108, 133–138. [Google Scholar] [CrossRef]
- Cao, J.; Xiao, G.; Xu, X.; Shen, D.; Jin, B. Study on carbonization of lignin by TG-FTIR and high-temperature carbonization reactor. Fuel Process. Technol. 2013, 106, 41–47. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Kishimoto, T.; Uraki, Y.; Ubukata, M. Chemical synthesis of β-O-4 type artificial lignin. Org. Biomol. Chem. 2006, 4, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Babij, N.R.; McCusker, E.O.; Whiteker, G.T.; Canturk, B.; Choy, N.; Creemer, L.C.; Amicis, C.V.D.; Hewlett, N.M.; Johnson, P.L.; Knobelsdorf, J.A.; et al. NMR chemical shifts of trace impurities: Industrially preferred solvents used in process and green chemistry. Org. Process Res. Dev. 2016, 20, 661–667. [Google Scholar] [CrossRef]
- Brebu, M.; Vasile, C. Thermal degradation of lignin—A review. Cell. Chem. Technol. 2010, 44, 353–363. [Google Scholar]
- Yan, Q.; Li, J.; Zhang, X.; Wang, C.; Zhang, J.; Cai, Z. Catalytic graphitization of kraft lignin to graphene-based structures with four different transitional metals. J. Nanoparticle Res. 2018, 20, 223. [Google Scholar] [CrossRef]
Sample Availability: Samples of the graphens are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe Resources | Solid Carbon | Liquid | Gas |
|---|---|---|---|
| Raw lignin | 36.5 | 24.2 | 39.3 |
| Fe–lignin–W | 34.3 | 19.6 | 46.1 |
| Fe–lignin–M | 33.1 | 19.0 | 47.9 |
| Fe–lignin–A | 31.4 | 17.5 | 51.1 |
| Fe–lignin–T | 30.8 | 17.5 | 51.7 |
| Samples | Sg (m2 g−1) |
|---|---|
| Raw lignin | 15.8 ± 2.5 |
| Fe–lignin–W | 59.1 ± 4.6 |
| Fe–lignin–M | 85.3 ± 3.5 |
| Fe–lignin–A | 110.6 ± 5.0 |
| Fe–lignin–T | 129.2 ± 2.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Q.; Cai, Z. Effect of Solvents on Fe–Lignin Precursors for Production Graphene-Based Nanostructures. Molecules 2020, 25, 2167. https://doi.org/10.3390/molecules25092167
Yan Q, Cai Z. Effect of Solvents on Fe–Lignin Precursors for Production Graphene-Based Nanostructures. Molecules. 2020; 25(9):2167. https://doi.org/10.3390/molecules25092167
Chicago/Turabian StyleYan, Qiangu, and Zhiyong Cai. 2020. "Effect of Solvents on Fe–Lignin Precursors for Production Graphene-Based Nanostructures" Molecules 25, no. 9: 2167. https://doi.org/10.3390/molecules25092167
APA StyleYan, Q., & Cai, Z. (2020). Effect of Solvents on Fe–Lignin Precursors for Production Graphene-Based Nanostructures. Molecules, 25(9), 2167. https://doi.org/10.3390/molecules25092167