Substituent Effects on NMR Spectroscopy of 2,2-Dimethylchroman-4-one Derivatives: Experimental and Theoretical Studies



Abstract
1. Introduction
2. Results and Discussion
2.1. Experimental NMR Chemical Shifts
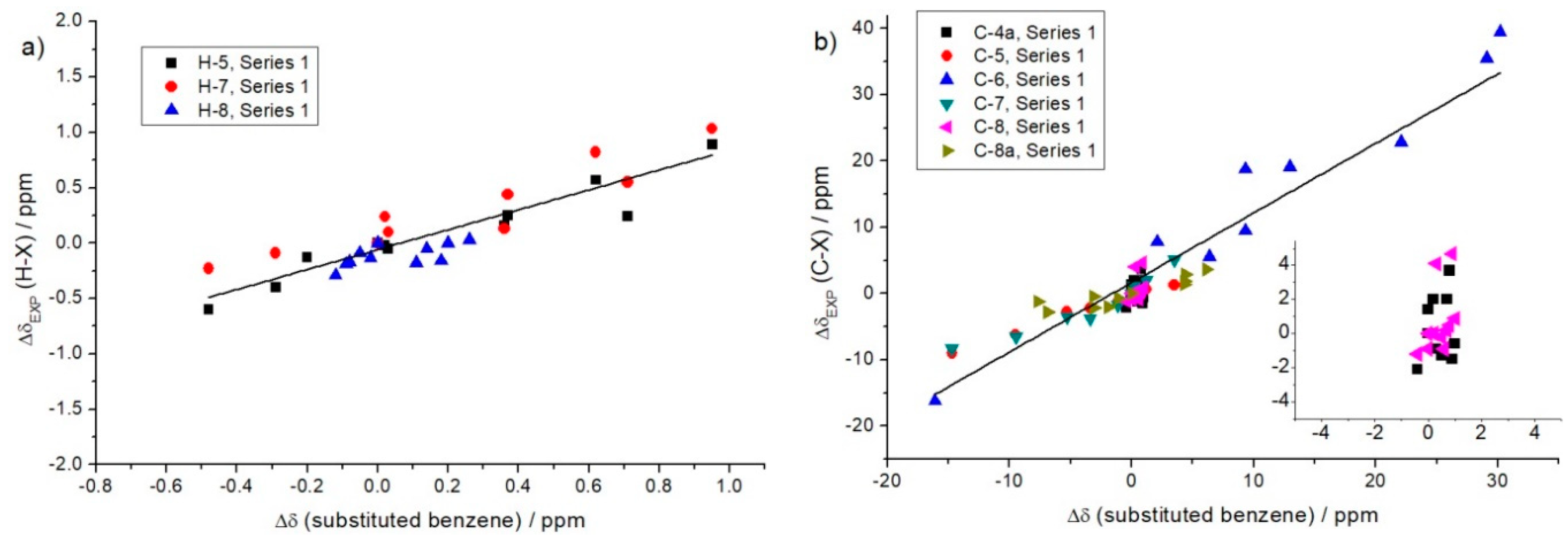
2.1.1. Lynch Correlation
2.1.2. Hammett Correlation
2.1.3. Experimental and Theoretical (DFT Calculations) Correlation
3. Materials and Methods
3.1. Spectroscopic Details
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ewing, D.F. Correlation Analysis in Chemistry: Recent Advances; Chapman, N.B., Shorter, J., Eds.; Plenum Press: New York, NY, USA, 1978. [Google Scholar]
- Hammett, L.P. Some Relations between Reaction Rates and Equilibrium Constants. Chem. Rev. 1935, 17, 125–136. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Ikezaki, A.; Ikene, T.; Nakamura, M. Electronic effects of para-substituents on the electron configuration of dicyano[meso-tetrakis(p-substituted phenyl)porphyrinato]iron(III) complexes. Inorg. Chim. Acta 2002, 335, 91–99. [Google Scholar] [CrossRef]
- Gorobets, N.Y.; Yermolayer, S.A.; Gurley, T.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Leadbeater, N.E. Difference between 1H NMR signals of primary amide protons as a simple spectral index of the amide intramolecular hydrogen bond strength. J. Phys. Org. Chem. 2012, 25, 287–295. [Google Scholar] [CrossRef]
- Bustos, C.; Alvarez-Thon, L.; Molins, E.; Moreno Villoslada, I.; Vallejos-Contreras, G.; Sánchez, C.; Zarate, X.; Mac-Leod Carey, D.; Schott, E. Tuning the molecular/electronic structure of new substituted pyrazoles: Synthesis, biological trials, theoretical approaches and Hammett correlations. J. Mol. Struct. 2018, 1171, 349–361. [Google Scholar] [CrossRef]
- Yuan, H.; Chen, P.-W.; Li, M.-Y.; Zhang, Y.; Peng, Z.-W.; Lin, W.; Paton, R.S.; Cao, C. Effects of substituents X and Y on the NMR chemical shifts of 2-(4-X phenyl)-5-Y pyrimidines. J. Mol. Struct. 2020, 1204, 127489. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; Wiley: Hoboken, NJ, USA, 2007; pp. 401–412. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry, Part. A: Structure and Mechanisms, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 335–344. [Google Scholar]
- Carroll, F.A. Perspectives on Structure and Mechanism in Organic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 1998; pp. 366–386. [Google Scholar]
- Markgraf, J.H.; Hong, L.; Richardson, D.P.; Schofield, M.H. Substituent effects on 15N and 13C NMR chemical shifts of 5-phenyl-1,3,4-oxathiazol-2-ones: A theoretical and spectroscopic study. Magn. Reson. Chem. 2007, 45, 985–988. [Google Scholar] [CrossRef]
- Lee, H.S.; Yu, J.S.; Lee, C.K. Use of Correlation of 1H and 13C Chemical Shifts of N-Arylsuccinanilic Acids, N-Arylsuccinimides, N-Arylmaleanilic Acids, and N-Arylmaleimides with the Hammett Substituent Constants for the Studies of Electronic Effects. Bull. Korean Chem. Soc. 2009, 30, 2351–2354. [Google Scholar]
- Kalyanasundaram, N.; Sakthinathan, S.P.; Suresh, R.; Kamalakkannan, D.; Joseph, S.J.; Vanangamudi, G.; Thirunarayanan, G. Spectral correlation analysis of Hammett substituent constants and biological activities of some (E)-1-(4-phenoxyphenyl)-3-phenylprop-2-en-1-ones. Int. Lett. Chem. Phys. Astron. 2014, 9, 23–47. [Google Scholar] [CrossRef]
- Raviola, C.; Ravelli, D.; Protti, S.; Albini, A.; Fagnoni, M. Conditions and Edges for the Photochemical Generation of Short-Lived Aryl Cations: A Computational Approach. Synlett 2015, 26, 471–478. [Google Scholar]
- Luo, S.; Wei, Z.; Spinney, R.; Yang, Z.; Chai, L.; Xiao, R. A novel model to predict gas–phase hydroxyl radical oxidation kinetics of polychlorinated compounds. Chemosphere 2017, 172, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Arsovski, V.M.; Bozic, B.D.; Mirkovic, J.M.; Vitnik, V.D.; Vitnik, Z.J.; Petrovic, S.D.; Uscumlic, G.S.; Mijin, D.Z. Computational and spectroscopic data correlation study of N,N′-bisarylmalonamides (Part II). J. Mol. Model. 2015, 21, 239. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, B.E. Theoretical Hammett Plot for the Gas-Phase Ionization of Benzoic Acid versus Phenol: A Computational Chemistry Lab Exercise. J. Chem. Ed. 2013, 90, 665–668. [Google Scholar] [CrossRef]
- Dean, F.M. Naturally Occuring Oxygen Ring Compounds; Butherworths: London, UK, 1963. [Google Scholar]
- Ellis, G.P.; Lockhart, I.M. Chromans and Tocopherols; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Green, G.; Evans, J.M.; Vong, A.K. Comprehensive Heterocyclic Chemistry II; McKillop, A., Ed.; Pergamon: Oxford, UK, 1996; Volume 5, pp. 469–500. [Google Scholar]
- Hodgetts, K.J.; Maragkou, K.I.; Wallace, T.W.; Wootton, C.R. Conjugate addition to 3-arylsulfinylchromones as a synthetic route to homochiral 2-substituted chromanones: Scope and limitations. Tetrahedron 2001, 57, 6793–6804. [Google Scholar] [CrossRef]
- Fridin-Saxim, M.; Pemberton, N.; Andersson, K.d.S.; Dayrager, C.; Friberg, A.; Grotli, M.; Luthman, K. Synthesis of 2-Alkyl-Substituted Chromone Derivatives Using Microwave Irradiation. J. Org. Chem. 2009, 74, 2755–2759. [Google Scholar] [CrossRef]
- Irsay, R.D. Pharmaceutical Method and Compositions Employing Substituted Chromanone Oximes and Chromanone Oxime Ethers. U.S. Patent 3678170, 18 July 1972. [Google Scholar]
- MacDonald, J.E.; Hysell, M.K.; Lui, Q.; Yu, D.; Ke, N.; Liu, G.; Li, H.Q.X.; Wong-Staal, F. Novel 3-Benzylidine and 3-Benzyl Substituted Chromanones. Patent WO2008067451, 5 June 2008. [Google Scholar]
- Takikawa, H.; Suzuki, K. Modified Chiral Triazolium Salts for Enantioselective Benzoin Cyclization of Enolizable Keto-Aldehydes: Synthesis of (+)-Sappanone B. Org. Lett. 2007, 9, 2713–2716. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Aldinger, C.E.; Beyer, T.A.; Bordner, J.; Bussolotti, D.F.; Inskeep, P.B.; Siegel, T.W. Hydantoin bioisosteres. In vivo active spiro hydroxy acetic acid aldose reductase inhibitors. J. Med. Chem. 1992, 35, 2169–2177. [Google Scholar] [CrossRef]
- Butler, J.D.; Conrad, W.E.; Lodewyk, M.W.; Fettinger, J.C.; Tantillo, D.J.; Kurth, M.J. Synthesis of Substituted Chromanones: An Organocatalytic Aldol/oxa-Michael Reaction. Org. Lett. 2010, 12, 3410–3413. [Google Scholar] [CrossRef]
- Pritchard, R.G.; Sheldrake, H.M.; Taylor, I.Z.; Wallace, T.W. Rapid stereoselective access to the tetracyclic core of puupehenone and related sponge metabolites using metal-free radical cyclisations of cyclohexenyl-substituted 3-bromochroman-4-ones. Tetrahedron Lett. 2008, 49, 4156–4159. [Google Scholar] [CrossRef]
- Meng, L.-G.; Liu, H.-F.; Wei, J.-L.; Gong, S.-N.; Xue, S. One-pot reaction of ortho-acylphenols and terminal alkynoates for synthesis of 2-alkyl-substituted chromanones. Tetrahedron Lett. 2010, 51, 1748–1750. [Google Scholar] [CrossRef]
- Iguchi, D.; Erra-Balsells, R.; Bonesi, S.M. Expeditious photochemical reaction toward the preparation of substituted chroman-4-ones. Tetrahedron Lett. 2014, 55, 4653–4656. [Google Scholar] [CrossRef]
- Morales, P.; Azofra, L.M.; Cumella, J.; Hernandez-Folgado, L.; Roldán, M.; Alkorta, I.; Jagerovic, N. Preparation of 2,2-dimethylchroman-4-ones from 5-alkyl-substituted resorcinols: Microwave-assisted synthesis and theoretical calculations. ARKIVOC 2014, 2014, 319–332. [Google Scholar]
- Iguchi, D. Fotoquímica de Algunos Ésteres y Amidas en Medio Homogéneo y Heterogéneo. Ph.D. Thesis, University of Buenos Aires, Buenos Aires, Argentina, 2015. [Google Scholar]
- Lynch, B.M. Proportionality relationships in the carbon-13 nuclear magnetic resonance spectra of para-disubstituted benzenes: A new interpretation of non-additive behavior. Can. J. Chem. 1977, 55, 541–547. [Google Scholar] [CrossRef]
- Spiesecke, H.; Schneider, W.G. Substituent Effects on the C13 and H1 Chemical Shifts in Monosubstituted Benzenes. J. Chem. Phys. 1961, 35, 731–738. [Google Scholar] [CrossRef]
- Maciel, G.E.; Natterstadt, J.J. Study of 13C Chemical Shifts in Substituted Benzenes. J. Chem. Phys. 1965, 42, 2427–2435. [Google Scholar] [CrossRef]
- Nelson, G.L.; Levy, G.C.; Cargioli, J.D. Solvent effects in carbon-13 nuclear magnetic resonance. Electronic perturbation of aromatic systems. J. Am. Chem. Soc. 1972, 94, 3089–3094. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; de Lima, R.A.; delle Monache, F. 13C nuclear magnetic resonance spectroscopy of 6- and 7-substituted coumarins. Correlation with Hammett constants. J. Chem. Soc. Perkin Trans. 1979, 2, 435–437. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds 1a–1k, 2a–2f and 3a–3f are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lynch correlations of 1H chemical shifts: ΔδEXP = a···Δδ(Benzene) + b | ||||
| Compounds | a | b | R2 | Hydrogen atoms |
| Series 1 | 0.90 | −0.06 | 0.80 | H-5; H-7; H-8 |
| Series 2 | 0.95 | −0.04 | 0.94 | H-5; H-6; H-8 |
| Series 3 | 0.94 | 0.00 | 0.98 | H-6; H-7; H-8 |
| Lynch correlations of 13C chemical shifts: ΔδEXP = a···Δδ(Benzene) + b | ||||
| Compounds | a | b | R2 | Carbon atoms |
| Series 1 | 1.05 | 1.7 | 0.92 | C-5; C-6; C-7; C-8a |
| Series 2 | 0.94 | −1.9 | 0.95 | C-4a; C-6; C-7; C-8 |
| Series 3 | 1.05 | −0.1 | 0.90 | C-4a; C-5; C-6; C-8 |
| Hammett correlations for 13C chemical shifts in para-positions (σp+): δ(C-X) = ρ···σp+ + δ0 | ||||
| Compounds | ρ | δ0 | R2 | Carbon atom |
| Series 1 | 2.8 | 160.0 | 0.41 | C-8a |
| Series 2 | 5.1 | 119.6 | 0.93 | C-4a |
| Series 3 | 4.1 | 118.8 | 0.62 | C-8 |
| Hammett correlations for 13C chemical shifts in meta-positions (σm): δ(C-X) = ρ···σm + δ0 | ||||
| Compounds | ρ | δ0 | R2 | Carbon atom |
| Series 1 | 6.6 | 119.9 | 0.77 | C-4a |
| No correlation | C-8 | |||
| Series 2 | No correlation | C-5 | ||
| 4.7 | 160.1 | 0.45 | C-8a | |
| Series 3 | No correlation | C-7 | ||
| No correlation | C-8a | |||
| Hammett correlations for 13C chemical shifts in meta-positions (σI): δ(C-X) = ρ···σI + δ0 | ||||
| Compounds | ρ | δ0 | R2 | Carbon atom |
| Series 1 | 5.8 | 120.0 | 0.49 | C-4a |
| No correlation | C-8 | |||
| Series 2 | No correlation | C-5 | ||
| 5.6 | 159.6 | 0.40 | C-8a | |
| Series 3 | No correlation | C-7 | ||
| No correlation | C-8a | |||
| δTHEOR = a···δEXP + b | ||||
| Hydrogen atoms type a | a | b | R2 | Comments |
| Ortho | 1.16 | −0.74 | 0.95 | Series 1 (H-5, H-7); Series 2 (H-6, H-8); Series 3 (H-6) |
| Meta | 1.08 | −0.23 | 0.96 | Series 1 (H-8); Series 2 (H-5); Series 3 (H-7) |
| Para | 0.96 | 0.54 | 0.98 | Series 3 (H-8) |
| Methylene | 1.17 | −0.42 | 0.71 | H-3 of all series |
| Carbon atoms type | a | b | R2 | Comments |
| Ipso | 0.88 | 23.93 | 0.75 | Series 1 (C-6); Series 2 (C-7); Series 3 (C-5) |
| Ortho | 1.04 | −0.71 | 0.93 | Series 2 (C-6, C-8); Series 3 (C-6, C-4a) |
| No correlation | Series 1 (C-5) | |||
| No correlation | Series 1 (C-7) | |||
| Meta | 1.11 | −8.78 | 0.99 | Series 1 (C-4a, C-8); Series 2 (C-5, C-8a); Series 3 (C-7, C-8a) |
| Para | 1.07 | −2.25 | 0.99 | Series 1 (C-8a); Series 2 (C-4a); Series 3 (C-8) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iguchi, D.; Ravelli, D.; Erra-Balsells, R.; Bonesi, S.M. Substituent Effects on NMR Spectroscopy of 2,2-Dimethylchroman-4-one Derivatives: Experimental and Theoretical Studies. Molecules 2020, 25, 2061. https://doi.org/10.3390/molecules25092061
Iguchi D, Ravelli D, Erra-Balsells R, Bonesi SM. Substituent Effects on NMR Spectroscopy of 2,2-Dimethylchroman-4-one Derivatives: Experimental and Theoretical Studies. Molecules. 2020; 25(9):2061. https://doi.org/10.3390/molecules25092061
Chicago/Turabian StyleIguchi, Daniela, Davide Ravelli, Rosa Erra-Balsells, and Sergio M. Bonesi. 2020. "Substituent Effects on NMR Spectroscopy of 2,2-Dimethylchroman-4-one Derivatives: Experimental and Theoretical Studies" Molecules 25, no. 9: 2061. https://doi.org/10.3390/molecules25092061
APA StyleIguchi, D., Ravelli, D., Erra-Balsells, R., & Bonesi, S. M. (2020). Substituent Effects on NMR Spectroscopy of 2,2-Dimethylchroman-4-one Derivatives: Experimental and Theoretical Studies. Molecules, 25(9), 2061. https://doi.org/10.3390/molecules25092061