
Synthesis and Cytotoxic Analysis of Novel Myrtenyl Grafted Pseudo-Peptides Revealed Potential Candidates for Anticancer Therapy

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
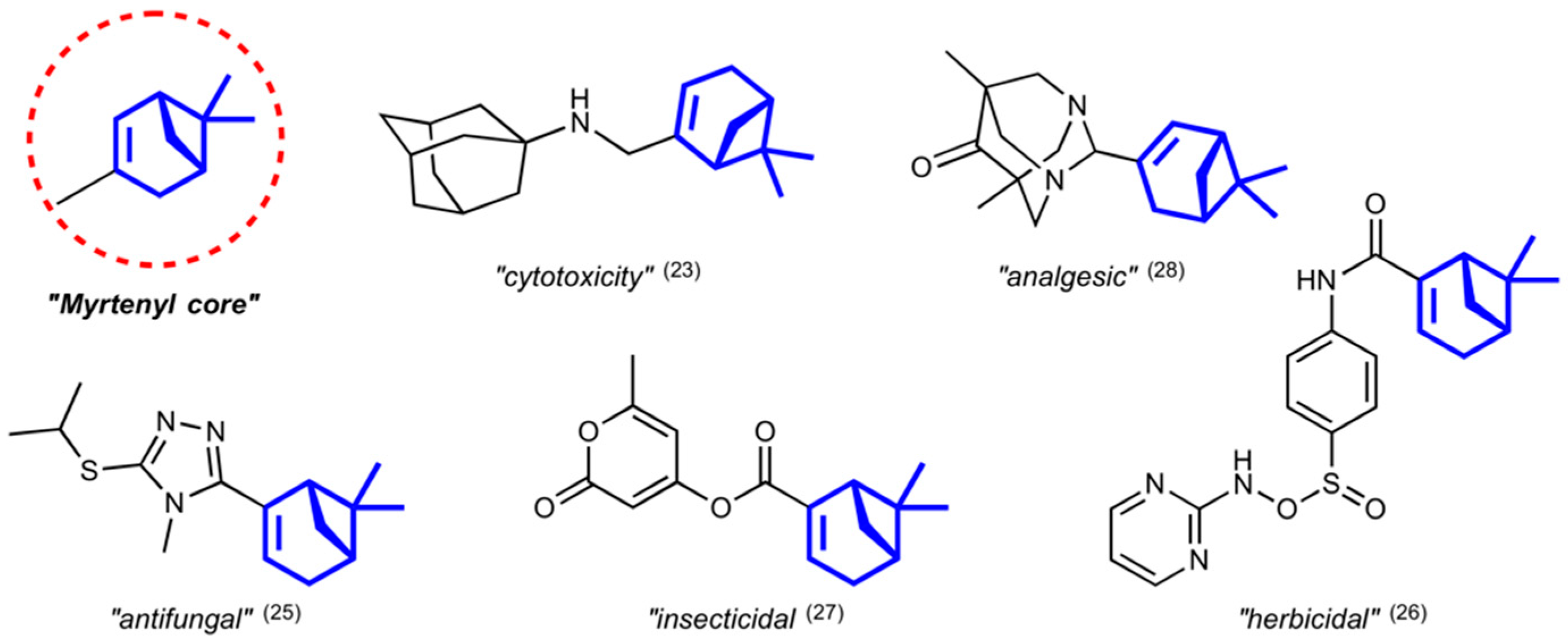
1. Introduction
2. Results
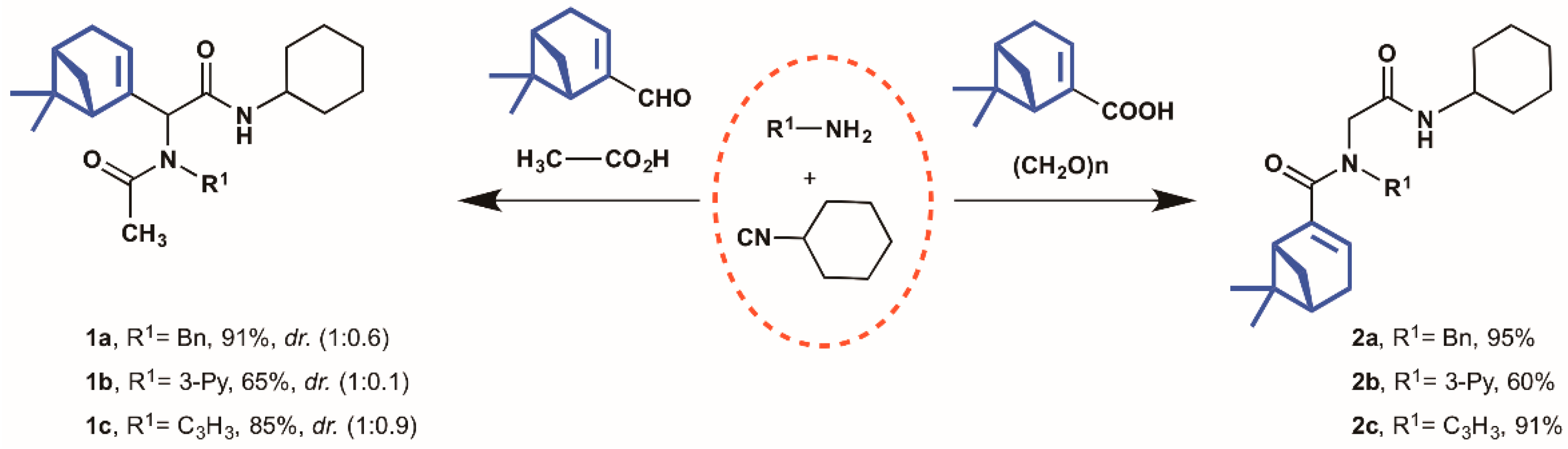
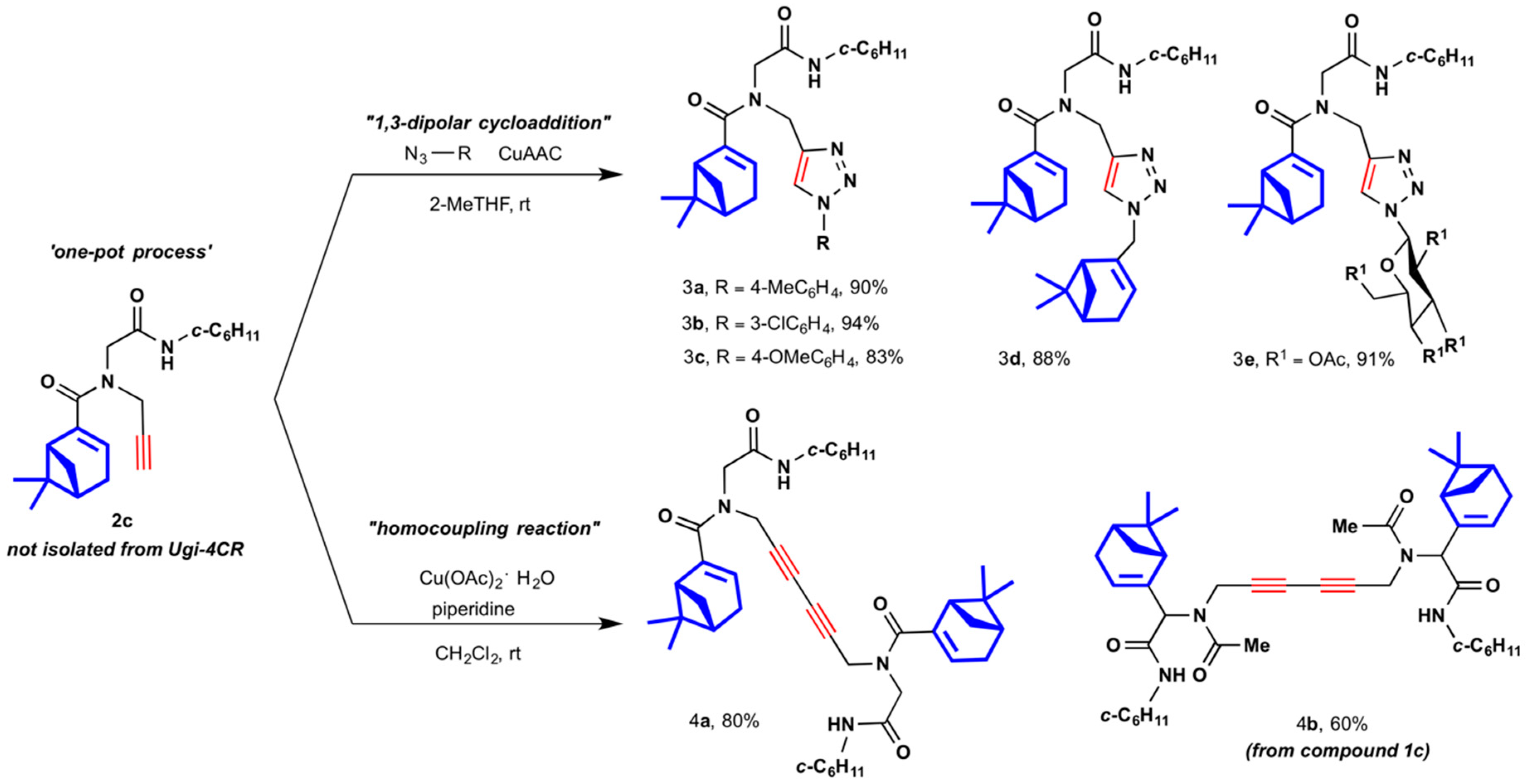
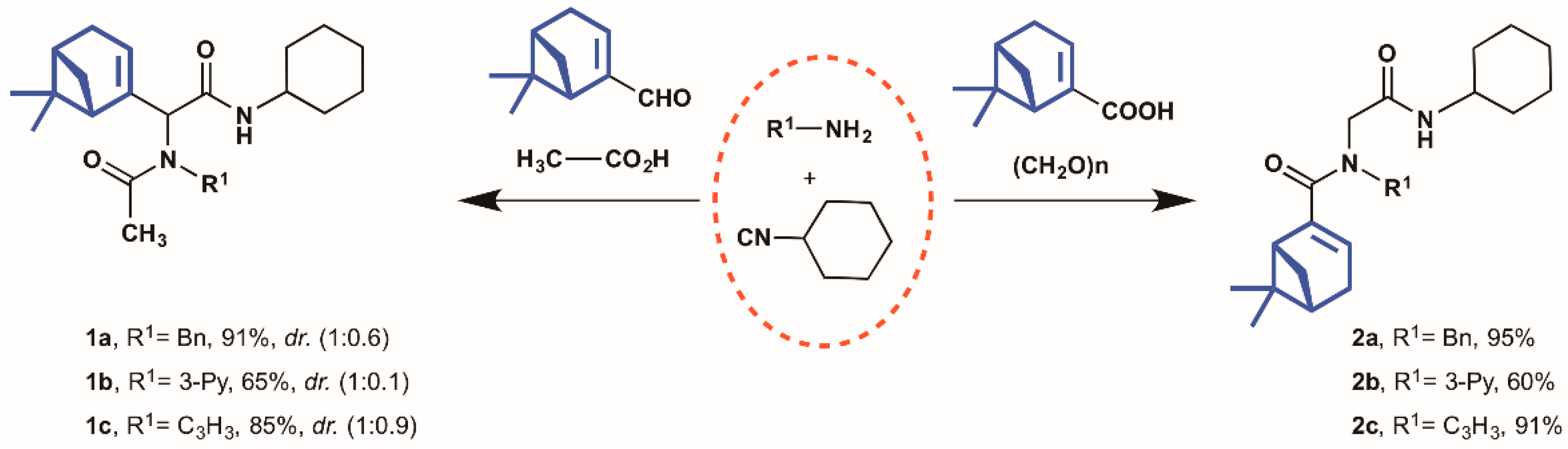
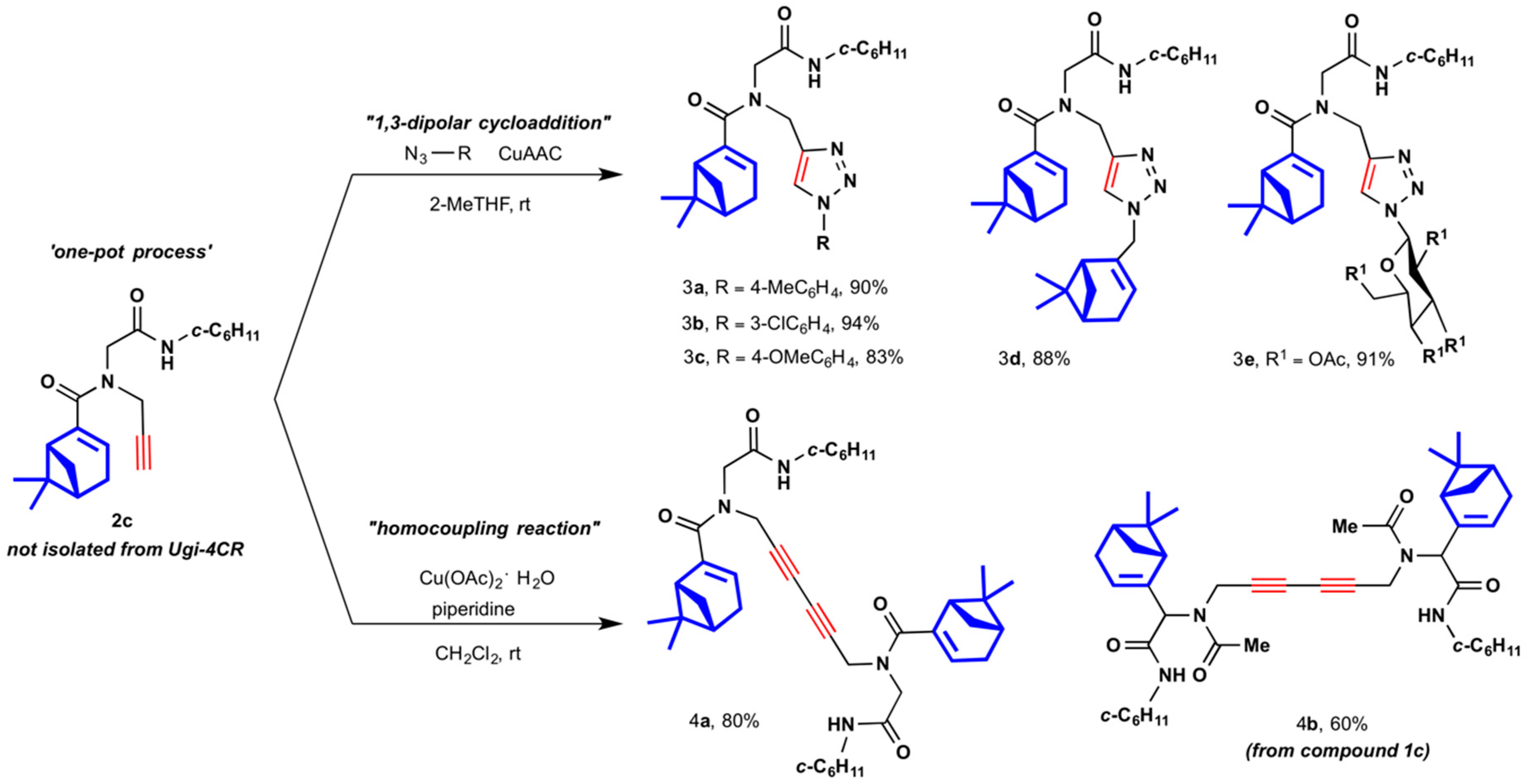
2.1. Synthesis of Myrtenyl Derivativess
2.2. Biological Assessment
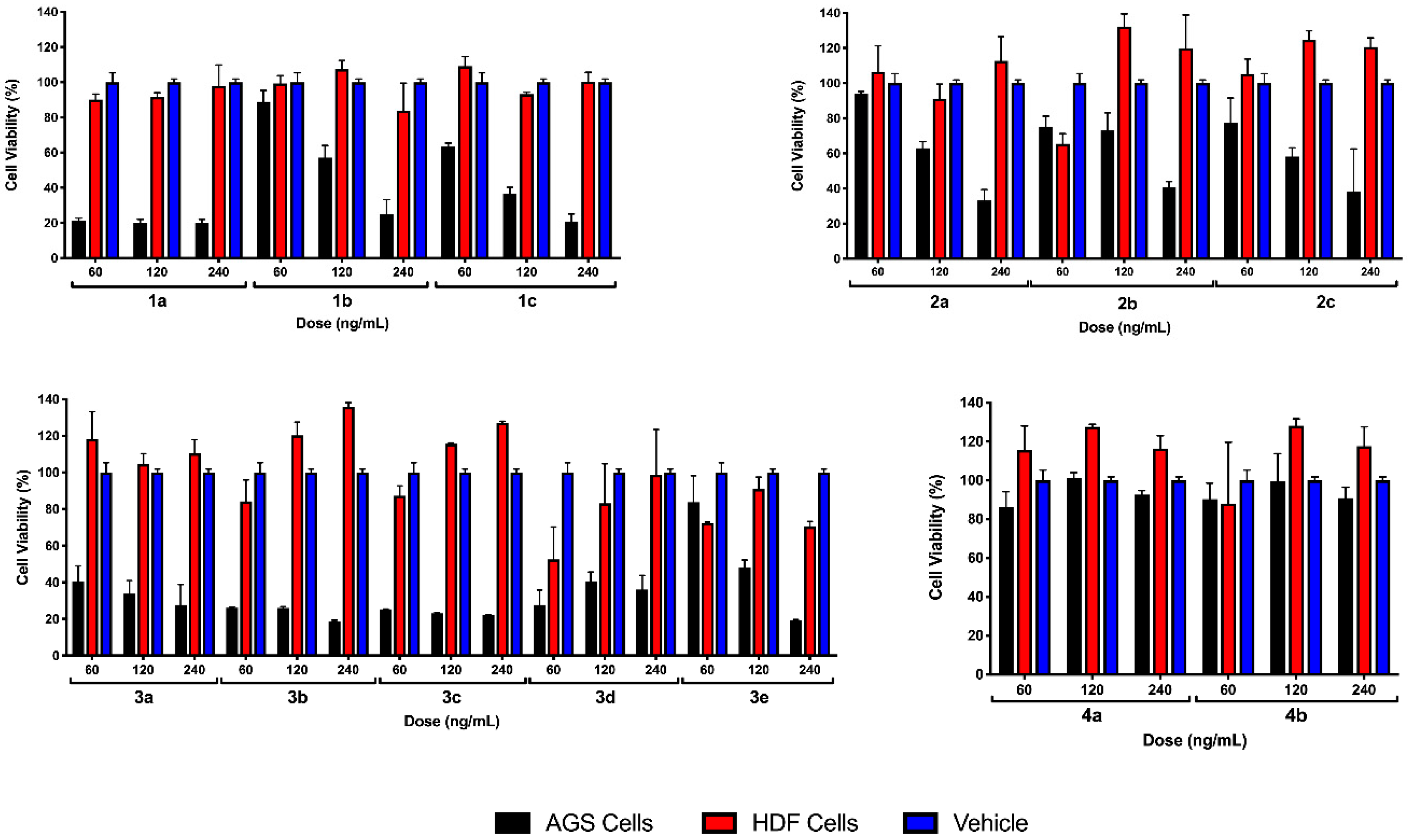
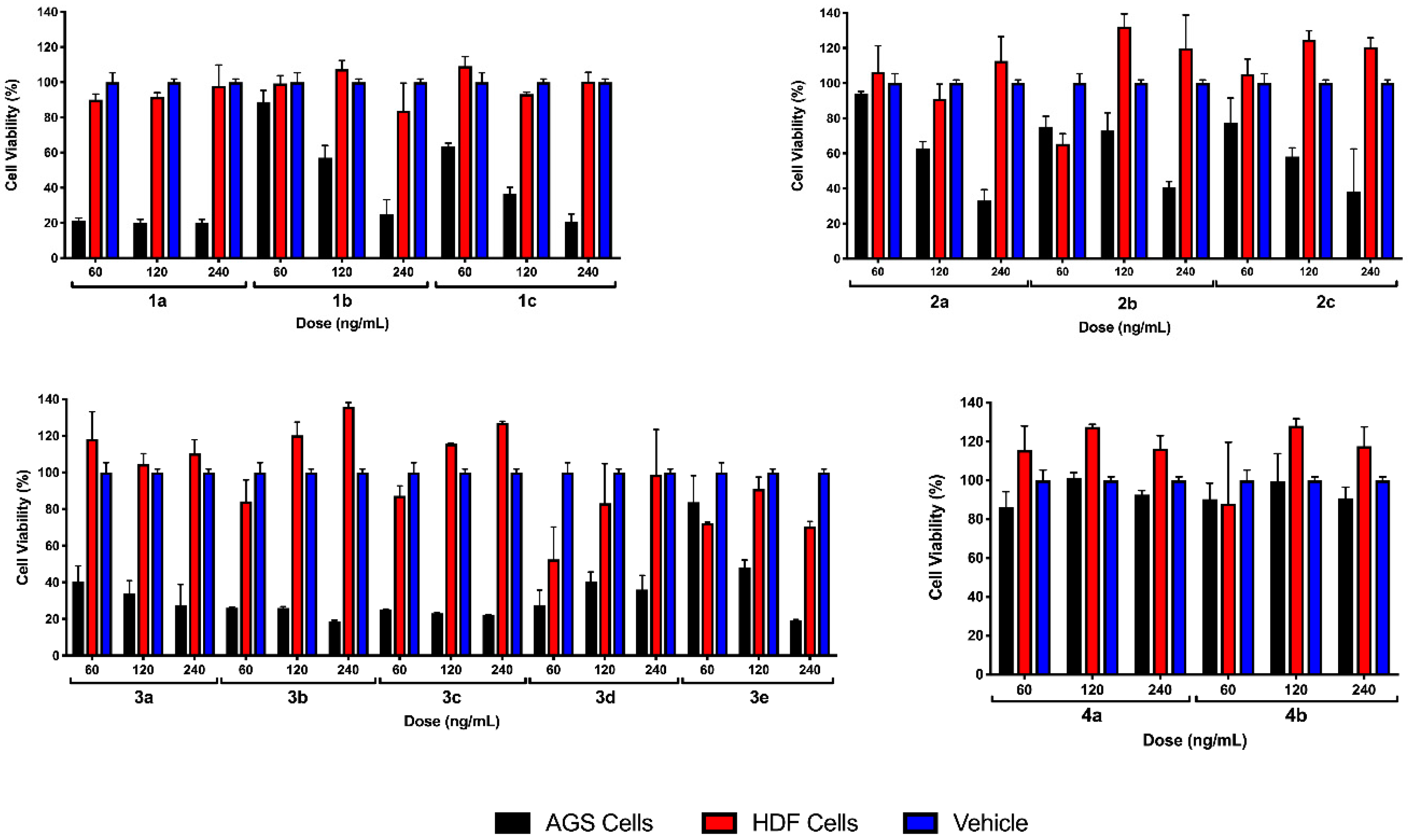
2.2.1. Preliminary Testing
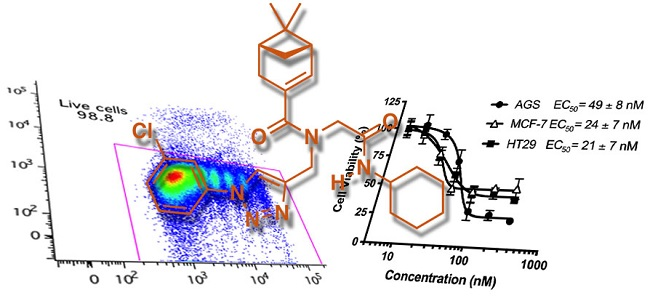
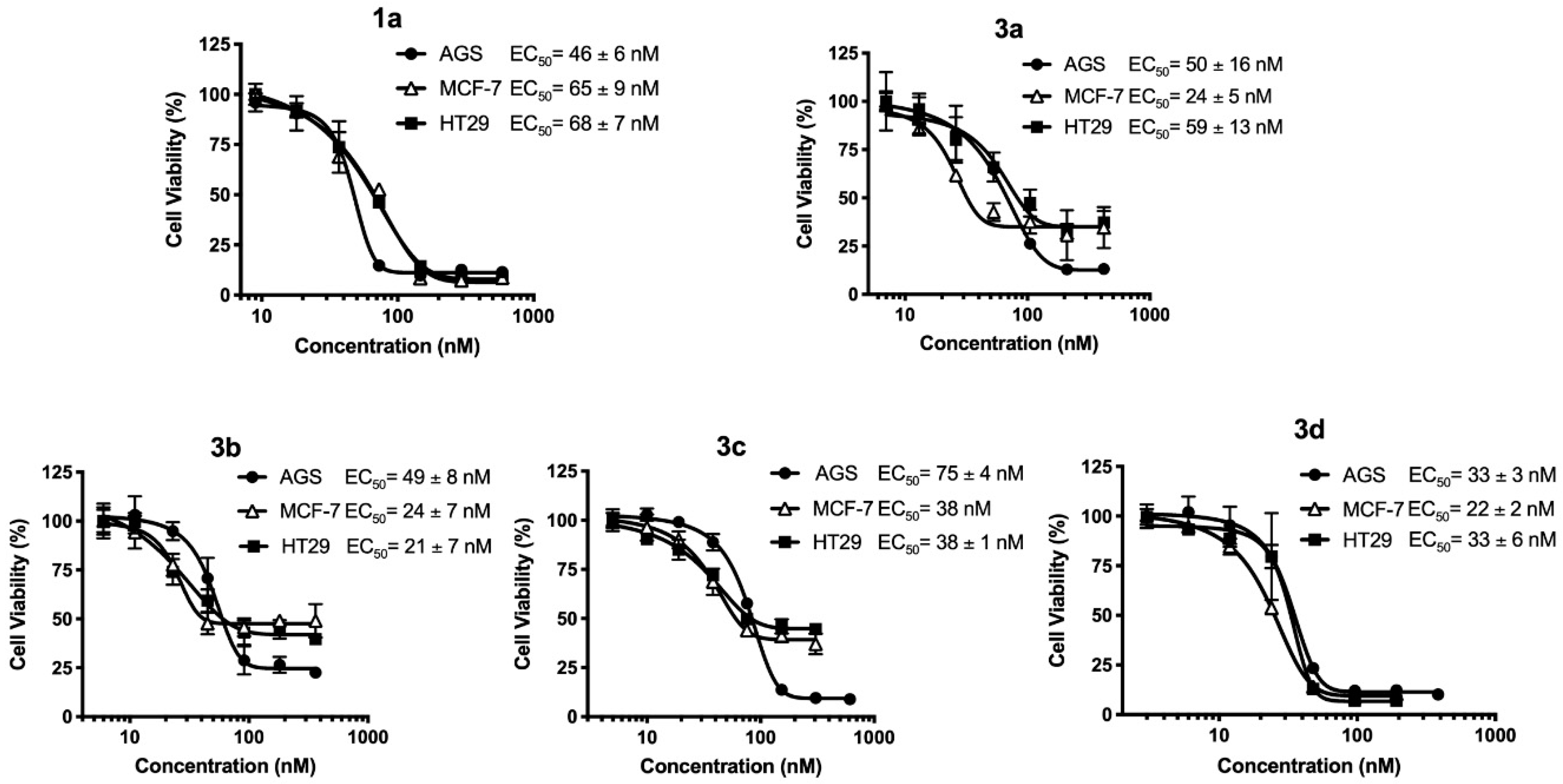
2.2.2. EC50 Determination on Myrtenyl Derivatives
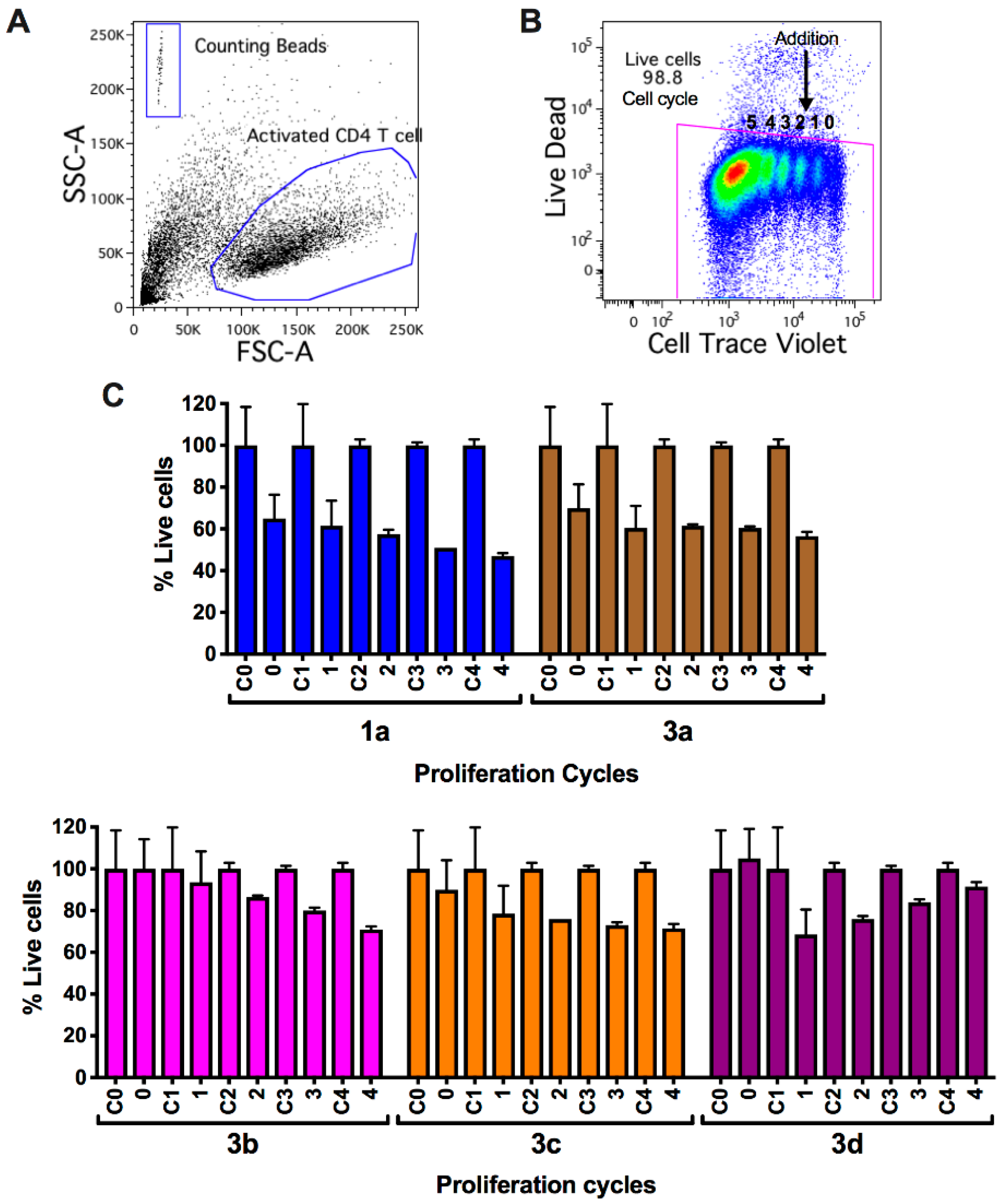
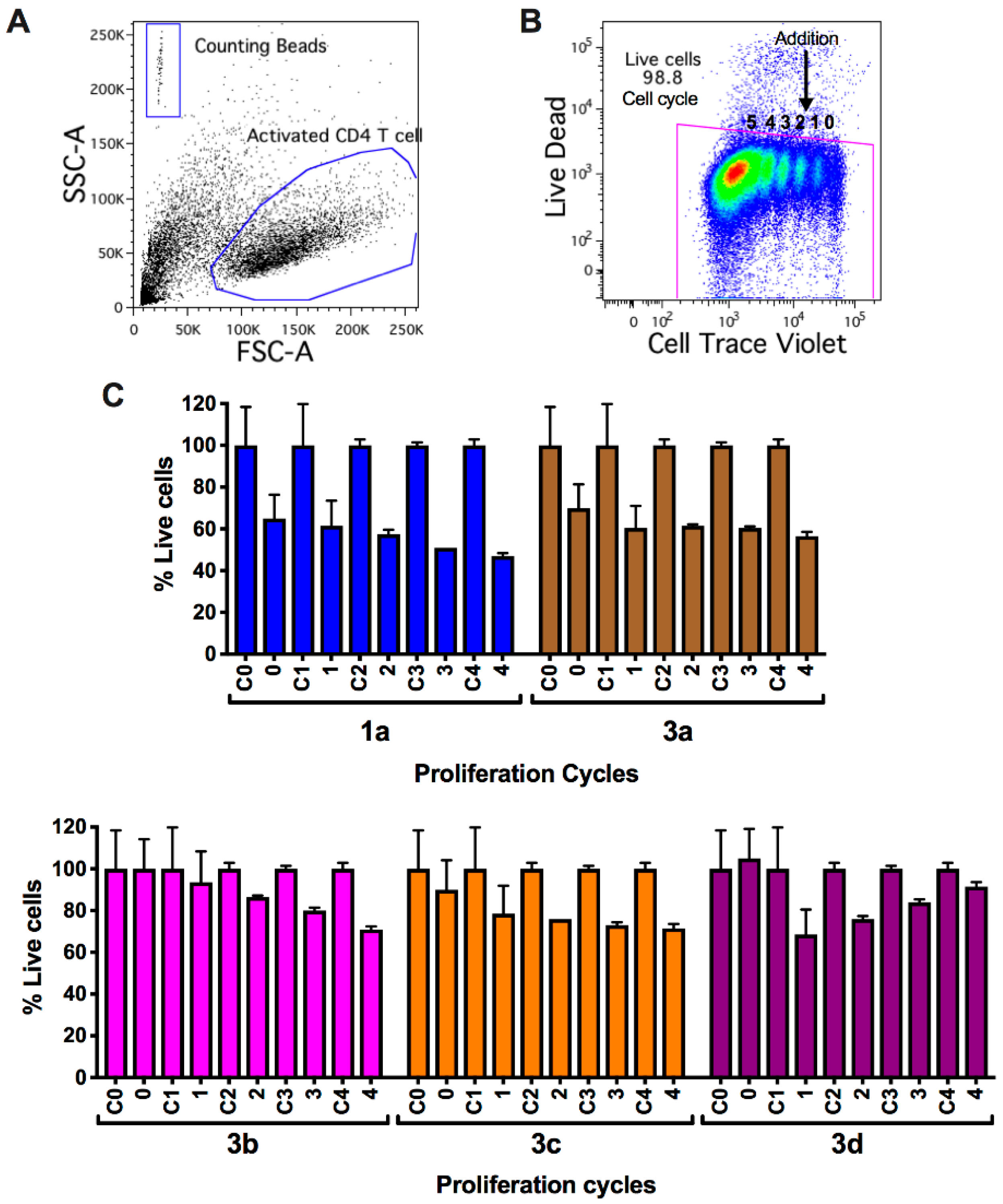
2.2.3. Viability and Cell Count Analysis of Myrtenyl Derivatives on CD4+ T Lymphocytes in Progressive Proliferative Cycles
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General
4.1.2. General Multicomponent Procedure (1a–2c)
4.1.3. One-Pot Multicomponent-Click General Procedure (3a–3e)
4.1.4. General Multicomponent Sequential Homocoupling Procedure (4a and 4b)
4.2. Biological Activity
4.2.1. Cell Culture
4.2.2. Cell Cytotoxicity Assay
4.2.3. Cell Cycle and Proliferation Assays
4.2.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mehta, K.; Gandhi, V.; Pathak, S.; Aggarwal, B.B.; Grover, R.K. Multi-targeted approach to cancer treatment: An international translational cancer research symposium. Anticancer Res. 2014, 34, 6791–6795. [Google Scholar] [PubMed]
- Bailon-Moscoso, N.; Romero-Benavides, J.C.; Ostrosky-Wegman, P. Development of anticancer drugs based on the hallmarks of tumor cells. Tumour Biol. 2014, 35, 3981–3995. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ja’fari, A.H.; Vila, R.; Freixa, B.; Tomi, F.; Casanova, J.; Costa, J.; Canigueral, S. Composition and antifungal activity of the essential oil from the rhizome and roots of Ferula hermonis. Phytochemistry 2011, 72, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Setzer, W.N.; Vogler, B.; Schmidt, J.M.; Leahy, J.G.; Rives, R. Antimicrobial activity of Artemisia douglasiana leaf essential oil. Fitoterapia 2004, 75, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Tellez, M.R.; Khan, I.A.; Kobaisy, M.; Schrader, K.K.; Dayan, F.E.; Osbrink, W. Composition of the essential oil of Lepidium meyenii (Walp). Phytochemistry 2002, 61, 149–155. [Google Scholar] [CrossRef]
- Bicas, J.L.; Dionisio, A.P.; Pastore, G.M. Bio-oxidation of terpenes: An approach for the flavor industry. Chem. Rev. 2009, 109, 4518–4531. [Google Scholar] [CrossRef]
- You, K.; Yin, D.; Mao, L.; Liu, P.; Luo, H.a. Catalytic Modulation on the Regioselectivity of the Photosensitized Oxidation of α-Pinene with Molecular Oxygen under Sodium Lamp Irradiation. Chinese J. Catal. 2011, 32, 1610–1616. [Google Scholar] [CrossRef]
- Becerra-Martínez, E.; Ayala-Mata, F.; Velázquez-Ponce, P.; Medina, M.E.; Jiménez-Vazquez, H.A.; Joseph-Nathan, P.; Zepeda, L.G. Nucleophilic additions on acetyldioxanes derived from (−)-(1R)-myrtenal used as chiral auxiliaries: Substituent effects on the stereochemical outcome. Tetrahedron Asymmetry 2017, 28, 1350–1358. [Google Scholar] [CrossRef]
- Chacón-García, L.; Lagunas-Rivera, S.; Pérez-Estrada, S.; Elena Vargas-Díaz, M.; Joseph-Nathan, P.; Tamariz, J.n.; Zepeda, L.G. New S,O-acetals from (1R)-(−)-myrtenal as chiral auxiliaries in nucleophilic additions. Tetrahedron Lett. 2004, 45, 2141–2145. [Google Scholar] [CrossRef]
- Vargas-Diaz, M.E.; Joseph-Nathan, P.; Tamariz, J.; Zepeda, L.G. Synthesis of a new (1R)-(−)-myrtenal-derived dioxadithiadodecacycle and its use as an efficient chiral auxiliary. Org. Lett. 2007, 9, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Díaz, M.E.; Mendoza-Figueroa, H.L.; Fragoso-Vázquez, M.J.; Ayala-Mata, F.; Joseph-Nathan, P.; Zepeda, L.G. Synthesis of acyldodecaheterocycles derived from (1R)-(–)-myrtenal and evaluation as chiral auxiliaries. Tetrahedron Asymmetry 2012, 23, 1588–1595. [Google Scholar] [CrossRef]
- Kamchonwongpaisan, S.; Nilanonta, C.; Tarnchompoo, B.; Thebtaranonth, C.; Thebtaranonth, Y.; Yuthavong, Y.; Kongsaeree, P.; Clardy, J. An antimalarial peroxide from Amomum krervanh Pierre. Tetrahedron Lett. 1995, 36, 1821–1824. [Google Scholar] [CrossRef]
- Kaufmann, D.; Dogra, A.K.; Wink, M. Myrtenal inhibits acetylcholinesterase, a known Alzheimer target. J. Pharm. Pharmacol. 2011, 63, 1368–1371. [Google Scholar] [CrossRef]
- Lokeshkumar, B.; Thamaraiselvan, R.; Mathiwos, D.; Balasubramanian, M.P. Anti-inflammatory effect of myrtenal against cytokines in experimental colon cancer. Aust. J. Sci. Technol. 2017, 1, 11–16. [Google Scholar]
- Babu, L.H.; Perumal, S.; Balasubramanian, M.P. Myrtenal, a natural monoterpene, down-regulates TNF-alpha expression and suppresses carcinogen-induced hepatocellular carcinoma in rats. Mol. Cell Biochem. 2012, 369, 183–193. [Google Scholar] [CrossRef]
- Baliga, M.S.; Jimmy, R.; Thilakchand, K.R.; Sunitha, V.; Bhat, N.R.; Saldanha, E.; Rao, S.; Rao, P.; Arora, R.; Palatty, P.L. Ocimum sanctum L (Holy Basil or Tulsi) and its phytochemicals in the prevention and treatment of cancer. Nutr. Cancer 2013, 65 (Suppl. 1), 26–35. [Google Scholar] [CrossRef]
- Hari Babu, L.; Perumal, S.; Balasubramanian, M.P. Myrtenal attenuates diethylnitrosamine-induced hepatocellular carcinoma in rats by stabilizing intrinsic antioxidants and modulating apoptotic and anti-apoptotic cascades. Cell. Oncol. 2012, 35, 269–283. [Google Scholar] [CrossRef]
- Lokeshkumar, B.; Sathishkumar, V.; Nandakumar, N.; Rengarajan, T.; Madankumar, A.; Balasubramanian, M.P. Anti-Oxidative Effect of Myrtenal in Prevention and Treatment of Colon Cancer Induced by 1, 2-Dimethyl Hydrazine (DMH) in Experimental Animals. Biomol. Ther. (Seoul) 2015, 23, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Martins, B.X.; Arruda, R.F.; Costa, G.A.; Jerdy, H.; de Souza, S.B.; Santos, J.M.; de Freitas, W.R.; Kanashiro, M.M.; de Carvalho, E.C.Q.; Sant’Anna, N.F.; et al. Myrtenal-induced V-ATPase inhibition—A toxicity mechanism behind tumor cell death and suppressed migration and invasion in melanoma. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1–12. [Google Scholar] [CrossRef]
- Ayyasamy, R.; Leelavinothan, P. Myrtenal alleviates hyperglycaemia, hyperlipidaemia and improves pancreatic insulin level in STZ-induced diabetic rats. Pharm. Biol. 2016, 54, 2521–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, A.; Pari, L.; Chandramohan, R.; Sheikh, B.A. Histopathological findings of the pancreas, liver, and carbohydrate metabolizing enzymes in STZ-induced diabetic rats improved by administration of myrtenal. J. Physiol. Biochem. 2014, 70, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Suslov, E.V.; Ponomarev, K.Y.; Rogachev, A.D.; Pokrovsky, M.A.; Pokrovsky, A.G.; Pykhtina, M.B.; Beklemishev, A.B.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F. Compounds Combining Aminoadamantane and Monoterpene Moieties: Cytotoxicity and Mutagenic Effects. Med. Chem. 2015, 11, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sintim, H.O. Dialkylamino-2,4-dihydroxybenzoic acids as easily synthesized analogues of platensimycin and platencin with comparable antibacterial properties. Chemistry 2011, 17, 3352–3357. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.S.; Duan, W.G.; Yang, L.X.; Huang, M.; Lei, F.H. Synthesis and Antifungal Activity of Novel Myrtenal-Based 4-Methyl-1,2,4-triazole-thioethers. Molecules 2017, 22, 193. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Duan, W.; Liu, H.; Ma, Y.; Lei, F. Synthesis and Bioactivity of N-(4-(N′-Substituted Sulfamoyl)Phenyl)Myrtenamides Containing a Heterocycle. Chem. Nat. Compd. 2018, 54, 56–62. [Google Scholar] [CrossRef]
- Tachibana, S.; Ohno, Y.; Fujihara, Y.; Okada, Y.; Sugiura, M.; Takagi, S.; Nomura, M. Synthesis and Physiological Activities of Monoterpene Carboxylic Acid Esters with Pyrones. J. Oleo Sci. 2006, 55, 181–189. [Google Scholar] [CrossRef]
- Ponomarev, K.; Pavlova, A.; Suslov, E.; Ardashov, O.; Korchagina, D.; Nefedov, A.; Tolstikova, T.y.; Volcho, K.; Salakhutdinov, N. Synthesis and analgesic activity of new compounds combining azaadamantane and monoterpene moieties. Med. Chem. Res. 2015, 24, 4146–4156. [Google Scholar] [CrossRef]
- Hernandez-Vazquez, E.; Chavez-Riveros, A.; Romo-Perez, A.; Ramirez-Apan, M.T.; Chavez-Blanco, A.D.; Morales-Barcenas, R.; Duenas-Gonzalez, A.; Miranda, L.D. Cytotoxic Activity and Structure-Activity Relationship of Triazole-Containing Bis(Aryl Ether) Macrocycles. ChemMedChem 2018, 13, 1193–1209. [Google Scholar] [CrossRef] [Green Version]
- Ingold, M.; Colella, L.; Hernandez, P.; Batthyany, C.; Tejedor, D.; Puerta, A.; Garcia-Tellado, F.; Padron, J.M.; Porcal, W.; Lopez, G.V. A Focused Library of NO-Donor Compounds with Potent Antiproliferative Activity Based on Green Multicomponent Reactions. ChemMedChem 2019, 14, 1669–1683. [Google Scholar] [CrossRef] [Green Version]
- Marcaccini, S.; Torroba, T. The use of the Ugi four-component condensation. Nat. Protoc. 2007, 2, 632–639. [Google Scholar] [CrossRef]
- Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C. Versuche mit Isonitrilen. Angew. Chem. 1959, 71, 386–388. [Google Scholar] [CrossRef]
- Domling, A.; Achatz, S.; Beck, B. Novel anti-tuberculosis agents from MCR libraries. Bioorg. Med. Chem. Lett. 2007, 17, 5483–5486. [Google Scholar] [CrossRef] [PubMed]
- Musonda, C.C.; Taylor, D.; Lehman, J.; Gut, J.; Rosenthal, P.J.; Chibale, K. Application of multi-component reactions to antimalarial drug discovery. Part 1: Parallel synthesis and antiplasmodial activity of new 4-aminoquinoline Ugi adducts. Bioorg. Med. Chem. Lett. 2004, 14, 3901–3905. [Google Scholar] [CrossRef] [PubMed]
- Nayyab, S.; O’Connor, M.; Brewster, J.; Gravier, J.; Jamieson, M.; Magno, E.; Miller, R.D.; Phelan, D.; Roohani, K.; Williard, P.; et al. Diamide Inhibitors of the Bacillus subtilis N-Acetylglucosaminidase LytG That Exhibit Antibacterial Activity. ACS Infect. Dis. 2017, 3, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Pando, O.; Stark, S.; Denkert, A.; Porzel, A.; Preusentanz, R.; Wessjohann, L.A. The Multiple Multicomponent Approach to Natural Product Mimics: Tubugis, N-Substituted Anticancer Peptides with Picomolar Activity. J. Am. Chem. Soc. 2011, 133, 7692–7695. [Google Scholar] [CrossRef]
- Richter, H.G.; Benson, G.M.; Blum, D.; Chaput, E.; Feng, S.; Gardes, C.; Grether, U.; Hartman, P.; Kuhn, B.; Martin, R.E.; et al. Discovery of novel and orally active FXR agonists for the potential treatment of dyslipidemia & diabetes. Bioorg. Med. Chem. Lett. 2011, 21, 191–194. [Google Scholar]
- Cioc, R.C.; Ruijter, E.; Orru, R.V.A. Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 2014, 16, 2958–2975. [Google Scholar] [CrossRef]
- Hein, C.D.; Liu, X.M.; Wang, D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm. Res. 2008, 25, 2216–2230. [Google Scholar] [CrossRef] [Green Version]
- Nwe, K.; Brechbiel, M.W. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother. Radiopharm. 2009, 24, 289–302. [Google Scholar] [CrossRef]
- Poonthiyil, V.; Lindhorst, T.K.; Golovko, V.B.; Fairbanks, A.J. Recent applications of click chemistry for the functionalization of gold nanoparticles and their conversion to glyco-gold nanoparticles. Beilstein J. Org. Chem. 2018, 14, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Naumann, K. Influence of chlorine substituents on biological activity of chemicals. J. Prakt. Chem. 1999, 341, 417–435. [Google Scholar] [CrossRef]
- Mendez, Y.; Perez-Labrada, K.; Gonzalez-Bacerio, J.; Valdes, G.; de los Chavez, M.A.; Osuna, J.; Charli, J.L.; Pascual, I.; Rivera, D.G. Combinatorial multicomponent access to natural-products-inspired peptidomimetics: Discovery of selective inhibitors of microbial metallo-aminopeptidases. ChemMedChem 2014, 9, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–c, 2a–c, 3a–e and 4a–b are available from the authors. |


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Concepción, O.; Belmar, J.; F. de la Torre, A.; M. Muñiz, F.; Pertino, M.W.; Alarcón, B.; Ormazabal, V.; Nova-Lamperti, E.; Zúñiga, F.A.; Jiménez, C.A. Synthesis and Cytotoxic Analysis of Novel Myrtenyl Grafted Pseudo-Peptides Revealed Potential Candidates for Anticancer Therapy. Molecules 2020, 25, 1911. https://doi.org/10.3390/molecules25081911
Concepción O, Belmar J, F. de la Torre A, M. Muñiz F, Pertino MW, Alarcón B, Ormazabal V, Nova-Lamperti E, Zúñiga FA, Jiménez CA. Synthesis and Cytotoxic Analysis of Novel Myrtenyl Grafted Pseudo-Peptides Revealed Potential Candidates for Anticancer Therapy. Molecules. 2020; 25(8):1911. https://doi.org/10.3390/molecules25081911
Chicago/Turabian StyleConcepción, Odette, Julio Belmar, Alexander F. de la Torre, Francisco M. Muñiz, Mariano W. Pertino, Barbara Alarcón, Valeska Ormazabal, Estefania Nova-Lamperti, Felipe A. Zúñiga, and Claudio A. Jiménez. 2020. "Synthesis and Cytotoxic Analysis of Novel Myrtenyl Grafted Pseudo-Peptides Revealed Potential Candidates for Anticancer Therapy" Molecules 25, no. 8: 1911. https://doi.org/10.3390/molecules25081911
APA StyleConcepción, O., Belmar, J., F. de la Torre, A., M. Muñiz, F., Pertino, M. W., Alarcón, B., Ormazabal, V., Nova-Lamperti, E., Zúñiga, F. A., & Jiménez, C. A. (2020). Synthesis and Cytotoxic Analysis of Novel Myrtenyl Grafted Pseudo-Peptides Revealed Potential Candidates for Anticancer Therapy. Molecules, 25(8), 1911. https://doi.org/10.3390/molecules25081911