Characterization of Ginkgo biloba Leaf Flavonoids as Neuroexocytosis Regulators

 , , ,
, , , 
Abstract
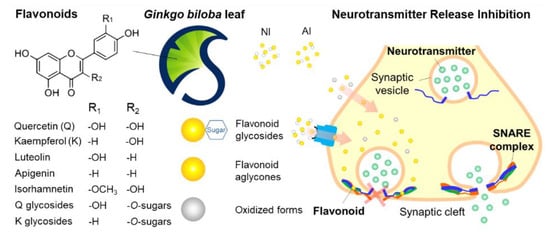
1. Introduction
2. Results and Discussion
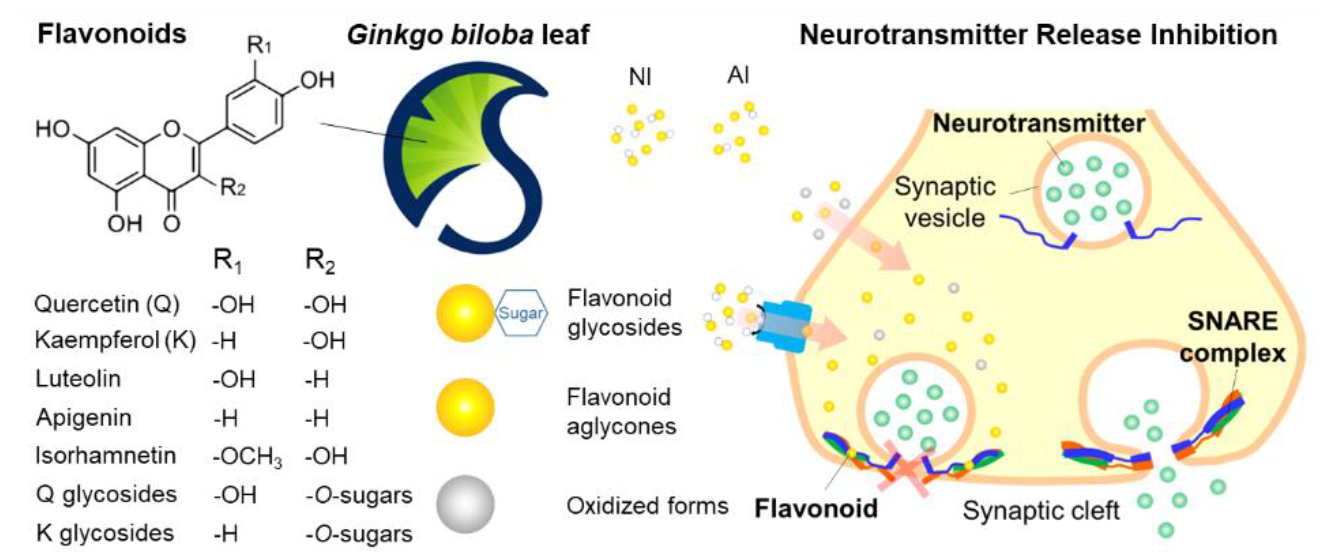
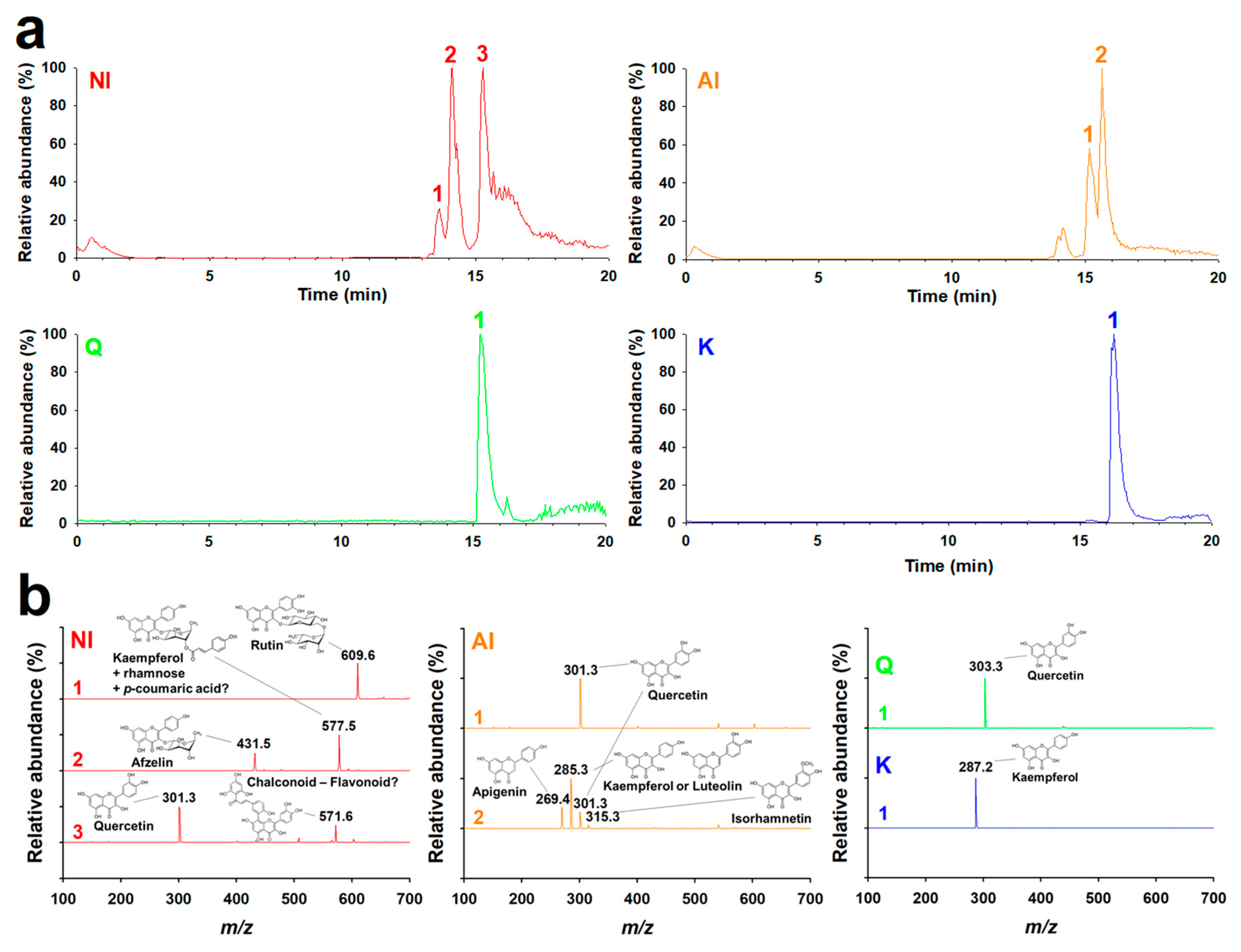
2.1. Identification of GBL Isolates
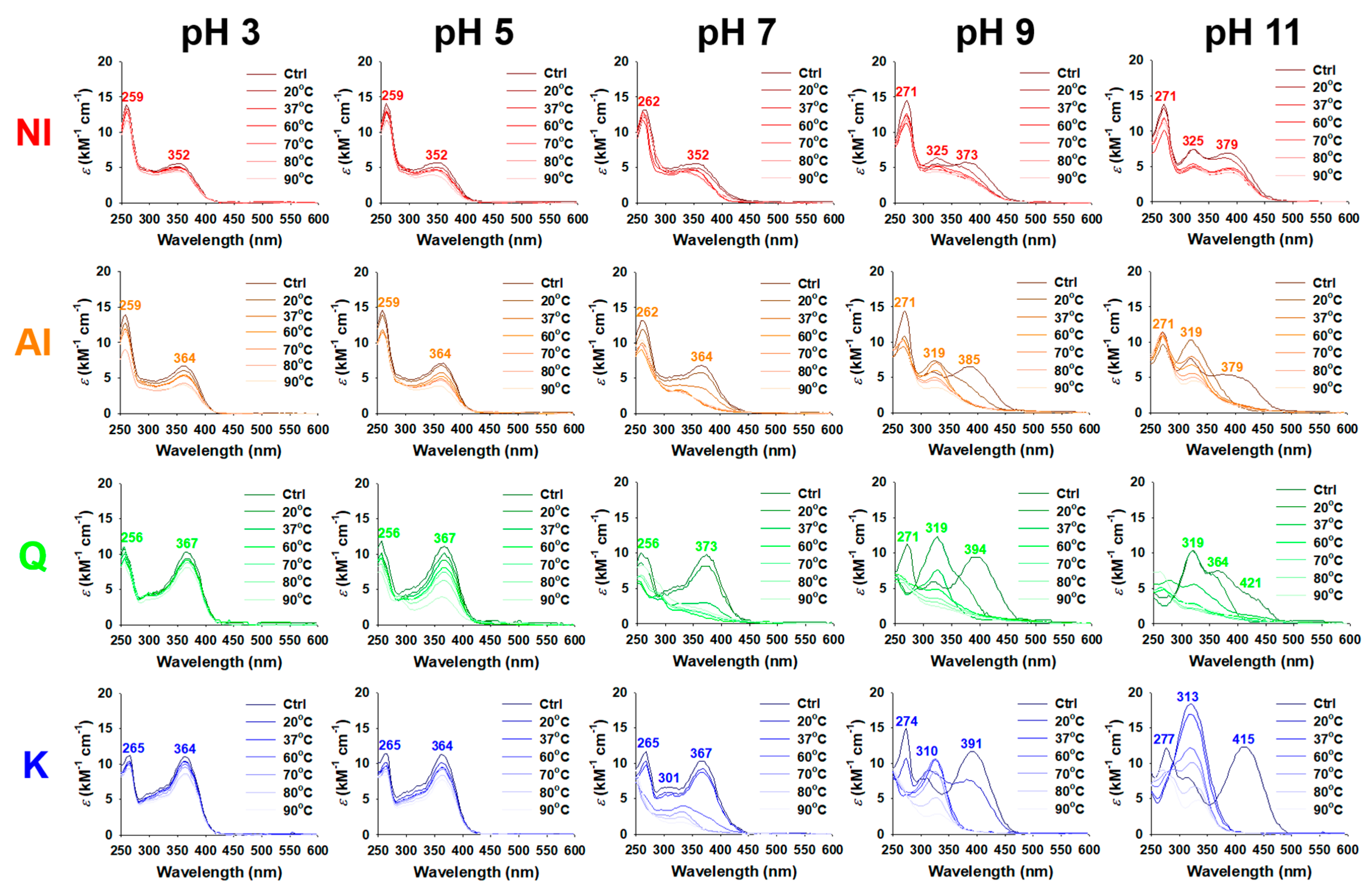
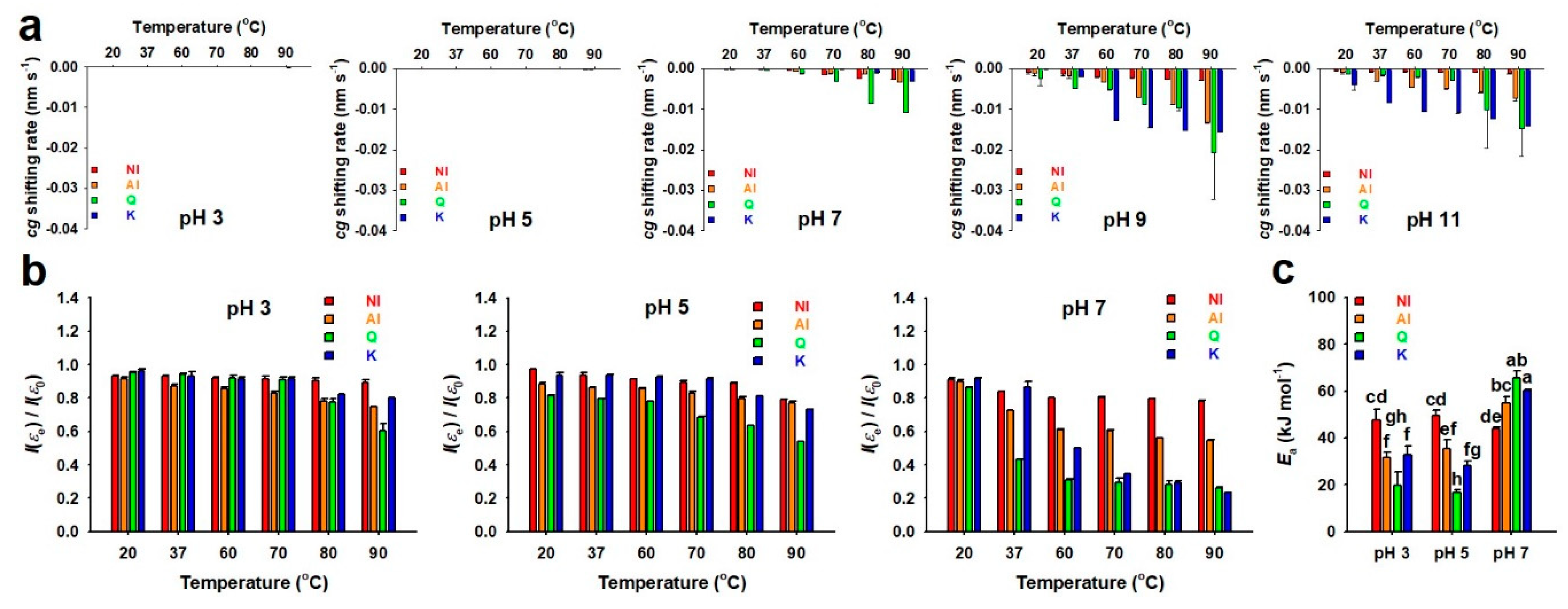
2.2. Thermal and pH Stabilities of GBL Isolates
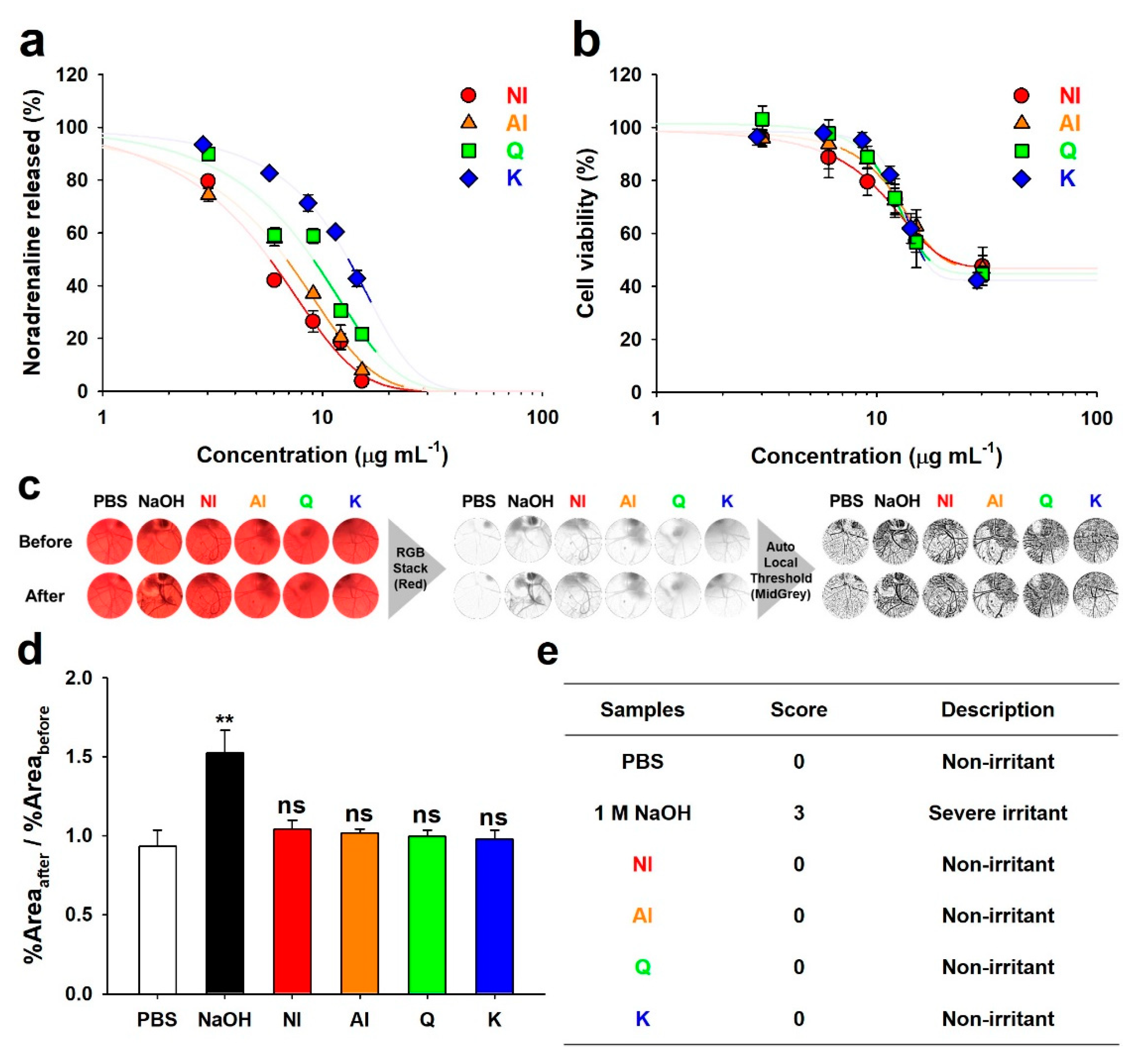
2.3. Neurotransmitter-Release Inhibitory Activity and Biocompatibility of GBL Isolates
2.4. Antioxidant and Anti-Inflammatory Activities of GBL Isolates
3. Materials and Methods
3.1. Chemicals
3.2. Isolation and Purification of Flavonoids from GBL Extracts
3.3. HPLC-MS/MS
3.4. Assessment of Thermal and pH Stability
3.5. Cell Cultivation
3.6. Norepinephrine Release Assay
3.7. Determination of Cell Viability
3.8. HET-CAM
3.9. Determination of Antioxidant Capacity and Anti-Inflammatory Effect
3.10. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- García-Lafuente, A.; Guillamón, E.; Villares, A.; Rostagno, M.A.; Martínez, J.A. Flavonoids as anti-inflammatory agents: implications in cancer and cardiovascular disease. Inflamm. Res. 2009, 58, 537–552. [Google Scholar] [CrossRef]
- Ravishankar, D.; Rajora, A.K.; Greco, F.; Osborn, H.M.I. Flavonoids as prospective compounds for anti-cancer therapy. Int. J. Biochem. Cell Biol. 2013, 45, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Cushnie, T.P.T.; Lamb, A.J. Recent advances in understanding the antibacterial properties of flavonoids. Int. J. Antimicrob. Agents 2011, 38, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Heo, P.; Park, J.-B.; Shin, Y.-K.; Kweon, D.-H. Visualization of SNARE-mediated hemifusion between giant unilamellar vesicles arrested by myricetin. Front. Mol. Neurosci. 2017, 10, 93. [Google Scholar] [CrossRef]
- Heo, P.; Yang, Y.; Han, K.Y.; Kong, B.; Shin, J.-H.; Jung, Y.; Jeong, C.; Shin, J.; Shin, Y.-K.; Ha, T.; et al. A chemical controller of SNARE-driven membrane fusion that primes vesicles for Ca2+-triggered millisecond exocytosis. J. Am. Chem. Soc. 2016, 138, 4512–4521. [Google Scholar] [CrossRef]
- Yang, Y.; Choi, J.K.; Jung, C.H.; Koh, H.J.; Heo, P.; Shin, J.Y.; Kim, S.; Park, W.-S.; Shin, H.-J.; Kweon, D.-H. SNARE-wedging polyphenols as small molecular botox. Planta Med. 2012, 78, 233–236. [Google Scholar] [CrossRef]
- Yang, Y.; Shin, J.Y.; Oh, J.-M.; Jung, C.H.; Hwang, Y.; Kim, S.; Kim, J.-S.; Yoon, K.-J.; Ryu, J.-Y.; Shin, J.; et al. Dissection of SNARE-driven membrane fusion and neuroexocytosis by wedging small hydrophobic molecules into the SNARE zipper. Proc. Natl. Acad. Sci. USA 2010, 107, 22145–22150. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Heo, P.; Kong, B.; Park, J.-B.; Jung, Y.; Shin, J.; Jeong, C.; Kweon, D.-H. Dynamic light scattering analysis of SNARE-driven membrane fusion and the effects of SNARE-binding flavonoids. Biochem. Biophys. Res. Commun. 2015, 465, 864–870. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum neurotoxins: biology, pharmacology, and toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, S.; Park, Y. Multifaceted therapeutic benefits of Ginkgo biloba L.: chemistry, efficacy, safety, and uses. J. Food Sci. 2008, 73, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, J.; Knipschild, P. Ginkgo biloba for cerebral insufficiency. Br. J. Clin. Pharmacol. 1992, 34, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, J.; Knipschild, P. Ginkgo biloba. Lancet 1992, 340, 1136–1139. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, L.J.; Ye, H.Y.; Gao, L.; Hou, W.; Tang, M.; Yang, G.; Zhong, Z.; Yuan, Y.; Peng, A. Isolation and purification of ginkgo flavonol glycosides from Ginkgo biloba leaves by high-speed counter-current chromatography. J. Sep. Sci. 2007, 30, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, G.L.; Riedl, K.M.; Schwartz, S.J. Effects of food formulation and thermal processing on flavones in celery and chamomile. Food Chem. 2013, 141, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Stengel, J. Mass spectrometric imaging of flavonoid glycosides and biflavonoids in Ginkgo biloba L. Phytochemistry 2016, 130, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Hasler, A.; Sticher, O.; Meier, B. Identification and determination of the flavonoids from Ginkgo biloba by high-performance liquid chromatography. J. Chromatogr. A 1992, 605, 41–48. [Google Scholar] [CrossRef]
- Weiz, G.; Breccia, J.D.; Mazzaferro, L.S. Screening and quantification of the enzymatic deglycosylation of the plant flavonoid rutin by UV–visible spectrometry. Food Chem. 2017, 229, 44–49. [Google Scholar] [CrossRef]
- Halbwirth, H. The creation and physiological relevance of divergent hydroxylation patterns in the flavonoid pathway. Int. J. Mol. Sci. 2010, 11, 595–621. [Google Scholar] [CrossRef]
- Cunha, V.R.R.; Constantino, V.R.L.; Ando, R.A. Raman spectroscopy and DFT calculations of para-coumaric acid and its deprotonated species. Vib. Spectrosc. 2012, 58, 139–145. [Google Scholar] [CrossRef]
- Amat, A.; Clementi, C.; Miliani, C.; Romani, A.; Sgamellotti, A.; Fantacci, S. Complexation of apigenin and luteolin in weld lake: a DFT/TDDFT investigation. Phys. Chem. Chem. Phys. 2010, 12, 6672–6684. [Google Scholar] [CrossRef]
- Zu, Y.; Li, C.; Fu, Y.; Zhao, C. Simultaneous determination of catechin, rutin, quercetin kaempferol and isorhamnetin in the extract of sea buckthorn (Hippophae rhamnoides L.) leaves by RP-HPLC with DAD. J. Pharm. Biomed. Anal. 2006, 41, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Sadik, O.A. Comparative analysis of quercetin oxidation by electrochemical, enzymatic, autoxidation, and free radical generation techniques: a mechanistic study. J. Agric. Food Chem. 2008, 56, 12081–12091. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.B.; Van Camp, J.; Vissenaekens, H.; Raes, K.; Smagghe, G.; Grootaert, C. Review on the use of cell cultures to study metabolism, transport, and accumulation of flavonoids: from mono-cultures to co-culture systems. Compr. Rev. Food Sci. Food Saf. 2015, 14, 741–754. [Google Scholar] [CrossRef]
- Yang, Y.; Oh, J.-M.; Heo, P.; Shin, J.Y.; Kong, B.; Shin, J.; Lee, J.-C.; Oh, J.S.; Park, K.W.; Lee, C.H.; et al. Polyphenols differentially inhibit degranulation of distinct subsets of vesicles in mast cells by specific interaction with granule-type-dependent SNARE complexes. Biochem. J. 2013, 450, 537–546. [Google Scholar] [CrossRef]
- Kim, D.-O.; Lee, C.Y. Comprehensive study on vitamin C equivalent antioxidant capacity (VCEAC) of various polyphenolics in scavenging a free radical and its structural relationship. Crit. Rev. Food Sci. Nutr. 2004, 44, 253–273. [Google Scholar] [CrossRef]
- Terao, J.; Murota, K.; Kawai, Y. Conjugated quercetin glucuronides as bioactive metabolites and precursors of aglycone in vivo. Food Funct. 2011, 2, 11–17. [Google Scholar] [CrossRef]
- Moro, C.; Palacios, I.; Lozano, M.; D’Arrigo, M.; Guillamón, E.; Villares, A.; Martínez, J.A.; García-Lafuente, A. Anti-inflammatory activity of methanolic extracts from edible mushrooms in LPS activated RAW 264.7 macrophages. Food Chem. 2012, 130, 350–355. [Google Scholar] [CrossRef]
- Lin, W.-W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Wang, J.; Fang, X.; Ge, L.; Cao, F.; Zhao, L.; Wang, Z.; Xiao, W. Antitumor, antioxidant and anti-inflammatory activities of kaempferol and its corresponding glycosides and the enzymatic preparation of kaempferol. PLoS ONE 2018, 13, e0197563. [Google Scholar] [CrossRef]
- Francisco, V.; Figueirinha, A.; Costa, G.; Liberal, J.; Lopes, M.C.; García-Rodríguez, C.; Geraldes, C.F.G.C.; Cruz, M.T.; Batista, M.T. Chemical characterization and anti-inflammatory activity of luteolin glycosides isolated from lemongrass. J. Funct. Food 2014, 10, 436–443. [Google Scholar] [CrossRef]
- Kim, H.K.; Cheon, B.S.; Kim, Y.H.; Kim, S.Y.; Kim, H.P. Effects of naturally occurring flavonoids on nitric oxide production in the macrophage cell line RAW 264.7 and their structure–activity relationships. Biochem. Pharmacol. 1999, 58, 759–765. [Google Scholar] [CrossRef]
- Kim, A.-R.; Shin, T.-S.; Lee, M.-S.; Park, J.-Y.; Park, K.-E.; Yoon, N.-Y.; Kim, J.-S.; Choi, J.-S.; Jang, B.-C.; Byun, D.-S. Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and anti-inflammatory properties. J. Agric. Food Chem. 2009, 57, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.Y.; Jeon, H.L.; Kim, J.; Kim, T.S.; Hong, Y.-H.; Jeong, M.K.; Park, K.-H.; Kim, B.-H.; Park, S.; Jang, W.-H. Two tiered approaches combining alternative test methods and minimizing the use of reconstructed human cornea-like epithelium tests for the evaluation of eye irritation potency of test chemicals. Toxicol. In Vitro 2020, 63, 104675. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, B.; Kay, G.; Matthews, K.H.; Knott, R.M.; Cairns, D. The hen’s egg chorioallantoic membrane (HET-CAM) test to predict the ophthalmic irritation potential of a cysteamine-containing gel: Quantification using Photoshop® and ImageJ. Int. J. Pharm. 2015, 490, 1–8. [Google Scholar] [CrossRef]
- NIH. ICCVAM-Recommended Test Method Protocol: Hen’s Egg Test–Chorioallantoic Membrane (HET-CAM) Test Method; No. 10-7553; National Institutes of Health (NIH): Durham, NC, USA, 2010; pp. B29–B38. [Google Scholar]
- Lee, B.H.; Nam, T.G.; Kim, S.Y.; Chun, O.K.; Kim, D.-O. Estimated daily per capita intakes of phenolics and antioxidants from coffee in the Korean diet. Food Sci. Biotechnol. 2019, 28, 269–279. [Google Scholar] [CrossRef]
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907. [Google Scholar] [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds (GBL NI, GBL AI, Q, and K) are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | EC50 (μg·mL−1) | CC50 (μg·mL−1) | EC50/CC50 |
|---|---|---|---|
| NI | 5.75 ± 0.10 a | 28.99 ± 6.07 a | 0.26 ± 0.07 a |
| AI | 6.84 ± 0.38 b | 23.31 ± 5.14 a | 0.30 ± 0.07 b |
| Quercetin | 9.05 ± 0.18 c | 17.75 ± 3.59 a | 0.52 ± 0.09 c |
| Kaempferol | 13.11 ± 0.45 d | 16.61 ± 1.09 a | 0.79 ± 0.03 d |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, C.; Park, J.-B.; Cho, S.; Kim, H.R.; Kim, Y.J.; Bae, H.; Kim, C.; Kang, H.; Jang, D.; Shin, Y.S.; et al. Characterization of Ginkgo biloba Leaf Flavonoids as Neuroexocytosis Regulators. Molecules 2020, 25, 1829. https://doi.org/10.3390/molecules25081829
Ban C, Park J-B, Cho S, Kim HR, Kim YJ, Bae H, Kim C, Kang H, Jang D, Shin YS, et al. Characterization of Ginkgo biloba Leaf Flavonoids as Neuroexocytosis Regulators. Molecules. 2020; 25(8):1829. https://doi.org/10.3390/molecules25081829
Chicago/Turabian StyleBan, Choongjin, Joon-Bum Park, Sora Cho, Hye Rin Kim, Yong Joon Kim, Hyungjin Bae, Chinhan Kim, Hakhee Kang, Davin Jang, Yong Sub Shin, and et al. 2020. "Characterization of Ginkgo biloba Leaf Flavonoids as Neuroexocytosis Regulators" Molecules 25, no. 8: 1829. https://doi.org/10.3390/molecules25081829
APA StyleBan, C., Park, J.-B., Cho, S., Kim, H. R., Kim, Y. J., Bae, H., Kim, C., Kang, H., Jang, D., Shin, Y. S., Kim, D.-O., Kim, H., & Kweon, D.-H. (2020). Characterization of Ginkgo biloba Leaf Flavonoids as Neuroexocytosis Regulators. Molecules, 25(8), 1829. https://doi.org/10.3390/molecules25081829