
Psoralen Derivatives as Inhibitors of Mycobacterium tuberculosis Proteasome

 , ,
, ,
Abstract
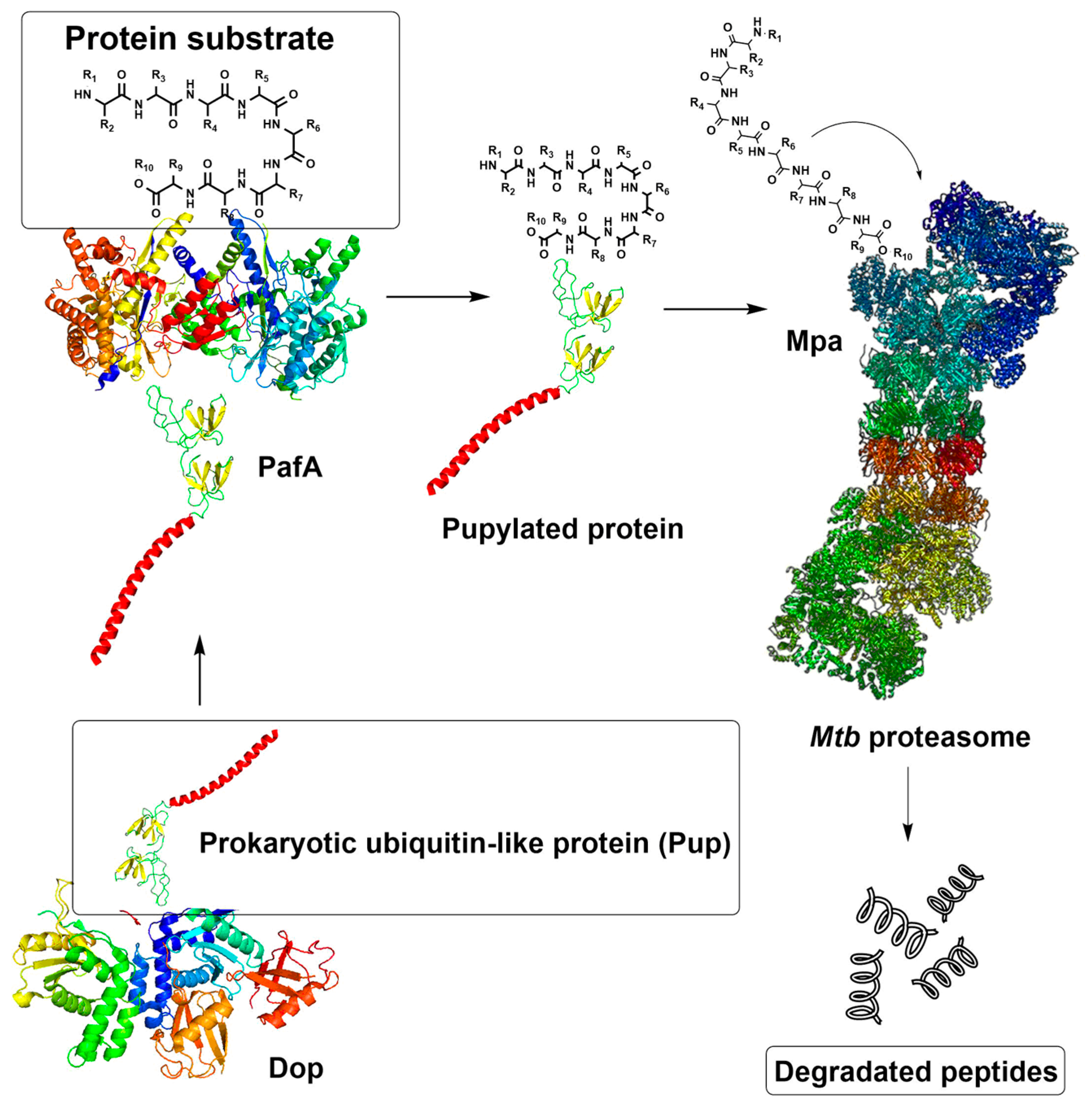
1. Introduction
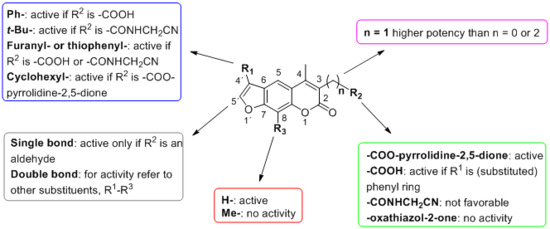
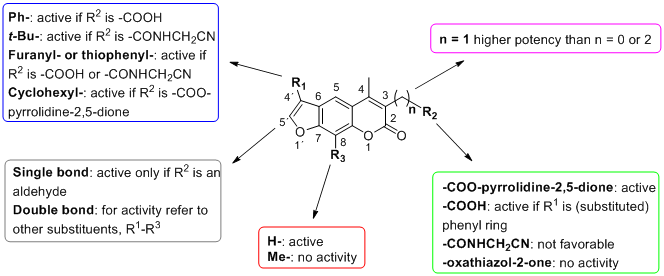
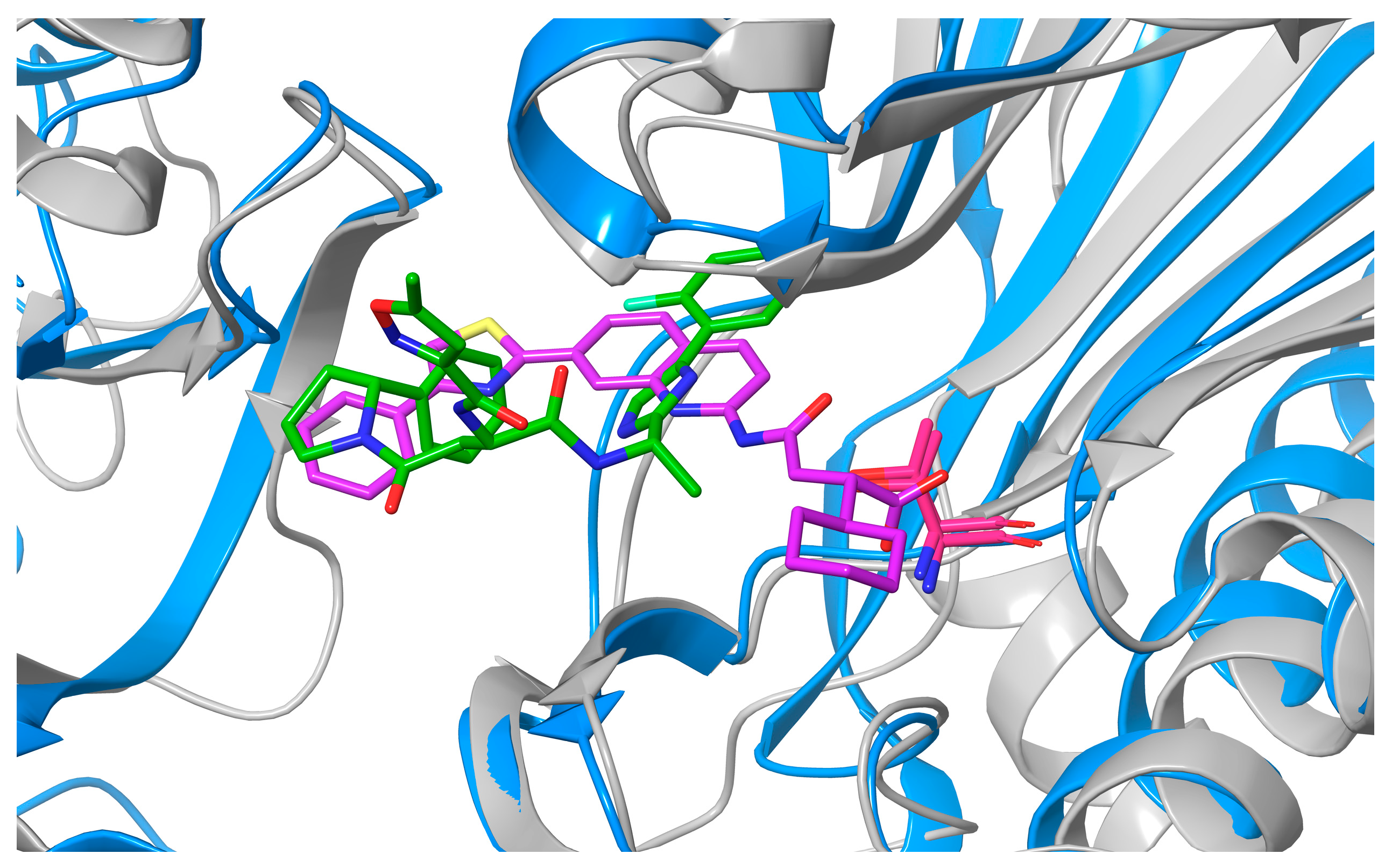
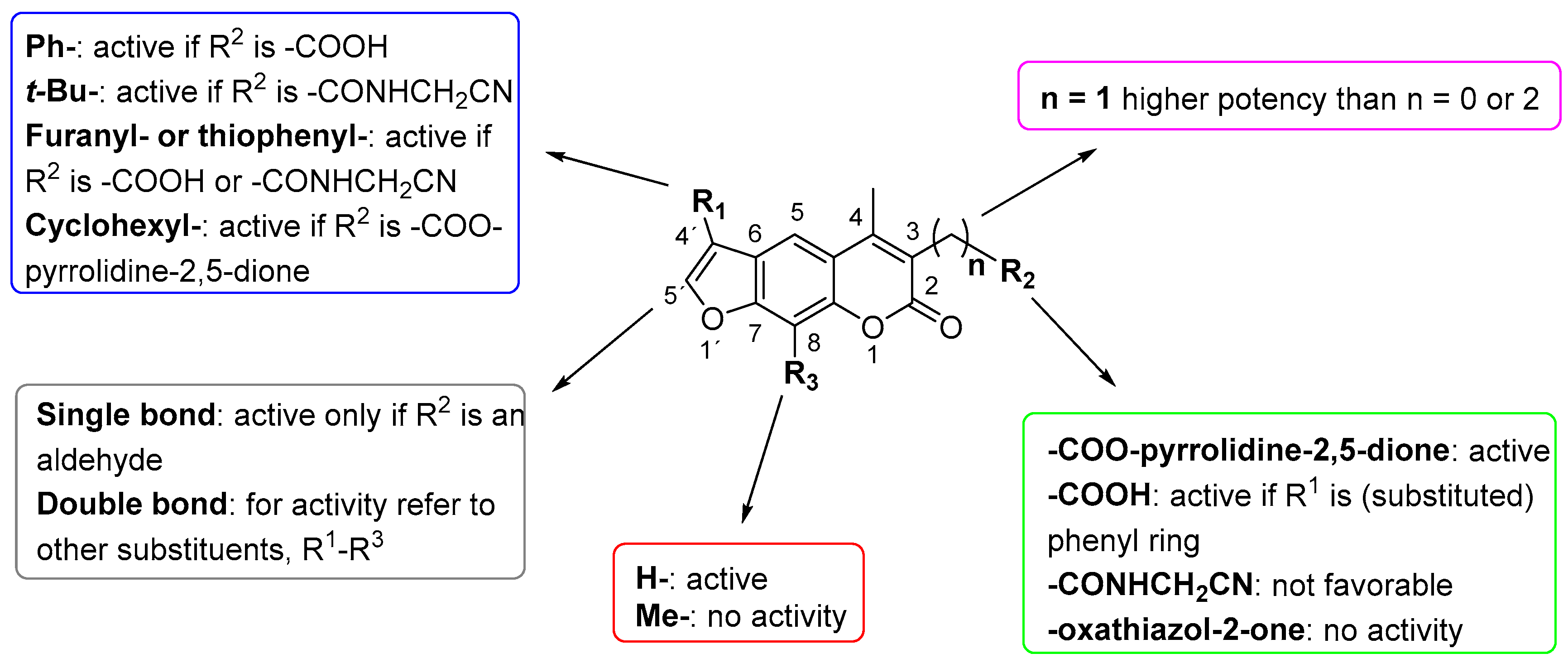
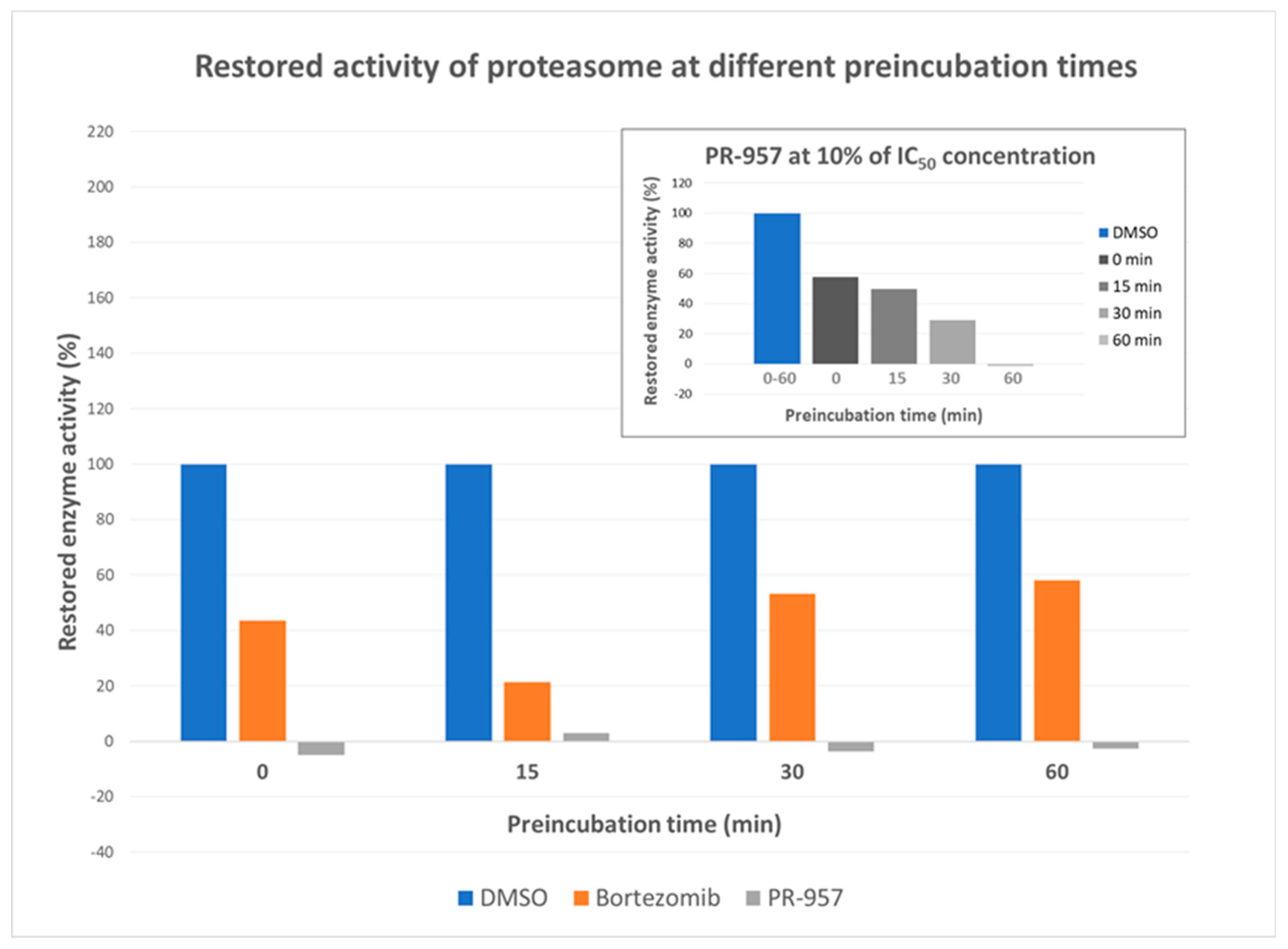
2. Results and Discussion
3. Materials and Methods
3.1. Isolation of the Enzyme
3.2. Enzymatic Assays
3.2.1. Determination of Ki and Km Values
3.2.2. Dilution Assays
3.2.3. Determination of kinact/Ki values for PR-957
3.3. Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hinkson, I.V.; Elias, J.E. The dynamic state of protein turnover: It’s about time. Trends Cell. Biol. 2011, 21, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell. Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Price, J.C.; Guan, S.; Burlingame, A.; Prusiner, S.B.; Ghaemmaghami, S. Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Dorrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and global influences on protein turnover in neurons and glia. eLife 2018, 7, e34202. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.H.; Darwin, K.H. Bacterial Proteasomes: Mechanistic and functional insights. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-16. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Samanovic, M.I.; Li, H.; Darwin, K.H. The pup-proteasome system of Mycobacterium tuberculosis. Sub-cell Biochem. 2013, 66, 267–295. [Google Scholar]
- Darwin, K.H. Prokaryotic ubiquitin-like protein (Pup), proteasomes and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 485–491. [Google Scholar] [CrossRef]
- Hu, G.; Lin, G.; Wang, M.; Dick, L.; Xu, R.M.; Nathan, C.; Li, H. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol. Microbiol. 2006, 59, 1417–1428. [Google Scholar] [CrossRef]
- Oczelik, D.; Barandun, J.; Schmitz, N.; Sutter, M.; Guth, E.; Damberger, F.F.; Allain, F.H.; Ban, N.; Weber-Ban, E. Structures of Pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat. Commun. 2012, 3, 1014–1024. [Google Scholar]
- Striebel, F.; Imkamp, F.; Sutter, M.; Steiner, M.; Mamedov, A.; Weber-Ban, E. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol. 2009, 16, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Darwin, K.H.; Li, H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat. Struct. Mol. Biol. 2010, 17, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Jastrab, J.B.; Zhang, S.; Kovach, A.; Zhao, G.; Darwin, K.H.; Li, H. Proteasome substrate capture and gate opening by the accessory factor PafE from Mycobacterium tuberculosis. J. Biol. Chem. 2018, 293, 4713–4723. [Google Scholar] [CrossRef] [PubMed]
- Delley, C.L.; Laederach, J.; Ziemski, M.; Bolten, M.; Boehringer, D.; Weber-Ban, E. Bacterial proteasome activator bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS ONE 2014, 9, e114348. [Google Scholar] [CrossRef] [PubMed]
- Zwickl, P.; Baumeister, W.; Steven, A. Dis-assembly lines: The proteasome and related ATPase-assisted proteases. Curr. Opin. Struct. Biol. 2000, 10, 242–250. [Google Scholar] [CrossRef]
- Orlowski, M.; Cardozo, C.; Michaud, C. Evidence for the presence of five distinct proteolytic components in the pituitary multicatalytic proteinase complex. Properties of two components cleaving bonds on the carboxyl side of branched chain and small neutral amino acids. Biochemistry 1993, 32, 1563–1572. [Google Scholar] [CrossRef]
- Lin, G.; Hu, G.; Tsu, C.; Kunes, Y.Z.; Li, H.; Dick, L.; Parsons, T.; Li, P.; Chen, Z.; Zwickl, P.; et al. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol. Microbiol. 2006, 59, 1405–1416. [Google Scholar] [CrossRef]
- Lin, G.; Tsu, C.; Dick, L.; Zhou, X.K.; Nathan, C. Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem. 2008, 283, 34423–34431. [Google Scholar] [CrossRef]
- Lin, G.; Chidawanyika, T.; Tsu, C.; Warrier, T.; Vaubourgeix, J.; Blackburn, C.; Gigstad, K.; Sintchak, M.; Dick, L.; Nathan, C. N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: Role of S3 and S1 binding pockets. J. Am. Chem. Soc. 2013, 135, 9968–9971. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Singh, P.K.; Fan, H.; Wang, R.; Sukenick, G.; Nathan, C.; Lin, G.; Li, H. Structural basis for the species-selective binding of N,C-capped dipeptides to the Mycobacterium tuberculosis proteasome. Biochemistry 2017, 56, 9246–9253. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; Kirk, C.J.; Basler, M. Proteasomes in immune cells: More than peptide producers? Nat. Rev. Immunol. 2010, 10, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Boubakri, H.; Seghezzi, N.; Duchateau, M.; Gominet, M.; Kofronova, O.; Benada, O.; Mazodier, P.; Pernodet, J.L. The absence of pupylation (prokaryotic ubiquitin-like protein modification) affects morphological and physiological differentiation in Streptomyces coelicolor. J. Bacteriol. 2015, 197, 3388–3399. [Google Scholar] [CrossRef]
- Lupoli, T.J.; Vaubourgeix, J.; Burns-Huang, K.; Gold, B. Targeting the proteostasis network for mycobacterial drug discovery. ACS Infect. Dis. 2018, 4, 478–498. [Google Scholar] [CrossRef]
- Gandotra, S.; Schnappinger, D.; Monteleone, M.; Hillen, W.; Ehrt, S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med. 2007, 13, 1515–1520. [Google Scholar] [CrossRef]
- Darwin, K.H.; Ehrt, S.; Gutierrez-Ramos, J.C.; Weich, N.; Nathan, C.F. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 2003, 302, 1963–1966. [Google Scholar] [CrossRef]
- Imkamp, F.; Ziemski, M.; Weber-Ban, E. Pupylation-dependent and -independent proteasomal degradation in mycobacteria. Biomol. Concepts 2015, 6, 285–301. [Google Scholar] [CrossRef]
- WHO Global Tuberculosis Report 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf?ua=1 (accessed on 14 February 2020).
- Zhan, W.; Hsu, H.-C.; Morgan, T.; Ouellette, T.; Burns-Huang, K.; Hara, R.; Wright, A.G.; Imaeda, T.; Okamoto, R.; Sato, K.; et al. Selective phenylimidazole-based inhibitors of the Mycobacterium tuberculosis proteasome. J. Med. Chem. 2019, 62, 9246–9253. [Google Scholar] [CrossRef]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting proteasomes in infectious organisms to combat disease. FEBS J. 2017, 284, 1503–1517. [Google Scholar] [CrossRef]
- Richy, N.; Sarraf, D.; Marechal, X.; Janmamode, N.; Le Guevel, R.; Genin, E.; Reboud-Ravaux, M.; Vidal, J. Structure-based design of human immuno- and constitutive proteasomes inhibitors. Eur. J. Med. Chem. 2018, 145, 570–587. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, C. An Overview of Bortezomib-Induced Neurotoxicity. Toxics 2015, 3, 294–303. [Google Scholar] [CrossRef]
- Ogorevc, E.; Schiffrer, E.S.; Sosič, I.; Gobec, S. A patent review of immunoproteasome inhibitors. Expert Opin. Ther. Pat. 2018, 28, 517–540. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Groll, M. Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chem. Int. Ed. 2012, 51, 8708–8720. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Groettrup, M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr. Opin. Chem. Biol. 2014, 23, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kasam, V.; Lee, N.R.; Kim, K.B.; Zhan, C.G. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg. Med. Chem. Lett. 2014, 24, 3614–3617. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Baur, R.; Le Chapelain, C.; Dubiella, C.; Heinemeyer, W.; Huber, E.M.; Groll, M. Structural elucidation of a nonpeptidic inhibitor specific for the human immunoproteasome. ChemBioChem 2017, 18, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Angelo, N.G.; Warren, J.D.; Nathan, C.F.; Lin, G. Oxathiazolones selectively inhibit the human immunoproteasome over the constitutive proteasome. ACS Med. Chem. Lett. 2014, 5, 405–410. [Google Scholar] [CrossRef]
- Bosc, E.; Nastri, J.; Lefort, V.; Valli, M.; Contiguiba, F.; Pioli, R.; Furlan, M.; da Bolzani, V.S.; El Amri, C.; Reboud-Ravaux, M. Piperlongumine and some of its analogs inhibit selectively the human immunoproteasome over the constitutive proteasome. Biochem. Biophys. Res. Commun. 2018, 496, 961–966. [Google Scholar] [CrossRef]
- Sosič, I.; Gobec, M.; Brus, B.; Knez, D.; Živec, M.; Konc, J.; Lešnik, S.; Ogrizek, M.; Obreza, A.; Žigon, D.; et al. Nonpeptidic selective inhibitors of the chymotrypsin-like (β5i) subunit of the immunoproteasome. Angew. Chem. Int. Ed. Engl. 2016, 55, 5745–5748. [Google Scholar] [CrossRef]
- Shannon Schiffrer, E.; Sosič, I.; Šterman, A.; Mravljak, J.; Mlinarič-Raščan, I.; Gobec, S.; Gobec, M. A focused structure–activity relationship study of psoralen-based immunoproteasome inhibitors. Med. Chem. Commun. 2019, 10, 1958–1965. [Google Scholar] [CrossRef]
- Inch. N.E.B. Transformation Protocol for BL21(DE3) Competent Cells (C2527). Available online: https://www.neb.com/protocols/1/01/01/transformation-protocol-for-bl21-de3-competent-cells-c2527 (accessed on 19 September 2019).
- Motulsky, H.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting; Oxford University Press: Oxford, NY, USA, 2004; p. 351. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | IC50 (µM) and (Hill Coefficient) | Ki and Type of Inhibition a | RA b at 10 µM (%) or Ki or IC50 (µM) against ß5i of IP (refs [41,42]) |
|---|---|---|---|---|
| Bortezomib |  | 0.11 ± 0.03 (0.76) | Ki = 133 ± 5 nM Mixed inhibition (R2 = 0.94, α = 0.85) (Slow) reversible inhibition | IC50 = 0.004 µM (ref [35]) |
| PR-957 |  | 2.2 ± 1.0 (0.91) | kinact/Ki = 96 ± 41 M−1·s−1 Ki = 5.2 ± 1.9 µM Noncompetitive inhibition (R2 = 0.83) Irreversible inhibition | IC50 = 0.015 ± 0.002 µM (ref [41]) |
| 1 |  | 39 ± 7 (0.69) | ND | Ki = 137 ± 33 µM |
| 2 |  | 15 ± 2 (1.75) | ND | 83% |
| 3 |  | 31 ± 5 (1.22) | ND | 53% |
| 4 |  | 4 ± 3 (1.59) | ND | Ki = 12.7 ± 3.7 µM |
| 5 |  | 17 ± 3 (0.70) | ND | 93% |
| 6 |  | 2.2 ± 0.3 (0.70) | ND | 82% |
| 7 |  | 8 ± 5 (0.42) | ND | 100% |
| 8 |  | 3.7 ± 1.5 (1.05) | Ki = 5.6 ± 20.8 µM Mixed inhibition (R2 = 0.54, α = 0.19) Reversible inhibition | 100% |
| 9 |  | 2 ± 2 (2.27) | ND | 66% |
| 10 |  | 3 ± 2 (4.46) | ND | 100% |
| 11 |  | 8.8 ± 1.0 (0.90) | Ki = 4.2 ± 2.1 µM Mixed inhibition (R2 = 0.91, α = 6.67) (Partially) reversible inhibition | IC50 = 0.94 ± 1.1 µM |
| 12 |  | 5 ± 2 (1.05) | ND | IC50 = 1.8 ± 0.4 µM |
| 13 |  | 3.2 ± 0.3 (0.90) | Ki = 1.1 ± 0.9 µM Mixed inhibition (R2 = 0.52, α = 6.94 × 10^16) (Partially) reversible inhibition | IC50 = 4.4 ± 0.1 µM |
| 14 |  | 9 ± 5 (2.42) | ND | IC50 = 6.9 ± 2.1 µM |
| 15 |  | 5.8 ± 2.1 (0.55) | Ki = 14.9 ± 45.0 µM Mixed inhibition (R2 = 0.77, α = 0.22) Reversible inhibition | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rožman, K.; Alexander, E.M.; Ogorevc, E.; Bozovičar, K.; Sosič, I.; Aldrich, C.C.; Gobec, S. Psoralen Derivatives as Inhibitors of Mycobacterium tuberculosis Proteasome. Molecules 2020, 25, 1305. https://doi.org/10.3390/molecules25061305
Rožman K, Alexander EM, Ogorevc E, Bozovičar K, Sosič I, Aldrich CC, Gobec S. Psoralen Derivatives as Inhibitors of Mycobacterium tuberculosis Proteasome. Molecules. 2020; 25(6):1305. https://doi.org/10.3390/molecules25061305
Chicago/Turabian StyleRožman, Kaja, Evan M. Alexander, Eva Ogorevc, Krištof Bozovičar, Izidor Sosič, Courtney C. Aldrich, and Stanislav Gobec. 2020. "Psoralen Derivatives as Inhibitors of Mycobacterium tuberculosis Proteasome" Molecules 25, no. 6: 1305. https://doi.org/10.3390/molecules25061305
APA StyleRožman, K., Alexander, E. M., Ogorevc, E., Bozovičar, K., Sosič, I., Aldrich, C. C., & Gobec, S. (2020). Psoralen Derivatives as Inhibitors of Mycobacterium tuberculosis Proteasome. Molecules, 25(6), 1305. https://doi.org/10.3390/molecules25061305