
Comparison of Molecular Recognition of Trimethyllysine and Trimethylthialysine by Epigenetic Reader Proteins

 , , and
, , and 
Abstract
1. Introduction
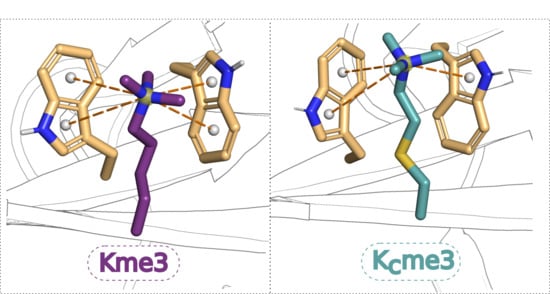
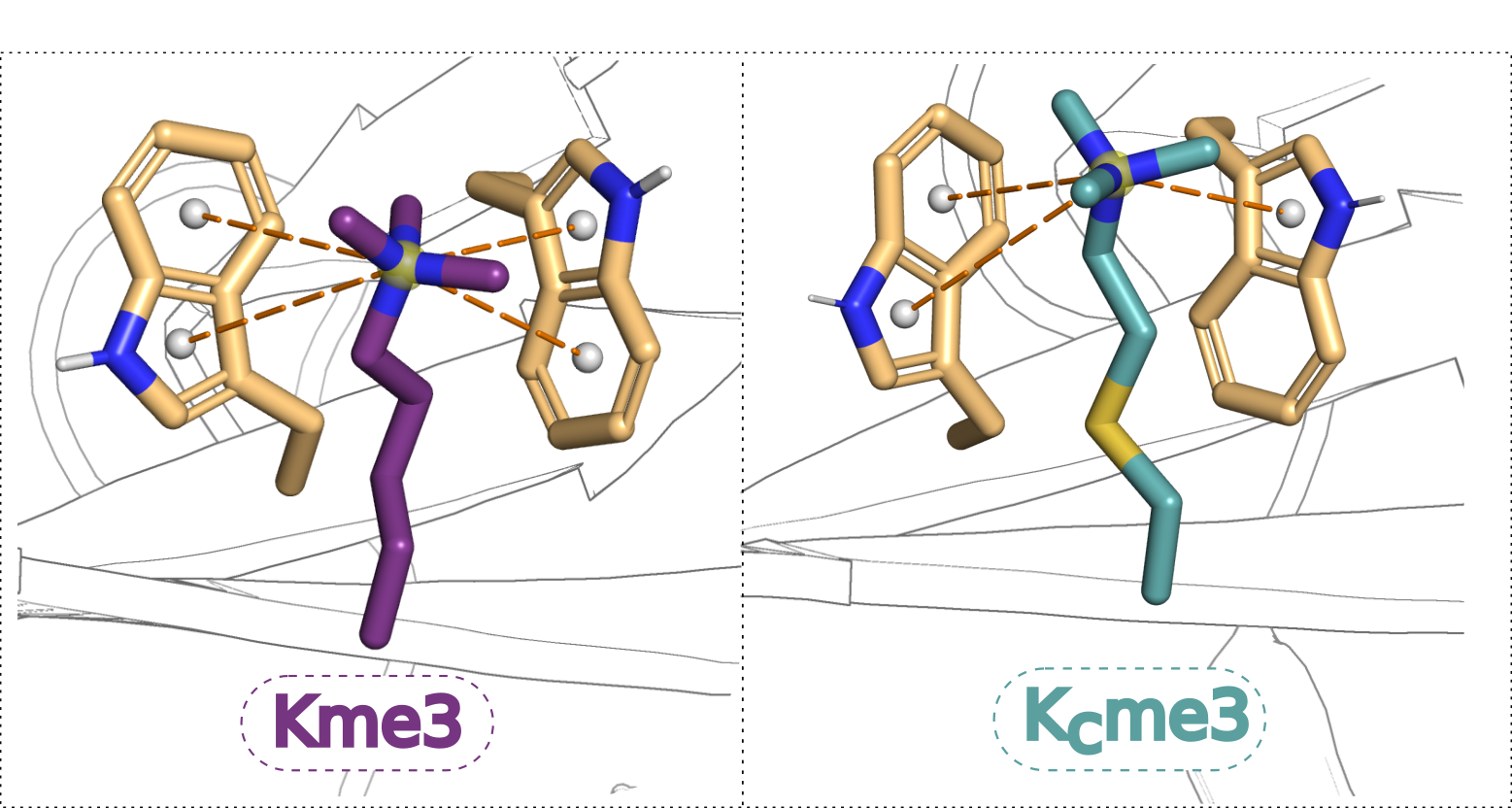
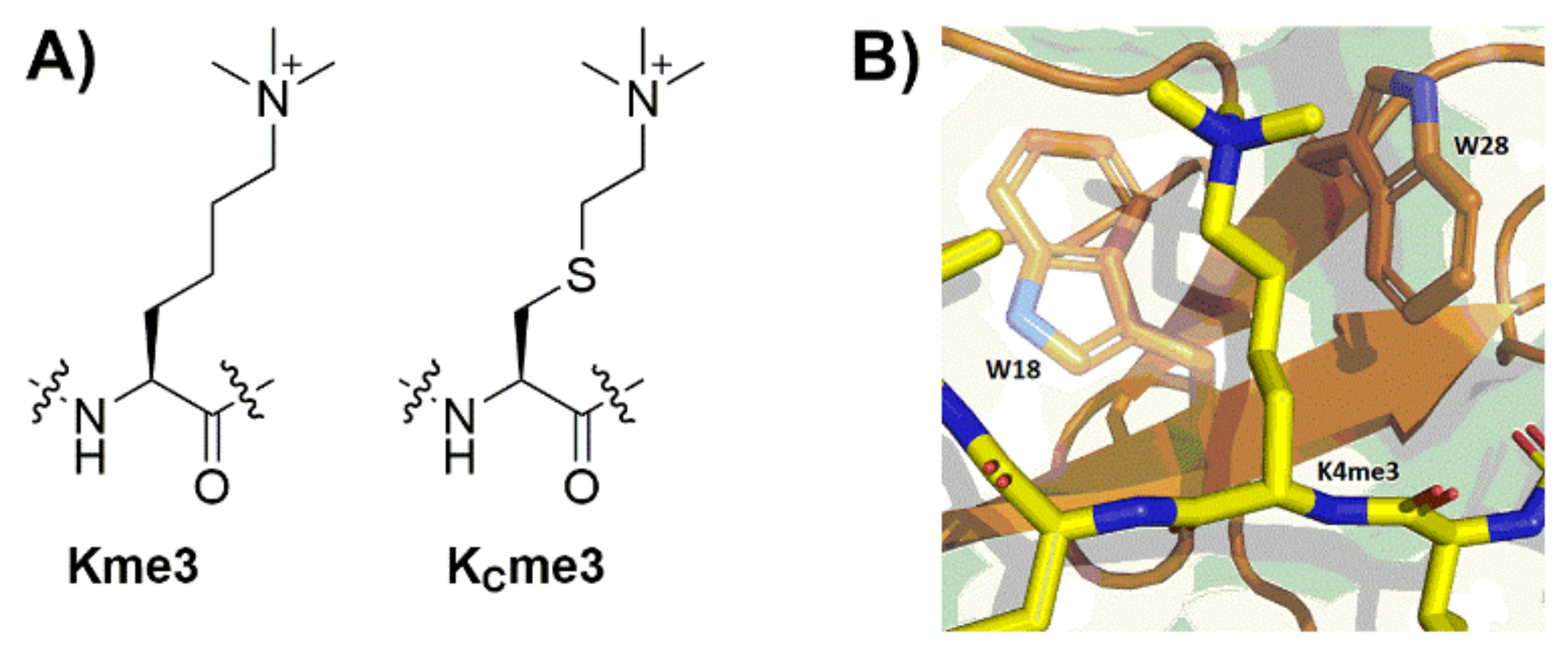
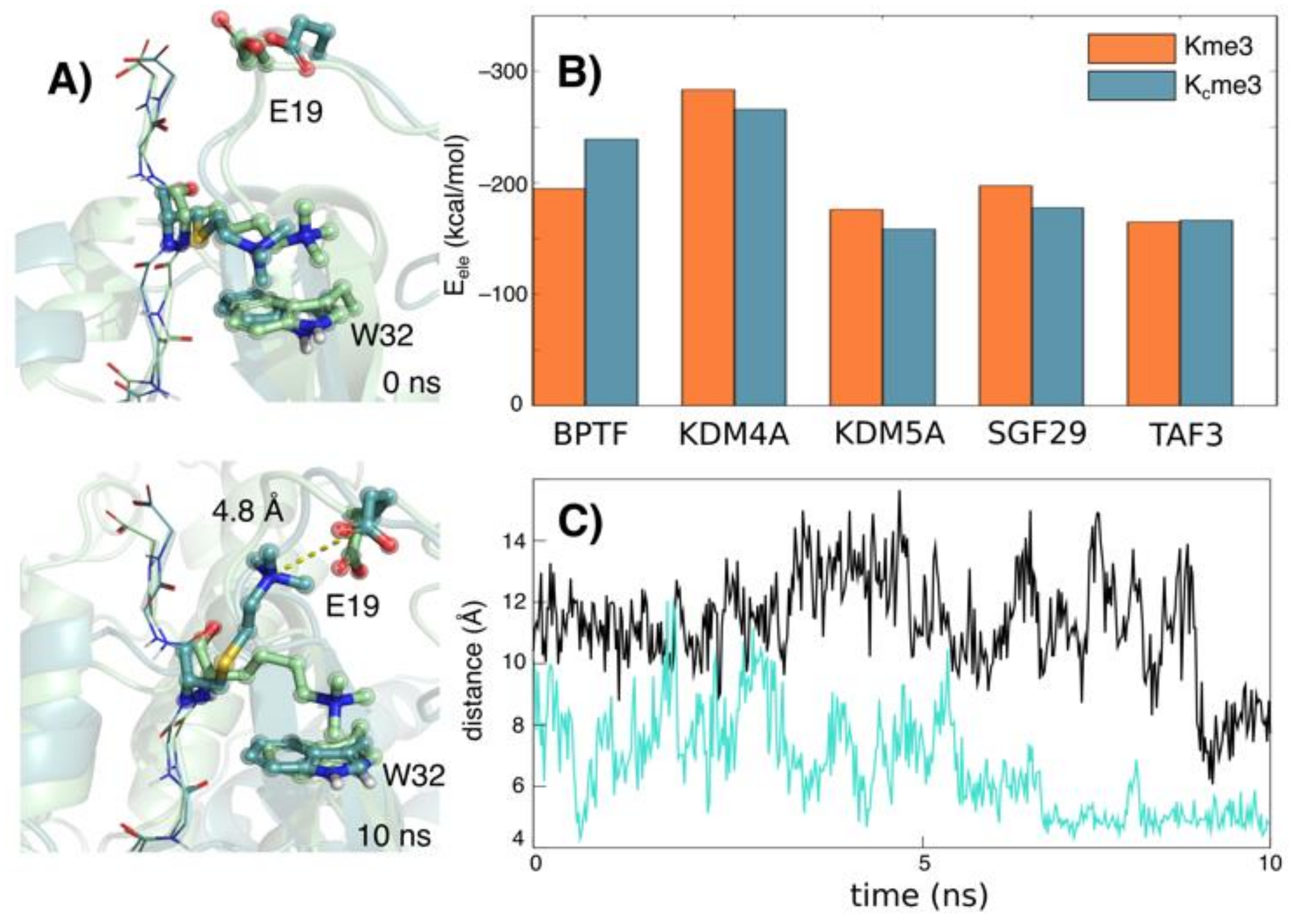
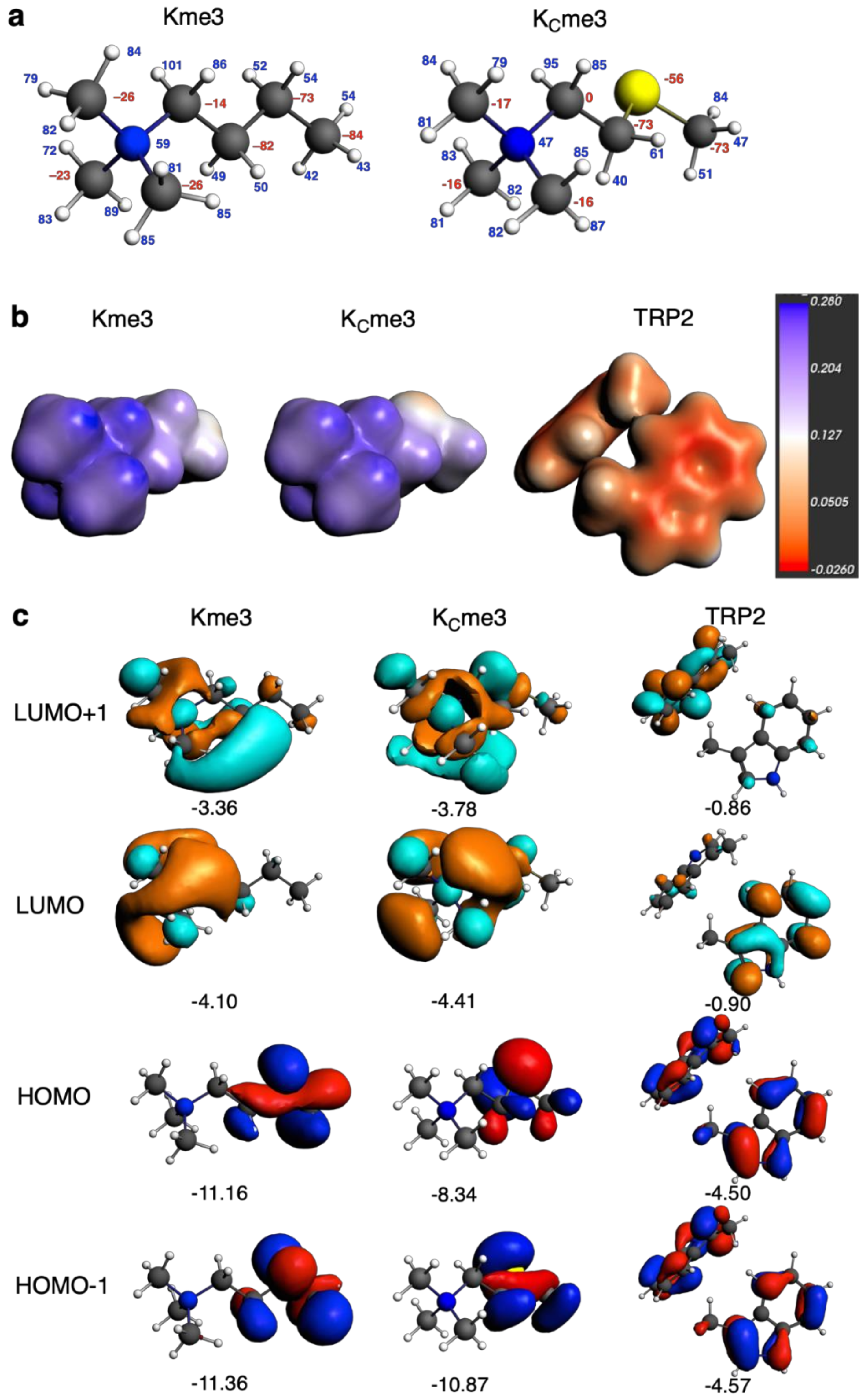
2. Results
3. Discussion
4. Materials and Methods
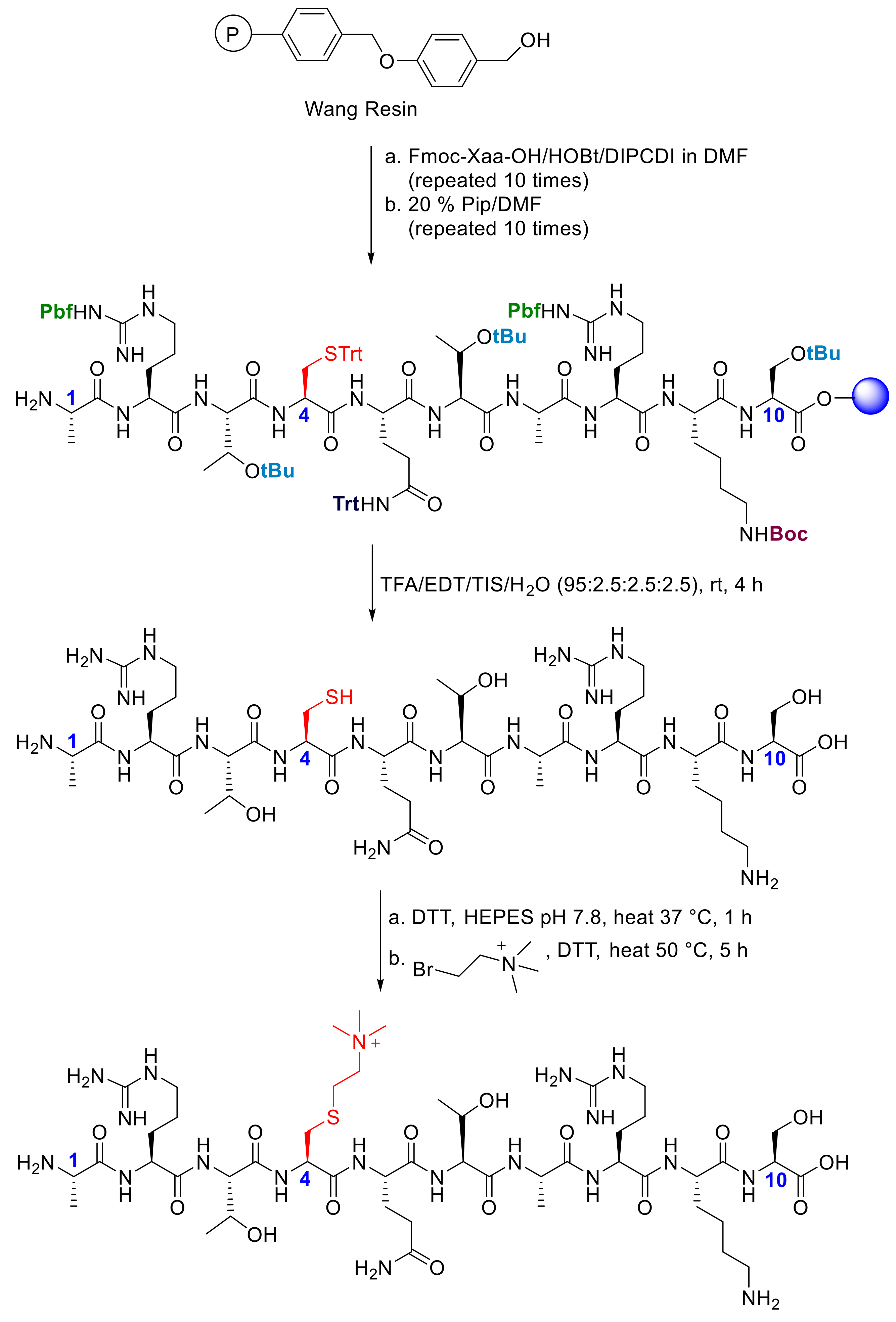
4.1. Preparation of H3K4me3 and H3KC4me3
4.2. Reader Domain expression and Purification
4.3. Isothermal Titration Calorimetry
4.4. Molecular Dynamics Simulations
4.5. Quantum Chemical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [PubMed]
- Black, J.C.; van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics. Mol. Cell 2012, 48, 491–507. [Google Scholar] [PubMed]
- Qian, C.; Zhou, M.M. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell. Mol. Life Sci. 2006, 63, 2755–2763. [Google Scholar] [PubMed]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar]
- Nadal, S.; Raj, R.; Mohammed, S.; Davis, B.G. Synthetic post-translational modifications of histones. Curr. Opin. Chem. Biol. 2018, 45, 35–47. [Google Scholar]
- Hughes, R.M.; Wiggins, K.R.; Khorasanizadeh, S.; Waters, M.L. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. USA 2007, 104, 11184–11188. [Google Scholar]
- Kamps, J.J.; Huang, J.; Poater, J.; Xu, C.; Pieters, B.J.G.E.; Dong, A.; Min, J.; Sherman, W.; Beuming, T.; Bickelhaupt, F.M.; et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 2015, 6, 8911. [Google Scholar]
- Al Temimi, A.H.K.; Belle, R.; Kumar, K.; Poater, J.; Betlem, P.; Pieters, B.J.G.E.; Paton, R.S.; Bickelhaupt, F.M.; Mecinovic, J. Recognition of shorter and longer trimethyllysine analogues by epigenetic reader proteins. Chem. Commun. 2018, 54, 2409–2412. [Google Scholar]
- Belle, R.; Al Temimi, A.H.K.; Kumar, K.; Pieters, B.J.G.E.; Tumber, A.; Dunford, J.E.; Johansson, C.; Oppermann, U.; Brown, T.; Schofield, C.J.; et al. Investigating d-lysine stereochemistry for epigenetic methylation, demethylation and recognition. Chem. Commun. 2017, 53, 13264–13267. [Google Scholar]
- Lang, K.; Chin, J.W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 2014, 114, 4764–4806. [Google Scholar]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [PubMed]
- Wang, Q.; Parrish, A.R.; Wang, L. Expanding the genetic code for biological studies. Chem. Biol. 2009, 16, 323–326. [Google Scholar] [PubMed]
- Hendrickson, T.L.; de Crecy-Lagard, V.; Schimmel, P. Incorporation of unnatural amino acids into proteins. Annu. Rev. Biochem. 2004, 73, 147–176. [Google Scholar] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Shimko, J.C.; North, J.A.; Bruns, A.N.; Poirier, M.G.; Ottesen, J.J. Prepartion of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 2011, 408, 187–204. [Google Scholar]
- Le, D.D.; Cortesi, A.T.; Myers, S.A.; Burlingame, A.L.; Fujimori, D.G. Site-specific and regiospecific installation of methylarginine analogs into recombinant histones and insights into effector protein binding. J. Am. Chem. Soc. 2013, 135, 2879–2882. [Google Scholar]
- Al Temimi, A.H.K.; van der Wekken-de Bruijne, R.; Proietti, G.; Guo, H.; Qian, P.; Mecinovic, J. γ-Thialysine versus lysine: An insight into the epigenetic methylation of histones. Bioconjugate Chem. 2019, 30, 1798–1804. [Google Scholar]
- Proietti, G.; Rainone, G.; Hintzen, J.C.J.; Mecinovic, J. Exploring the histone acylome through incorporation of γ-thialysine on histone tails. Bioconjugate Chem. 2020, 31, 844–851. [Google Scholar]
- Huang, R.; Holbert, M.A.; Tarrant, M.K.; Curtet, S.; Colquhoun, D.R.; Dancy, B.M.; Dancy, D.C.; Hwang, Y.; Tang, Y.; Meeth, K.; et al. Site-specific introduction of acetyl-lysine mimic into peptides and proteins by cysteine alkylation. J. Am. Chem. Soc. 2010, 132, 9986–9987. [Google Scholar] [PubMed]
- Simon, M.D.; Chu, F.; Racki, L.R.; Cecile, C.; Burlingame, A.L.; Panning, B.; Narlikar, G.J.; Shokat, K.M. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 2007, 128, 1003–1012. [Google Scholar] [PubMed]
- Pieters, B.J.G.E.; Hintzen, J.C.J.; Grobben, Y.; Al Temimi, A.H.K.; Kamps, J.J.A.G.; Mecinovic, J. Installation of Trimethyllysine analogs on intact histones via cysteine alkylation. Bioconjugate Chem. 2019, 30, 952–958. [Google Scholar]
- Seeliger, D.; Soeroes, S.; Klingberg, R.; Schwarzer, D.; Grubmuller, H.; Fischle, W. Quantitative assessment of protein interaction with methyl-lysine analogues by hybrid computational and experimental approaches. ACS Chem. Biol. 2012, 7, 150–154. [Google Scholar] [PubMed]
- Chen, Z.; Notti, R.Q.; Ueberheide, B.; Ruthenburg, A.J. Quantitative and structural assessment of histone methyllysine analogue engagement by cognate binding proteins reveals affinity decrements relative to those of native counterparts. Biochemistry 2018, 57, 300–304. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Gallivan, J.P.; Dougherty, D.A. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar]
- Motherwell, W.B.; Moreno, R.B.; Pavlakos, I.; Arendorf, J.R.T.; Arif, T.; Tizzard, G.J.; Coles, S.J.; Aliev, A.E. Noncovalent interactions of π systems with sulfur: The atomic chameleon of molecular recognition. Angew. Chem. Int. Ed. 2018, 57, 1193–1198. [Google Scholar]
- Petraglia, R.; Corminboeuf, C. A Caveat on SCC-DFTB and noncovalent interactions involving sulfur atoms. J. Chem. Theory Comput. 2013, 9, 3020–3025. [Google Scholar]
- van der Lubbe, S.C.C.; Fonseca Guerra, C. The nature of hydrogen bonds: A delineation of the role of different energy components on hydrogen bond strengths and lengths. Chem. Asian J. 2019, 14, 2760–2769. [Google Scholar]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar]
- Pieters, B.J.G.E.; Belle, R.; Mecinovic, J. The effect of length on histone H3K4me3 on recognition by reader proteins. ChemBioChem 2013, 14, 2408–2412. [Google Scholar]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Christallogr. D. 2010, 66, 12–21. [Google Scholar]
- Peters, M.B.; Yang, Y.; Wang, B.; Fusti-Molnar, L.; Weaver, M.N.; Merz Jr, K.M. Structural survey of zinc containing proteins and the development of the zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar]
- Case, D.A.; Darden, T.A., III. AMBER11; University of California: San Francisco, CA, USA, 2010. [Google Scholar]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamic of peptides: The frictional dependence of isomerization rates of N-acetyalanyl-N’-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar]
- Ryckeart, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical intergration of the cartesian equations of a motion ofa system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Wang, H.; Fang, J.; Gao, X. The optimal particle-mesh interpolation basis. J. Chem. Phys. 2017, 124107. [Google Scholar]
- Philips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar]
- Miller III, B.R.; McGee Jr, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, N.; Roitberg, A.E. MMBPSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar]
- Fonseca Guerra, C.; van der Wijst, T.; Poater, J.; Swart, M.; Bickelhaupt, F.M. Adenine versus guanine quartets in aqueous solution: Dispersion-corrected DFT study on the differences in pi-stacking and hydrogen-bonding behavior. Theor. Chem. Acc. 2010, 125, 245–252. [Google Scholar]
- van der Wijst, T.; Fonseca Guerra, C.; Swart, M.; Bickelhaupt, F.M.; Lippert, B.A. A ditopic ion-pair receptor based on stacked nucleobase quartets. Angew. Chem. Int. Ed. 2009, 121, 3285–3287. [Google Scholar]
- Padial, J.S.; de Gelder, R.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Mecinovic, J. Stabilisation of 2,6-diarylpyridinium cation by through-space polar-pi interactions. Chem-Eur. J. 2014, 20, 6268–6271. [Google Scholar]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 19, 2315–2372. [Google Scholar]
- Brauer, B.; Kesharwani, M.K.; Kozuch, S.; Martin, J.M.L. The S66 × 8 benchmark for noncovalent interactions revisited: Explicitly correlated ab initio methods and density functional theory. Phys. Chem. Chem. Phys. 2016, 18, 20905–20925. [Google Scholar]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S.A. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar]
- Bickelhaupt, F.M.; Baerends, E.J. Reviews in Computational Chemistry; Wiley-VCH: New York, NY, USA, 2000; Volume 15, pp. 1–86. [Google Scholar]
- Bickelhaupt, F.M.; Diefenbach, A.; de Visser, S.P.; de Koning, L.J.; Nibberning, N.M.N. Nature of the three-electron bond in H2S∴SH2+. J. Phys. Chem. A 1998, 102, 9549–9553. [Google Scholar]
- Wolters, L.P.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory. WIREs. Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar]
- Fonseca Guerra, C.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of Mulliken, Bader, Hirshfeld, Weindhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H3K4me3 | H3KC4me3 | |||||||
|---|---|---|---|---|---|---|---|---|
| Kd (μM) | ΔG° (kcal mol−1) | ΔH° (kcal mol−1) | −TΔS° (kcal mol−1) | Kd (μM) | ΔG° (kcal mol−1) | ΔH° (kcal mol−1) | −TΔS° (kcal mol−1) | |
| KDM5APHD | 0.071 ± 0.008 | −9.7 ± 0.1 | −10.7 ± 0.1 | 1.0 ± 0.1 | 0.15 ± 0.2 | −9.3 ± 0.1 | −9.6 ± 0.1 | 0.3 ± 0.1 |
| TAF3PHD | 0.084 ± 0.012 | −9.6 ± 0.1 | −10.7 ± 0.1 | 1.1 ± 0.1 | 0.042 ± 0.007 | −10.1 ± 0.1 | −10.8 ± 0.1 | 0.7 ± 0.1 |
| BPTFPHD | 1.9 ± 0.2 | −7.8 ± 0.1 | −12.4 ± 0.1 | 4.6 ± 0.1 | 3.8 ± 0.5 | −7.4 ± 0.1 | −9.0 ± 0.1 | 1.6 ± 0.1 |
| SGF29TTD | 2.6 ± 0.3 | −7.6 ± 0.1 | −8.0 ± 0.1 | 0.4 ± 0.1 | 6.1 ± 0.7 | −7.1 ± 0.1 | −5.8 ± 0.1 | −1.3 ± 0.2 |
| KDM4ATTD | 6.6 ± 0.8 | −7.1 ± 0.1 | −13.0 ± 0.2 | 5.9 ± 0.2 | 3.1 ± 0.6 | −7.5 ± 0.1 | −14.8 ± 0.2 | 7.3 ± 0.2 |
| TRP2–Kme3 2 | TRP2–KCme3 3 | |
|---|---|---|
| ΔE(aq) | −10.2 | −8.1 |
| ΔEstrain(aq) | 0.1 | 2.6 |
| ΔEint(aq) | −10.3 | −10.7 |
| ΔEint(desolv) | 17.3 | 18.8 |
| ΔEint | −27.6 | −29.5 |
| ΔEPauli | 20.8 | 24.3 |
| ΔVelstat | −15.0 | −17.0 |
| ΔEoi | −13.0 | −14.4 |
| ΔEdisp | −20.4 | −22.3 |
| d(HMe-CTRP−6MR) | 2.88 | 2.94 |
| d(HMe-CTRP−5MR) | 2.78 | 2.88 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hintzen, J.C.J.; Poater, J.; Kumar, K.; Al Temimi, A.H.K.; Pieters, B.J.G.E.; Paton, R.S.; Bickelhaupt, F.M.; Mecinović, J. Comparison of Molecular Recognition of Trimethyllysine and Trimethylthialysine by Epigenetic Reader Proteins. Molecules 2020, 25, 1918. https://doi.org/10.3390/molecules25081918
Hintzen JCJ, Poater J, Kumar K, Al Temimi AHK, Pieters BJGE, Paton RS, Bickelhaupt FM, Mecinović J. Comparison of Molecular Recognition of Trimethyllysine and Trimethylthialysine by Epigenetic Reader Proteins. Molecules. 2020; 25(8):1918. https://doi.org/10.3390/molecules25081918
Chicago/Turabian StyleHintzen, Jordi C. J., Jordi Poater, Kiran Kumar, Abbas H. K. Al Temimi, Bas J. G. E. Pieters, Robert S. Paton, F. Matthias Bickelhaupt, and Jasmin Mecinović. 2020. "Comparison of Molecular Recognition of Trimethyllysine and Trimethylthialysine by Epigenetic Reader Proteins" Molecules 25, no. 8: 1918. https://doi.org/10.3390/molecules25081918
APA StyleHintzen, J. C. J., Poater, J., Kumar, K., Al Temimi, A. H. K., Pieters, B. J. G. E., Paton, R. S., Bickelhaupt, F. M., & Mecinović, J. (2020). Comparison of Molecular Recognition of Trimethyllysine and Trimethylthialysine by Epigenetic Reader Proteins. Molecules, 25(8), 1918. https://doi.org/10.3390/molecules25081918