Free and Immobilized Lecitase™ Ultra as the Biocatalyst in the Kinetic Resolution of (E)-4-Arylbut-3-en-2-yl Esters


 ,
,  , and
, and 
Abstract

1. Introduction
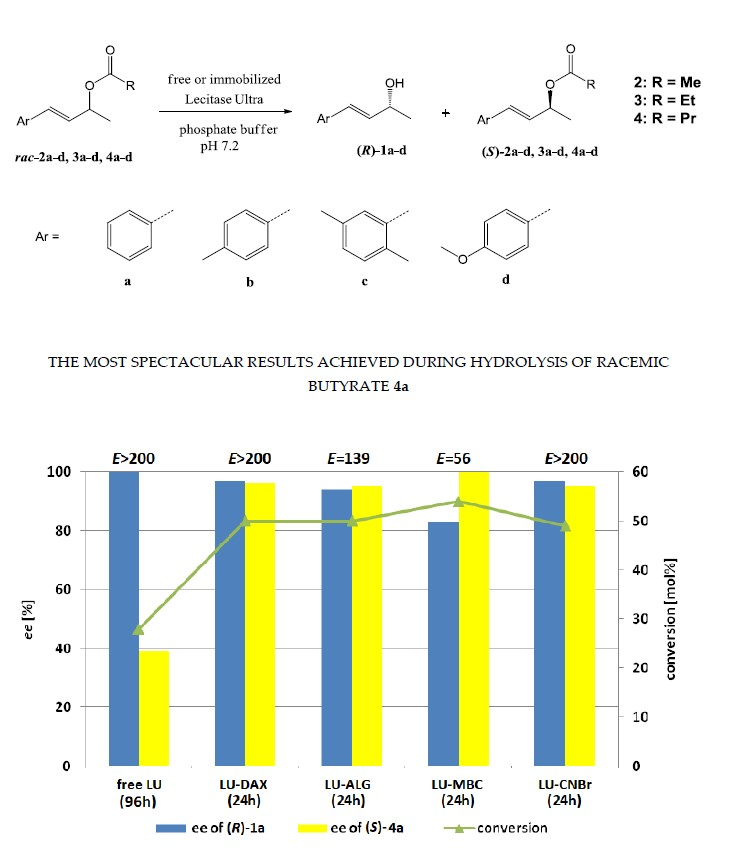
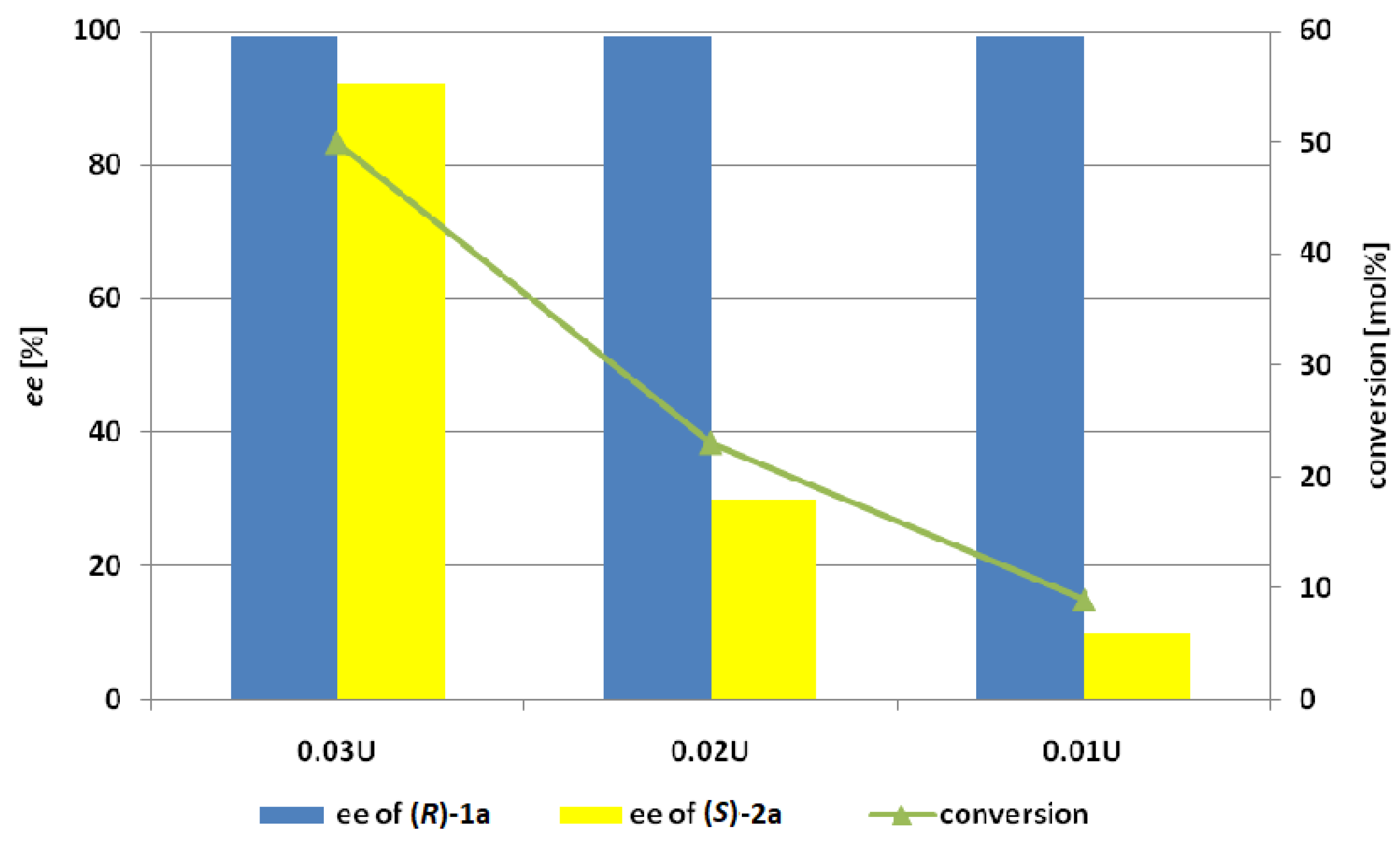
2. Results and Discussion
3. Materials and Methods
3.1. Enzyme and Chemicals
3.2. Analysis
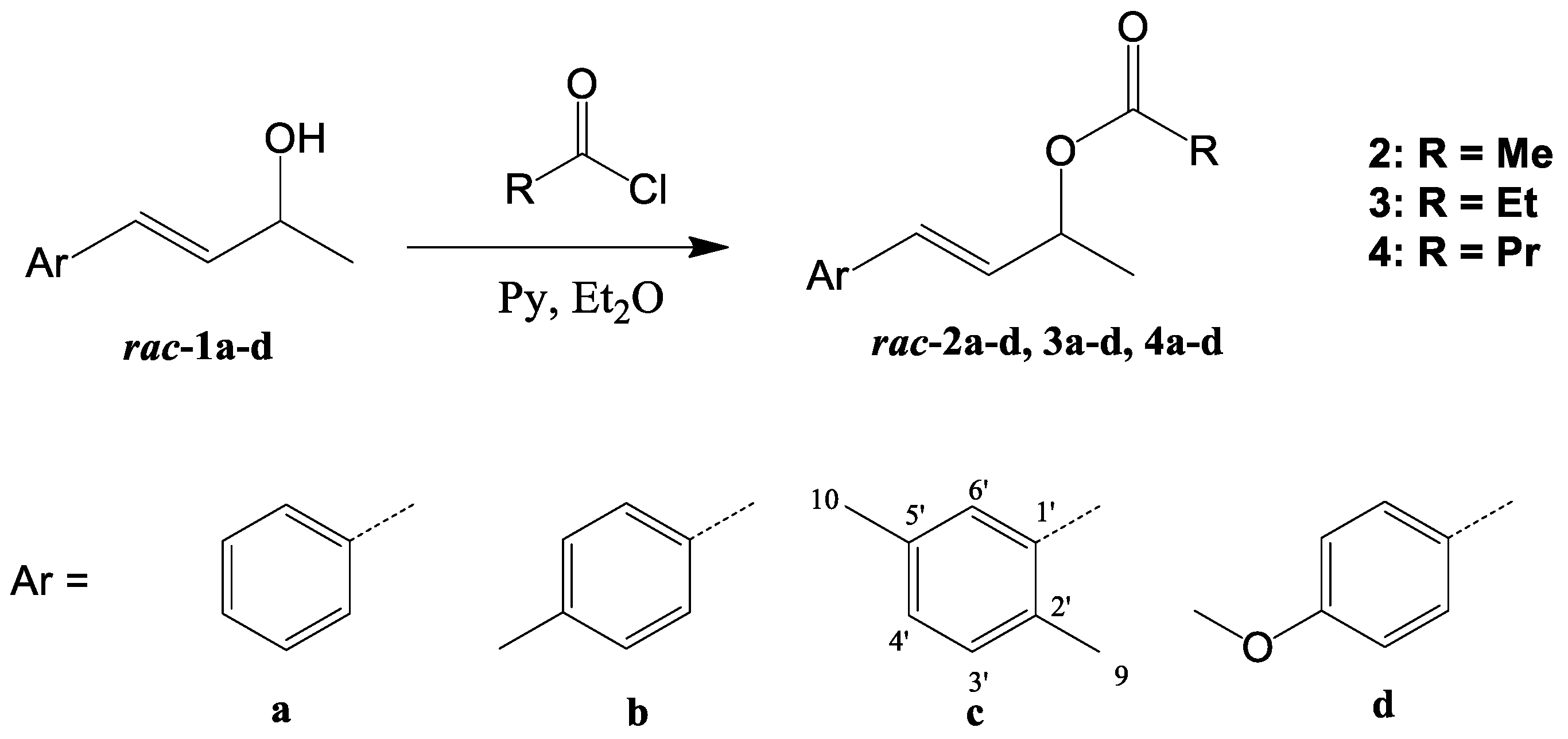
3.3. Synthesis of Racemic Esters 2a–d, 3a–d, and 4a–d—General Procedure
3.4. Determination of Enzyme Activity
3.5. Immobilization of Lecitase™ Ultra by Entrapment in Calcium Alginate
3.6. Immobilization of Lecitase™ Ultra by Adsorption on Polyacrylic Resin (Supelite™ DAX-8)
3.7. Immobilization of Lecitase™ Ultra by Covalent Bonds on Cyanogen Bromide-Activated Agarose (CNBr)
3.8. Immobilization of Lecitase™ Ultra by Covalent Bonds on Modified Bacterial Cellulose (MBC)
3.9. Hydrolysis of Esters 2a–d, 3a–d and 4a–d Catalyzed by free or Immobilized Lecitase™ Ultra
3.9.1. General Procedure
3.9.2. The Effect of Dosage of Lecitase Ultra immobilized Cyanogen Bromide-Activated Agarose (LU-CNBr)
3.9.3. Isolation of Products Obtained by Hydrolysis of Esters 4a–4c—General Procedure
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Virgen-Ortíz, J.J.; dos Santos, J.C.S.; Ortiz, C.; Berenguer-Murcia, Á.; Barbosa, O.; Rodrigues, R.C.; Fernandez-Lafuente, R. Lecitase Ultra: A phospholipase with great potential in biocatalysis. Mol. Catal. 2019, 473, 110405. [Google Scholar] [CrossRef]
- Yang, B.; Wang, Y.H.; Yang, J.G. Optimization of enzymatic degumming process for rapeseed oil. JAOCS J. Am. Oil Chem. Soc. 2006, 83, 653–658. [Google Scholar] [CrossRef]
- Manjula, S.; Jose, A.; Divakar, S.; Subramanian, R. Degumming rice bran oil using phospholipase-A1. Eur. J. Lipid Sci. Technol. 2011, 113, 658–664. [Google Scholar] [CrossRef]
- Lamas, D.L.; Crapiste, G.H.; Constenla, D.T. Changes in quality and composition of sunflower oil during enzymatic degumming process. LWT Food Sci. Technol. 2014, 58, 71–76. [Google Scholar] [CrossRef]
- Sampaio, K.A.; Zyaykina, N.; Wozniak, B.; Tsukamoto, J.; De Greyt, W.; Stevens, C.V. Enzymatic degumming: Degumming efficiency versus yield increase. Eur. J. Lipid Sci. Technol. 2015, 117, 81–86. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; Zhao, G.; Wang, P.; Wang, L.; Wu, H.; Fang, X.; Sun, X.; Wu, X.; Zheng, Z. Enhancing the performance of a phospholipase A1 for oil degumming by bio-imprinting and immobilization. J. Mol. Catal. B Enzym. 2016, 123, 122–131. [Google Scholar] [CrossRef]
- Baeza-Jiménez, R.; López-Martínez, L.X.; Otero, C.; Kim, I.H.; García, H.S. Enzyme-Catalysed hydrolysis of phosphatidylcholine for the production of lysophosphatidylcholine. J. Chem. Technol. Biotechnol. 2013, 88, 1859–1863. [Google Scholar] [CrossRef]
- Lim, C.W.; Kim, B.H.; Kim, I.H.; Lee, M.W. Modeling and optimization of phospholipase A1-catalyzed hydrolysis of phosphatidylcholine using response surface methodology for lysophosphatidylcholine production. Biotechnol. Prog. 2015, 31, 35–41. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, Y.; Wang, X. Enzymatic preparation of L-α-glycerylphosphorylcholine in an aqueous medium. Eur. J. Lipid Sci. Technol. 2012, 114, 1254–1260. [Google Scholar] [CrossRef]
- Bang, H.J.; Kim, I.H.; Kim, B.H. Phospholipase A1-catalyzed hydrolysis of soy phosphatidylcholine to prepare L-α-glycerylphosphorylcholine in organic-aqueous media. Food Chem. 2016, 190, 201–206. [Google Scholar] [CrossRef]
- Gerits, L.R.; Pareyt, B.; Masure, H.G.; Delcour, J.A. Native and enzymatically modified wheat (Triticum aestivum L.) endogenous lipids in bread making: A focus on gas cell stabilization mechanisms. Food Chem. 2015, 172, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Melis, S.; Pauly, A.; Delcour, J.A. Impact of lipases with different substrate specificity in wheat flour separation on the properties of the resultant gluten. J. Cereal Sci. 2017, 77, 291–296. [Google Scholar] [CrossRef]
- Melis, S.; Pauly, A.; Gerits, L.R.; Pareyt, B.; Delcour, J.A. Lipases as processing aids in the separation of wheat flour into gluten and starch: Impact on the lipid population, gluten agglomeration, and yield. J. Agric. Food Chem. 2017, 65, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Gofferjé, G.; Motulewicz, J.; Stäbler, A.; Herfellner, T.; Schweiggert-Weisz, U.; Flöter, E. Enzymatic degumming of crude jatropha oil: Evaluation of impact factors on the removal of phospholipids. JAOCS J. Am. Oil Chem. Soc. 2014, 91, 2135–2141. [Google Scholar] [CrossRef]
- Li, Y.; Huang, Y.; Du, W.; Dai, L.; Liu, D. Combined phospholipase and lipase catalysis for biodiesel production from phospholipids-containing oil. Biotechnol. Bioprocess Eng. 2015, 20, 965–970. [Google Scholar] [CrossRef]
- Moharana, T.R.; Byreddy, A.R.; Puri, M.; Barrow, C.; Rao, N.M. Selective enrichment of omega-3 fatty acids in oils by phospholipase A1. PLoS ONE 2016, 11, e0151370. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, M.; Song, K.; Wang, L.; Tang, S.; Riley, W.W. Partial hydrolysis of soybean oil by phospholipase A1 (Lecitase Ultra). Food Chem. 2010, 121, 1066–1072. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Hu, C.; Cao, Q.; Yang, X.; Zhao, M. Preparation of diacylglycerol-enriched oil from free fatty acids using Lecitase Ultra-catalyzed esterification. JAOCS J. Am. Oil Chem. Soc. 2011, 88, 1557–1565. [Google Scholar] [CrossRef]
- Liu, N.; Wang, Y.; Zhao, Q.; Cui, C.; Fu, M.; Zhao, M. Immobilisation of Lecitase® Ultra for production of diacylglycerols by glycerolysis of soybean oil. Food Chem. 2012, 134, 301–307. [Google Scholar] [CrossRef]
- Liu, N.; Wang, Y.; Zhao, Q.; Zhao, M. Production of palm oil-based diacylglycerol using LecitaseTM Ultra-catalyzed glycerolysis and molecular distillation. Food Sci. Biotechnol. 2014, 23, 365–371. [Google Scholar] [CrossRef]
- Kim, I.H.; Garcia, H.S.; Hill, C.G. Phospholipase A1-catalyzed synthesis of phospholipids enriched in n-3 polyunsaturated fatty acid residues. Enzym. Microb. Technol. 2007, 40, 1130–1135. [Google Scholar] [CrossRef]
- Kim, I.H.; Garcia, H.S.; Hill, C.G. Synthesis of structured phosphatidylcholine containing n-3 PUFA residues via acidolysis mediated by immobilized phospholipase A1. JAOCS J. Am. Oil Chem. Soc. 2010, 87, 1293–1299. [Google Scholar] [CrossRef]
- Baeza-Jiménez, R.; Noriega-Rodríguez, J.A.; García, H.S.; Otero, C. Structured phosphatidylcholine with elevated content of conjugated linoleic acid: Optimization by response surface methodology. Eur. J. Lipid Sci. Technol. 2012, 114, 1261–1267. [Google Scholar] [CrossRef]
- Gan, L.J.; Wang, X.Y.; Yang, D.; Zhang, H.; Shin, J.A.; Hong, S.T.; Park, S.H.; Lee, K.T. Emulsifying properties of lecithin containing different fatty acids obtained by immobilized Lecitase Ultra-catalyzed reaction. JAOCS J. Am. Oil Chem. Soc. 2014, 91, 579–590. [Google Scholar] [CrossRef]
- Li, X.; Chen, J.F.; Yang, B.; Li, D.M.; Wang, Y.H.; Wang, W.F. Production of structured phosphatidylcholine with high content of DHA/EPA by immobilized phospholipase A1-catalyzed transesterification. Int. J. Mol. Sci. 2014, 15, 15244–15258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; No, D.S.; Kim, B.H.; Garcia, H.S.; Kim, Y.; Kim, I.H. Immobilized phospholipase A1-catalyzed modification of phosphatidylcholine with n-3 polyunsaturated fatty acid. Food Chem. 2014, 157, 132–140. [Google Scholar] [CrossRef]
- Xi, X.; Feng, X.; Shi, N.; Ma, X.; Lin, H.; Han, Y. Immobilized phospholipase A1-catalyzed acidolysis of phosphatidylcholine from Antarctic krill (Euphausia superba) for docosahexaenoic acid enrichment under supercritical conditions. J. Mol. Catal. B Enzym. 2016, 126, 46–55. [Google Scholar] [CrossRef]
- Gumel, A.M.; Annuar, M.S.M. Thermomyces lanuginosus lipase-catalyzed synthesis of natural flavor esters in a continuous flow microreactor. 3 Biotech 2016, 6, 24. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Filice, M.; Terreni, M.; Guisan, J.M.; Fernandez-Lafuente, R.; Palomo, J.M. Lecitase® Ultra as regioselective biocatalyst in the hydrolysis of fully protected carbohydrates. Strong modulation by using different immobilization protocols. J. Mol. Catal. B Enzym. 2008, 51, 110–117. [Google Scholar] [CrossRef]
- Ghanem, A.; Aboul-Enein, H.Y. Application of lipases in kinetic resolution of racemates. Chirality 2005, 17, 1–15. [Google Scholar] [CrossRef]
- Carvalho, A.C.L.D.M.; Fonseca, T.D.S.; De Mattos, M.C.; De Oliveira, M.D.C.F.; De Lemos, T.M.L.G.; Molinari, F.; Romano, D.; Serra, I. Recent advances in lipase-mediated preparation of pharmaceuticals and their intermediates. Int. J. Mol. Sci. 2015, 16, 29682–29716. [Google Scholar] [CrossRef] [PubMed]
- Angajala, G.; Pavan, P.; Subashini, R. Lipases: An overview of its current challenges and prospectives in the revolution of biocatalysis. Biocatal. Agric. Biotechnol. 2016, 7, 257–270. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Palomo, J.M.; Guisan, J.M.; Fernandez-Lafuente, R. Effect of the immobilization protocol in the activity, stability, and enantioselectivity of Lecitase® Ultra. J. Mol. Catal. B Enzym. 2007, 47, 99–104. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Cabrera, Z.; Godoy, C.; Fernandez-Lafuente, R.; Palomo, J.M.; Guisan, J.M. Interfacially activated lipases against hydrophobic supports: Effect of the support nature on the biocatalytic properties. Process Biochem. 2008, 43, 1061–1067. [Google Scholar] [CrossRef]
- Mishra, M.K.; Kumaraguru, T.; Sheelu, G.; Fadnavis, N.W. Lipase activity of Lecitase® Ultra: Characterization and applications in enantioselective reactions. Tetrahedron Asymmetry 2009, 20, 2854–2860. [Google Scholar] [CrossRef]
- Dos Santos, J.C.S.; Garcia-Galan, C.; Rodrigues, R.C.; de Sant’ Ana, H.B.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Improving the catalytic properties of immobilized Lecitase via physical coating with ionic polymers. Enzym. Microb. Technol. 2014, 60, 1–8. [Google Scholar] [CrossRef]
- Garcia-Galan, C.; Barbosa, O.; Hernandez, K.; Dos Santos, J.C.S.; Rodrigues, R.C.; Fernandez-Lafuente, R. Evaluation of styrene-divinylbenzene beads as a support to immobilize lipases. Molecules 2014, 19, 7629–7645. [Google Scholar] [CrossRef]
- Rueda, N.; Dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Barbosa, O.; Fernandez-Lafuente, R. Improved performance of lipases immobilized on heterofunctional octyl-glyoxyl agarose beads. RSC Adv. 2015, 5, 11212–11222. [Google Scholar] [CrossRef]
- Mishra, M.K.; Harini, M.; Kumaraguru, T.; Lakshmi Prasanna, T.; Fadnavis, N.W. A porous vessel bioreactor for gel entrapped biocatalysts: Kinetic resolution of trans-methyl (4-methoxyphenyl)glycidate by Lecitase® Ultra in gelatin organogel (Gelozyme). J. Mol. Catal. B Enzym. 2011, 71, 56–62. [Google Scholar] [CrossRef]
- Kumaraguru, T.; Harini, T.; Basetty, S. Immobilization of Lecitase® Ultra on recyclable polymer support: Application in resolution of trans-methyl(4-methoxyphenyl)glycidate in organic solvents. Tetrahedron Asymmetry 2017, 28, 1612–1617. [Google Scholar] [CrossRef]
- Cabrera, Z.; Fernandez-Lorente, G.; Palomo, J.M.; Guisan, J.M.; Fernandez-Lafuente, R. Asymmetric hydrolysis of dimethyl 3-phenylglutarate catalyzed by Lecitase Ultra®. Effect of the immobilization protocol on its catalytic properties. Enzym. Microb. Technol. 2008, 43, 531–536. [Google Scholar] [CrossRef]
- Garcia-Galan, C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Zhao, Y.; Noreen, S.; Shah, S.Z.H.; Bharagava, R.N.; Iqbal, H.M.N. Modifying bio-catalytic properties of enzymes for efficient biocatalysis: A review from immobilization strategies viewpoint. Biocatal. Biotransform. 2019, 37, 159–182. [Google Scholar] [CrossRef]
- Di Cosimo, R.; Mc Auliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef] [PubMed]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef]
- Sheelu, G.; Kavitha, G.; Fadnavis, N.W. Efficient immobilization of Lecitase in gelatin hydrogel and degumming of rice bran oil using a spinning basket reactor. JAOCS J. Am. Oil Chem. Soc. 2008, 85, 739–748. [Google Scholar] [CrossRef]
- Yu, D.; Jiang, L.; Li, Z.; Shi, J.; Xue, J.; Kakuda, Y. Immobilization of phospholipase A1 and its application in soybean oil degumming. JAOCS J. Am. Oil Chem. Soc. 2012, 89, 649–656. [Google Scholar] [CrossRef]
- da Silva, F.B.; de Morais Júnior, W.G.; da Silva, C.V.; Vieira, A.T.; Batista, A.C.F.; de Faria, A.M.; Assunção, R.M.N. Preparation and characterization of cellulose triacetate as support for Lecitase Ultra immobilization. Molecules 2017, 22, 1930. [Google Scholar] [CrossRef]
- Drozd, R.; Szymańska, M.; Rakoczy, R.; Junka, A.; Szymczyk, P.; Fijałkowski, K. Functionalized magnetic bacterial cellulose beads as carrier for Lecitase® Ultra immobilization. Appl. Biochem. Biotechnol. 2019, 187, 176–193. [Google Scholar] [CrossRef]
- Liu, N.; Fu, M.; Wang, Y.; Zhao, Q.; Sun, W.; Zhao, M. Immobilization of LecitaseTM® Ultra onto a novel polystyrene DA-201 resin: Characterization and biochemical properties. Appl. Biochem. Biotechnol. 2012, 168, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.S.; Garcia-Galan, C.; Danelli, D.; Paludo, N.; Barbosa, O.; Rodrigues, R.C.; Fernandez-Lafuente, R. Use of Lecitase Ultra immobilized on styrene-divinylbenzene beads as catalyst of esterification reactions: Effects of ultrasounds. Catal. Today 2015, 255, 27–32. [Google Scholar] [CrossRef]
- Yu, D.; Ma, Y.; Xue, S.J.; Jiang, L.; Shi, J. Characterization of immobilized phospholipase A1 on magnetic nanoparticles for oil degumming application. LWT Food Sci. Technol. 2013, 50, 519–525. [Google Scholar] [CrossRef]
- Gonçalves, K.M.; Júnior, I.I.; Papadimitriou, V.; Zoumpanioti, M.; Leal, I.C.R.; De Souza, R.O.M.A.; Cordeiro, Y.; Xenakis, A. Nanoencapsulated Lecitase Ultra and Thermomyces lanuginosus lipase, a comparative structural study. Langmuir 2016, 32, 6746–6756. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galan, C.; Dos Santos, J.C.S.; Barbosa, O.; Torres, R.; Pereira, E.B.; Corberan, V.C.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Tuning of Lecitase features via solid-phase chemical modification: Effect of the immobilization protocol. Process Biochem. 2014, 49, 604–616. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Gliszczyńska, A.; Czarnecka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Białońska, A. Chiral δ-iodo-γ-lactones derived from cuminaldehyde, 2,5-dimethylbenzaldehyde and piperonal: Chemoenzymatic synthesis and antiproliferative activity. Tetrahedron Asymmetry 2016, 27, 227–237. [Google Scholar] [CrossRef]
- Gładkowski, W.; Włoch, A.; Pawlak, A.; Sysak, A.; Białońska, A.; Mazur, M.; Mituła, P.; Maciejewska, G.; Obmińska-Mrukowicz, B.; Kleszczyńska, H. Preparation of enantiomeric β-(2′,5′-dimethylphenyl)bromolactones, their antiproliferative activity and effect on biological membranes. Molecules 2018, 23, 3035. [Google Scholar] [CrossRef]
- Brenna, E.; Caraccia, N.; Fuganti, C.; Fuganti, D.; Grasselli, P. Enantioselective synthesis of β-substituted butyric acid derivatives via orthoester Claisen rearrangement of enzymatically resolved allylic alcohols: Application to the synthesis of (R)-(-)-baclofen. Tetrahedron Asymmetry 1997, 8, 3801–3805. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Grasselli, P.; Serra, S. Enzyme-mediated synthesis of (S)-and (R)-Verapamil. Eur. J. Org. Chem. 2001, 1349–1357. [Google Scholar] [CrossRef]
- Leśniarek, A.; Chojnacka, A.; Gładkowski, W. Application of Lecitase® Ultra-catalyzed hydrolysis to the kinetic resolution of (E)-4-phenylbut-3-en-2-yl esters. Catalysts 2018, 8, 423. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Wawrzeńczyk, C. Synthesis of halolactones with methoxyphenyl ring. Przem. Chem. 2011, 5, 918–922. [Google Scholar]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and anticancer activity of novel halolactones with β-aryl substituents from simple aromatic aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Gładkowski, W.; Gliszczyńska, A.; Siepka, M.; Czarnecka, M.; Maciejewska, G. Kinetic resolution of (E)-4-(2′,5′-dimethylphenyl)-but-3-en-2-ol and (E)-4-(benzo[d][1′,3′]dioxol-5′-yl)-but-3-en-2-ol through lipase-catalyzed transesterification. Tetrahedron Asymmetry 2015, 26, 702–709. [Google Scholar] [CrossRef]
- Mahdi, B.A.; Bhattacharya, A.; Gupta, A. Enhanced lipase production from Aeromonas sp. S1 using Sal deoiled seed cake as novel natural substrate for potential application in dairy wastewater treatment. J. Chem. Technol. Biotechnol. 2012, 87, 418–426. [Google Scholar] [CrossRef]
- Ghanem, A.; Schurig, V. Lipase-catalyzed access to enantiomerically pure (R)-and (S)-trans-4-phenyl-3-butene-2-ol. Tetrahedron Asymmetry 2003, 14, 57–62. [Google Scholar] [CrossRef]
- Thalén, L.K.; Sumic, A.; Bogár, K.; Norinder, J.; Persson, A.K.; Bäckvall, J.E. Enantioselective synthesis of α-methyl carboxylic acids from readily available starting materials via chemoenzymatic dynamic kinetic resolution. J. Org. Chem. 2010, 75, 6842–6847. [Google Scholar]
- Horn, A.; Kazmaier, U. Stereoselective modification of N-(α-Hydroxyacyl)-glycinesters via palladium-catalyzed allylic alkylation. Org. Lett. 2019, 21, 4595–4599. [Google Scholar] [CrossRef]
- Akai, S.; Hanada, R.; Fujiwara, N.; Kita, Y.; Egi, M. One-Pot synthesis of optically active allyl esters via lipase-vanadium combo catalysis. Org. Lett. 2010, 12, 4900–4903. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Białońska, A. Convenient chemoenzymatic route to optically active β-aryl-δ-iodo-γ-lactones and β-aryl-γ-iodo-δ-lactones with the defined configurations of stereogenic centers. Eur. J. Org. Chem. 2015, 2015, 605–615. [Google Scholar] [CrossRef]
- Skrobiszewski, A.; Gładkowski, W.; Maciejewska, G.; Wawrzeńczyk, C. Chemoenzymatic synthesis of trans-β-aryl-δ-hydroxy-γ-lactones and enzymatic kinetic resolution of their racemic mixtures. Molecules 2016, 21, 1552. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, H.K.; Park, J. (S)-selective dynamic kinetic resolution of allylic alcohols by enzyme-metal bicatalysis. Bull. Korean Chem. Soc. 2007, 28, 2096–2098. [Google Scholar]
- Pencreac’h, G.; Baratti, J.C. Hydrolysis of p-nitrophenyl palmitate in n-heptane by the Pseudomonas cepacia lipase: A simple test for the determination of lipase activity in organic media. Enzyme 1996, 18, 417–422. [Google Scholar]
- Blandino, A.; Macías, M.; Cantero, D. Formation of calcium alginate gel capsules: Influence of sodium alginate and CaCl2 concentration on gelation kinetics. J. Biosci. Bioeng. 1999, 88, 686–689. [Google Scholar] [CrossRef]
- Egger, D.; Wehtje, E.; Adlercreutz, P. Characterization and optimization of phospholipase A2 catalyzed synthesis of phosphatidylcholine. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1997, 1343, 76–84. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2a–2d, 3a–3d, 4a–4d are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Substrate | R | Co-Solvent | Buffer | t [h] | c [%]1 | ees [%] | eep [%] | E2 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2a | Me | acetone | Tris/HCl3 | 120 | 46 | 25 | 29 | 2 |
| 2 | 3a | Et | acetone | Tris/HCl | 120 | 42 | 52 | 71 | 10 |
| 3 | 4a | Pr | acetone | Tris/HCl | 168 | 47 | 77 | 88 | 36 |
| 4 | 2a | Me | acetone | phosphate4 | 168 | 51 | 50 | 49 | 5 |
| 5 | 3a | Et | acetone | phosphate | 144 | 45 | 70 | 84 | 24 |
| 6 | 4a | Pr | acetone | phosphate | 96 | 28 | 39 | >99 | >200 |
| 7 | 2a | Me | DMF | phosphate | 168 | 49 | 28 | 29 | 2 |
| 8 | 3a | Et | DMF | phosphate | 144 | 46 | 68 | 80 | 18 |
| 9 | 4a | Pr | DMF | phosphate | 96 | 52 | 39 | 88 | 23 |
| 10 | 2a | Me | DMSO | phosphate | 168 | 42 | 30 | 41 | 3 |
| 11 | 3a | Et | DMSO | phosphate | 144 | 27 | 22 | 59 | 5 |
| 12 | 4a | Pr | DMSO | phosphate | 96 | 53 | 69 | 87 | 30 |

| Entry | Substrate | Ar | R | t [h] | c [%]1 | ees [%] | eep [%] | E2 |
|---|---|---|---|---|---|---|---|---|
| 1 | 2b |  | Me | 72 | 43 | 9 | 12 | 1 |
| 2 | 3b |  | Et | 96 | 30 | 28 | 64 | 6 |
| 3 | 4b |  | Pr | 120 | 30 | 37 | 85 | 18 |
| 4 | 2c |  | Me | 144 | 41 | 42 | 61 | 6 |
| 5 | 3c |  | Et | 144 | 45 | 65 | 80 | 18 |
| 6 | 4c |  | Pr | 96 | 32 | 36 | 77 | 11 |
| 7 | 2d |  | Me | 6 | 59 | 0 | 0 | 0 |
| 8 | 3d |  | Et | 6 | 56 | 0 | 0 | 0 |
| 9 | 4d |  | Pr | 96 | 65 | 0 | 0 | 0 |
| Entry | Carrier | Biocatalyst | Activity [U/ g of support] |
|---|---|---|---|
| 1 | Supelite™ DAX-8 | LU-DAX | 0.06 |
| 2 | Sodium Alginate | LU-ALG | 0.04 |
| 3 | Modified Bacterial Celulose | LU-MBC | 0.045 |
| 4 | Cyanogen Bromide-Activated Agarose | LU-CNBr | 0.06 |

| Entry | Substrate | Ar | t [h] | c [%]1 | ees [%] | eep [%] | E2 |
|---|---|---|---|---|---|---|---|
| 1 | 4b |  | 24 | 48 | 89 | 98 | >200 |
| 2 | 4c |  | 48 | 50 | 99 | 99 | >200 |
| 3 | 4d |  | 72 | 56 | 0 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leśniarek, A.; Chojnacka, A.; Drozd, R.; Szymańska, M.; Gładkowski, W. Free and Immobilized Lecitase™ Ultra as the Biocatalyst in the Kinetic Resolution of (E)-4-Arylbut-3-en-2-yl Esters. Molecules 2020, 25, 1067. https://doi.org/10.3390/molecules25051067
Leśniarek A, Chojnacka A, Drozd R, Szymańska M, Gładkowski W. Free and Immobilized Lecitase™ Ultra as the Biocatalyst in the Kinetic Resolution of (E)-4-Arylbut-3-en-2-yl Esters. Molecules. 2020; 25(5):1067. https://doi.org/10.3390/molecules25051067
Chicago/Turabian StyleLeśniarek, Aleksandra, Anna Chojnacka, Radosław Drozd, Magdalena Szymańska, and Witold Gładkowski. 2020. "Free and Immobilized Lecitase™ Ultra as the Biocatalyst in the Kinetic Resolution of (E)-4-Arylbut-3-en-2-yl Esters" Molecules 25, no. 5: 1067. https://doi.org/10.3390/molecules25051067
APA StyleLeśniarek, A., Chojnacka, A., Drozd, R., Szymańska, M., & Gładkowski, W. (2020). Free and Immobilized Lecitase™ Ultra as the Biocatalyst in the Kinetic Resolution of (E)-4-Arylbut-3-en-2-yl Esters. Molecules, 25(5), 1067. https://doi.org/10.3390/molecules25051067