Abstract
The enzymatic synthesis of nucleoside analogues has been shown to be a sustainable and efficient alternative to chemical synthesis routes. In this study, dihalogenated nucleoside analogues were produced by thermostable nucleoside phosphorylases in transglycosylation reactions using uridine or thymidine as sugar donors. Prior to the enzymatic process, ideal maximum product yields were calculated after the determination of equilibrium constants through monitoring the equilibrium conversion in analytical-scale reactions. Equilibrium constants for dihalogenated nucleosides were comparable to known purine nucleosides, ranging between 0.071 and 0.081. To achieve 90% product yield in the enzymatic process, an approximately five-fold excess of sugar donor was needed. Nucleoside analogues were purified by semi-preparative HPLC, and yields of purified product were approximately 50% for all target compounds. To evaluate the impact of halogen atoms in positions 2 and 6 on the antiproliferative activity in leukemic cell lines, the cytotoxic potential of dihalogenated nucleoside analogues was studied in the leukemic cell line HL-60. Interestingly, the inhibition of HL-60 cells with dihalogenated nucleoside analogues was substantially lower than with monohalogenated cladribine, which is known to show high antiproliferative activity. Taken together, we demonstrate that thermodynamic calculations and small-scale experiments can be used to produce nucleoside analogues with high yields and purity on larger scales. The procedure can be used for the generation of new libraries of nucleoside analogues for screening experiments or to replace the chemical synthesis routes of marketed nucleoside drugs by enzymatic processes.
1. Introduction
The standard production route for nucleoside analogues in the pharmaceutical industry is still chemical synthesis. It usually involves protection–deprotection steps and the use of hazardous chemicals and solvents. Furthermore, chemical synthesis routes for purine nucleoside analogues usually show low selectivity and finally lead to low product yields [1,2]. Due to the drawbacks of chemical synthesis routes, alternative methods have been developed. Enzymatic synthesis is environmentally more friendly, highly selective, and efficient [3]. The most commonly applied enzyme-catalyzed reaction for the preparation of nucleosides and their analogues is the one-pot transglycosylation with pyrimidine and purine nucleoside phosphorylases [4]. Here, the sugar moiety is exchanged between two nucleobases in the presence of inorganic phosphate.
So far, mainly nucleoside phosphorylases (NP) of mesophilic organisms have been used for the synthesis of nucleoside analogues. However, enzymes from thermophilic bacteria or archaea are of increasing interest, as they are active over a wide temperature and pH range, which facilitates the solubility of substrates. Furthermore, mass transfer is improved due to a decreased viscosity and an increase in diffusion coefficient and flux at higher temperatures [5,6]. In addition, due to the ease of their purification, thermophilic enzymes are cost-effective biocatalysts for industrial processes [6].
Nucleoside analogues form a valuable class of drugs that have been widely used for more than 50 years for several indications such as cancer, viral, or protozoal infections [7,8,9]. Purine nucleoside analogues such as cladribine, fludarabine, or clofarabine are marketed drugs for the treatment of hematological malignancies [8]. These compounds are derivatives of adenosine and show modifications in the base and/or the sugar moiety. Cladribine, fludarabine, and clofarabine have a halogen atom at position 2 of the nucleobase, which was shown to be an important prerequisite for deaminase resistance and, therefore, its increased intracellular activity [10]. These compounds all share a similar mechanism of action, such as the inhibition of DNA synthesis, inhibition of DNA repair, and accumulation of DNA strand breaks.
Biological properties of enzymatically produced nucleoside analogues have been studied before. Analogues of 2′-deoxyribosides were produced using mesophilic nucleoside deoxyribosyltransferase from Lactobacillus leichmannii [11,12]. In a first attempt, 12 analogues of 2′-deoxyadenosine were enzymatically produced at a 100 to 400 mg scale with an average yield of 64% [11]. In a second study, 8-substituted purine nucleoside analogues were synthesized with yields of <10 to 70% [12]. Of the compounds produced, 2-chloro and 2-bromo analogues of 2′-deoxyadenosine were good inhibitors in tumor cell lines [11,12].
In the present study, the enzymatic synthesis of 2,6-dichloropurine riboside (3a), 2,6-dichloropurine deoxyriboside (3b), 6-chloro-2-fluoropurine riboside (3c), and 6-chloro-2-fluoropurine deoxyriboside (3d) with thermostable nucleoside phosphorylases was optimized based on thermodynamic equilibrium state calculations [13]. Equilibrium constants for dihalogenated nucleosides were calculated and used to determine optimum reactions to reach 90% or 95% product yields. The transferability of the results obtained on an analytical scale to up-scaling experiments was analyzed. Furthermore, we evaluated the cytotoxic activity of dihalogenated nucleoside analogues in a hematologic tumor cell line. While 2-halogenated nucleoside analogues were shown to have an increased stability and activity due to a resistance to deaminases [10], dihalogenated compounds have not been studied before.
2. Results
2.1. Optimization of the Synthesis of Dihalogenated Nucleosides Based on Thermodynamic Calculations
Transglycosylation reactions were employed to produce 2,6-dichloropurine (2a) and 6-chloro-2-fluoropurine (2b) containing nucleosides using uridine (1a) and thymidine (1b) as sugar donors (Scheme 1). Pyrimidine nucleoside phosphorylase (PyNP, EC 2.4.2.2) and purine nucleoside phosphorylase (PNP, EC 2.4.2.1) from a thermophilic organism were applied as biocatalysts.
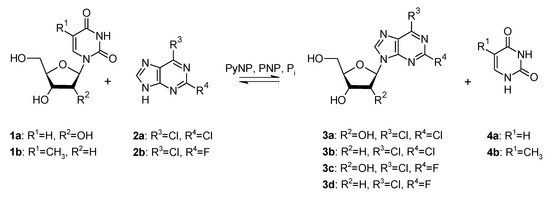
Scheme 1.
Transglycosylation reaction to produce dihalogenated purine nucleosides using thermostable nucleoside phosphorylases as biocatalysts and uridine (1a) and thymidine (1b) as sugar donors. 2a—2,6-dichloropurine, 2b—6-chloro-2-fluoropurine, 3a—2,6-dichloropurine riboside, 3b—2,6-dichloropurine deoxyriboside, 3c—6-chloro-2-fluoropurine riboside, 3d—6-chloro-2-fluoropurine deoxyriboside, 4a—uracil, 4b—thymine, PyNP—pyrimidine nucleoside phosphorylase, PNP—purine nucleoside phosphorylase, Pi—inorganic phosphate.
To efficiently produce 2a- and 2b-containing nucleosides in a transglycosylation reaction, equilibrium state thermodynamic calculations were carried out [13]. We have recently shown that nucleoside phosphorolysis is a reversible endothermic reaction under tight thermodynamic control [14]. Consequently, it is possible to predict the equilibrium states of transglycosylation reactions, if the equilibrium constants of phosphorolysis of both participating nucleosides are known [13]. However, for 3a–d, equilibrium constants of phosphorolysis have not been described before. Thus, the equilibrium states of analytical-scale reactions were used to calculate these values for 3a–d, employing previously published equations (for more detail, please see reference [14], the respective Supplementary Materials [15,16], as well as the externally hosted supplementary material of this publication [17]). The recently described equilibrium constants of the sugar donors 1a and 1b [14] served as the basis for these calculations. Analytical-scale reactions were performed with equal concentrations of base and sugar donor of 5 mM and 2 mM of phosphate. At equilibrium, product formation was between 55% (3b) and 60% (3a and 3c).
Thus, the calculated equilibrium constants (K2) were in the range of 0.071 (3d) to 0.081 (3a) (Table 1). These values fit well with previously reported equilibrium constants of the phosphorolysis of other purine nucleosides [14]. Equilibrium constants were slightly higher for 2a-derived nucleosides (3a and 3b) compared to 2b-derived nucleosides (3c and 3d) and for deoxyribonucleosides in comparison to ribonucleosides (Table 1).

Table 1.
Equilibrium state thermodynamic calculations were used to determine appropriate reaction conditions for the maximum conversion of 3a–d.
Employing these equilibrium constants, optimal reaction conditions to provide 90% or 95% conversion were calculated. Sugar donor excess was predicted to be in the range of 4.4 to 6.1 to reach 90% product yield and 8.8 to 12.5 to obtain 95% product formation (Table 1). In accordance with the equilibrium constants of 1a and 1b, a higher sugar donor excess was estimated to be needed for the synthesis of deoxyribonucleosides compared to the corresponding ribonucleosides.
Analytical-scale experiments with the selected conditions confirmed the theoretical predictions. Product formation of 90% and 95% was observed under the tested reaction conditions for all target compounds (Figure 1). However, the time to reach equilibrium differed between ribonucleosides and deoxyribonucleosides. While equilibrium was reached within 1 h for 3b and 3d, equilibrium was only observed after 6 h for 3a and 3c (Figure 1). Therefore, for semi-preparative-scale, reactions with ribonucleosides and deoxyribonucleosides were stopped after 6 h and 2 h, respectively. In semi-preparative experiments, conditions were used to reach 90% product yield. Doubling of the amount of 1a and 1b would have been needed to reach 95% product formation. However, high quantities of the sugar donor would increase the substrate cost and the ecological burden, as a lot of sugar donor is wasted. In addition, it complicates purification, as an efficient product separation is no longer ensured.
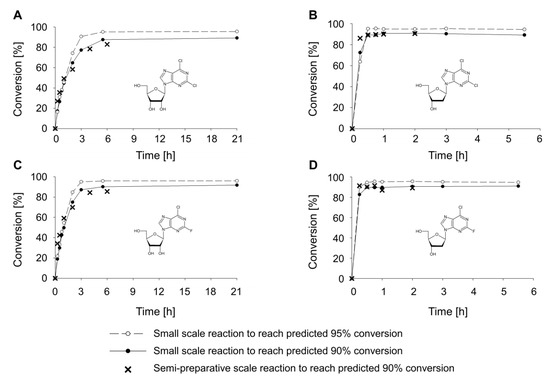
Figure 1.
Synthesis of 3a (A), 3b (B), 3c (C), and 3d (D) in transglycosylation reactions at small and semi-preparative scale using thermostable nucleoside phosphosporylases PyNP 02 and PNP 02. Equilibrium state calculations served as a basis to determine reaction conditions and were confirmed by reactions performed at small and semi-preparative scale. Base concentrations of 5 mM and sugar donor concentrations of 22 to 62.5 mM (Table S3) were used. Substrate concentrations were chosen based on thermodynamic calculations. Thermostable nucleoside phosphorylases were added to a final concentration of 0.1 mg mL−1 each. Enzymatic reactions were performed in 0.5 mM potassium phosphate buffer at pH 7.5 using a reaction temperature of 40 °C. Conversion was calculated based on formation of the product nucleoside from the halogenated base.
2.2. Synthesis of Dihalogenated Nucleosides in Semi-Preparative Scale
Dihalogenated nucleosides were produced at 50-mL scale to validate the efficiency and specificity of the synthesis and purification process. Products were purified by HPLC. Recovery of the sugar donor was validated for 3b as an example. Appropriate reaction conditions were employed to reach 90% product yield.
Enzymatic transglycosylation reactions led to conversions of 83% (3a), 92% (3b), 90% (3c), and 92% (3d) (Figure 1, Table S1). This was in good accordance with small-scale experiments. The time course of product formation was also well comparable with small-scale studies. Equilibrium was reached within 1 h for 3b and 3d and mainly within 6 h for 3a and 3c (Figure 1). Except for the expected reaction products uracil (4a) or thymine (4b), no further by-products were observed by HPLC analysis in any of the reactions.
Products synthesized in transglycosylation reactions were purified by semi-preparative HPLC. Proteins were removed by filtration as a prerequisite for this step. Product loss during protein removal and HPLC purification was in the range of 18% (3a) to 31% (3b and 3d) (Table S1). Still, this step was required to guarantee high product purity. Freeze-drying yielded the target compounds as white powders. The highest losses in product quantity (28% to 32%) were observed in this step (Table S1). Losses can be reduced by collecting more of the product during HPLC purification and by conducting a second round of freeze-drying.
In total, 34 mg, 36 mg, 32 mg, and 32 mg of purified powder were obtained for 3a, 3b, 3c, and 3d, respectively (Table S1). These values correspond to a recovery of approximately 50% for each target compound (Table S1). Product purity was greater than 96%, and structures were confirmed by NMR (Table S2, Figure S1 and Figure S2).
Since the sugar donors used can be a cost factor for the enzymatic synthesis of nucleoside analogues, the recovery of 1b was analyzed using the synthesis of 3b as an example. In the enzymatic reaction, 369 mg of 1b was added. The transglycosylation reaction resulted in 74 mg (20%) conversion to 4b. After protein removal and HPLC purification, 251 mg of the remaining 1b were recovered. In the freeze-drying process 22% of 1b was lost and in total, 195 mg 1b (53% yield) was recaptured from the initial 369 mg added as the starting material.
2.3. Effect of Dihalogenated Nucleoside Analogues on Cell Growth
Dihalogenated nucleosides 3a–d were tested for their ability to inhibit the growth of HL-60 cells in culture. Monohalogenated cladribine, which is known to efficiently inhibit HL-60 cells [18,19], was used as positive control. While for cladribine an IC50 value of 0.22 µM [± 0.11 µM] was determined, 50% of inhibition by the dihalogenated nucleoside analogues was observed for concentrations between 10 and 100 µM (Figure 2). The inhibition of growth of the control cell line HEK293 was in the similar range of nucleoside analogue concentrations. No unspecific growth inhibition in the control cell line HEK293 was detected for cladribine. Hence, nucleoside analogues with halogen atoms in positions 2 and 6 are no suitable inhibitors of leukemia cell lines.
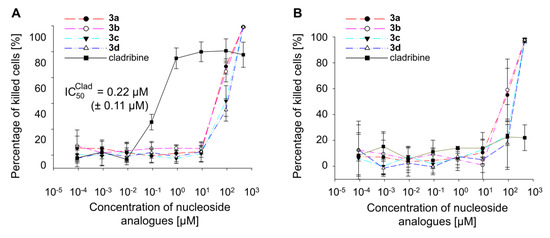
Figure 2.
Biological activity of dihalogenated nucleoside analogues in the leukemia cell line HL-60 (A) or the control cell line HEK293 (B). Nucleoside analogues were serially diluted, and the percentage of killed cells was determined by XTT assay.
3. Discussion
The current study shows the feasibility of optimizing the enzymatic synthesis of modified nucleosides based on initial experiments and thermodynamic calculations to determine equilibrium constants. Dihalogenated nucleoside analogues 3a–d were formed with percentages of conversion of 90% to 95% at small scale and 83% to 92% at semi-preparative scale. Yields of purified product were approximately 50%.
Dihalogenated purine bases have been studied before as substrates for NPs from extreme environments. Both 2a and 2b were converted to the respective riboside and deoxyriboside nucleosides by a thermostable PNP from Geobacillus thermoglucosidasius with conversions of 54% to 69% [20]. A sugar donor-to-nucleobase ratio of 3.34 in the presence of 3.34 equivalents of phosphate (compared to the base concentration) was used. Using an immobilized PyNP of Thermus thermophilus and the same PNP, Zhou and colleagues later observed increasing product formation with reduced phosphate concentrations [21]. A negative impact on the product yield of phosphate equivalents above 0.3 in comparison to the nucleobase was recently confirmed by thermodynamic studies on transglycosylation reactions [13,22]. The advantages of the application of thermodynamic equilibrium state calculations in transglycosylation reactions is convincingly confirmed in the present study, which also represents the first example of the fruitful application of this methodology in practice. After calculating the equilibrium constants based on a single analytical reaction each, reaction conditions were designed to reach product yields of 90% and 95%. Small- and semi-preparative-scale experiments employing the improved conditions confirmed the calculated predictions. Hence, under optimized conditions, higher product yields were observed compared to previous studies [20,21].
Besides the optimization of the enzymatic synthesis process by thermodynamic calculation, the present study also investigated the impact of a chlorine atom at position 6 of the nucleoside with respect to its inhibitory effect on leukemic cells. This was based on the observation that halogenation is an important prerequisite for anticancer activity [3,23,24], since amino groups make the compound susceptible to deamination by adenosine deaminase (ADA) [25,26]. The addition of a chlorine or fluorine atom in position 2 of adenosine strongly increased its stability, as observed for the very potent leukemia drugs cladribine and fludarabine [23,26]. Interestingly, the dihalogenated analogues tested in the present study with cell line HL-60 showed less growth inhibition than the monohalogenated cladribine. An explanation for the low activity could involve the uptake of the dihalogenated nucleosides, the activation by nucleoside and nucleotide kinases, or the interaction with the intracellular targets. The uptake of cladribine involves equilibrative nucleoside transporter 1 and 2 as well as concentrative nucleoside transporter 3 [27], which might not recognize dihalogenated nucleosides. Most of the known nucleoside analogue drugs including cladribine are only active as the respective di- or triphosphorylated nucleotides, and activation in vivo is often insufficient [28]. Hence, a lack of activity might be explained by human kinases not efficiently phosphorylating dihalogenated nucleosides. Intracellular targets of cladribine have been recently well summarized by Freyer and colleagues [29]. Ribonucleotide reductase is one of the main targets of cladribine. Its inhibition leads to a depletion of deoxyadenosine and hence to an imbalance within the deoxyribonucleotide pool. This leads to a number of other effects such as endonuclease activation and double-strand DNA breaks. Additionally, the inhibition of adenosine deaminase [30] or RNA polymerase [31] by cladribine were described before. However, further studies are needed to better understand the limitations of dihalogenated nucleosides in the inhibition of leukemia cell lines.
Although the dihalogenated nucleoside analogues showed less growth inhibition in leukemia cell line HL-60 than their monohalogenated counterparts, they may also serve as valuable starting material for the preparation of a broad palette of modified nucleosides [32,33]. Dihalogenated nucleoside analogues have a much higher solubility than monohalogenated analogues [20] and can therefore serve as precursors for the synthesis of new compounds or approved substances in industrial scale.
4. Materials and Methods
4.1. General Information
All chemicals and solvents were of analytical grade or higher and purchased, if not stated otherwise, from Sigma-Aldrich (Steinheim, Germany), Carl Roth (Karlsruhe, Germany), TCI Deutschland (Eschborn, Germany), and VWR (Darmstadt, Germany). Cladribine was obtained from TCI with a purity >98%. Water was purified and deionized by an EASYpure II purification system to reach a resistivity of 18.2 MΩ cm at 25 °C (Werner, Leverkusen, Germany). Thermostable nucleoside phosphorylases (PyNP 02 [E-PyNP-0002] and PNP 02 [E-PNP-0002]) were provided by BioNukleo and used as recommended by the manufacturer. Nucleoside phosphorylases were heterologously expressed in E. coli and originate from thermophilic bacteria with temperature optima at 60 °C. Enzymes were purified by affinity chromatography. Enzyme concentrations of 0.1 mg mL−1 were used for PyNP and PNP. PyNP 02 and PNP 02 had activities of 40 U mL−1 and 60 U mL−1 at 40 °C and pH 9 for their natural substrates uridine and adenosine, respectively, as determined by the UV spectroscopy-based assay described recently [34]. Cell lines HEK-239 and HL-60 were purchased from the German Collection of Microorganisms and Cell Cultures GmbH. All experimental and calculated data are available free of charge from an external online repository [17].
4.2. Optimization of the Enzymatic Synthesis of Dihalogenated Purine Nucleosides
Thermodynamic calculations were shown to allow for a reliable calculation of product yields in nucleoside phosphorylase catalyzed transglycosylation reactions based on equilibrium constants of phosphorolysis [13,22]. Equilibrium constants for the phosphorolysis of uridine (1a) and thymidine (1b) were previously described to be 0.18 and 0.125, respectively, at 40 °C [14]. To estimate equilibrium constants K2 for the synthesis of the dihalogenated nucleoside analogues, enzymatic reactions were performed with 5 mM of sugar donor (1a or 1b), 5 mM of the purine base 2a or 2b) in 2 mM potassium phosphate (pH 7.5) using 0.1 mg mL−1 of the biocatalysts PyNP 02 and PNP 02. Reactions were performed at 40 °C for 20 h. To analyze sugar donor cleavage and product formation, samples were analyzed by HPLC. First, 30 µL were removed from the reaction mixture, and an equal volume of methanol was added to stop the reaction. After centrifugation at 21,500 g for 10 min, the supernatant was diluted 1:10 and transferred to HPLC vials.
Solving the system of equilibrium constraints given by the laws of mass action of the first and second (reverse) phosphorolysis of the transglycosylation sequence for these values, as described recently [13,14], yielded equilibrium constants of phosphorolysis for 3a–d. Based on these equilibrium constants, the sugar donor excess needed to reach 90% and 95% product formation was calculated [13]. Sugar donor excess was calculated to be between 4.4 and 6.1 to reach 90% product yield and 8.8 and 12.5 to guarantee 95% product formation (Table S3). Reactions were performed to verify theoretical calculations at 1 mL scale. Base concentrations of 5 mM and enzyme concentration of 0.1 mg mL−1 for PyNP 02 and PNP 02 were used in 0.5 mM potassium phosphate buffer (pH 7.5). The reaction temperature was 40 °C. Regular samples were taken to analyze sugar donor cleavage and product formation by HPLC. A 30 µL sample was removed from the reaction mixture, and an equal volume of methanol was added to stop the reaction. After centrifugation at 21,500 g for 10 min, the supernatant was diluted 1:10 and transferred to HPLC vials.
4.3. Enzymatic Production of Dihalogenated Purine Nucleosides
To produce dihalogenated nucleoside analogues, base concentrations of 5 mM and sugar donor concentrations of 22 to 30.5 mM were applied in a reaction volume of 50 mL. Thermostable pyrimidine and purine nucleoside phosphorylase PyNP 02 and PNP 02 were added in a concentration of 0.1 mg mL−1 each. Enzymatic reactions were performed in 0.5 mM potassium phosphate buffer at pH 7.5 using a reaction temperature of 40 °C. Reactions were stirred at 100 rpm. Regular samples were taken to monitor enzymatic reactions by HPLC as described above.
The reactions were stopped by enzyme removal after 2 h and 6 h for deoxyriboside and riboside nucleosides, respectively. Protein was removed by filtration using centrifugal filter units with a cut-off of 30 kDa (Millipore, Darmstadt, Germany) at room temperature. Filtrate was stored at 4 °C until purification by semi-preparative HPLC.
Analytical HPLC was performed to determine product quantity after each step. Product loss per step was determined based on the product quantity of step 1 (e.g., after enzymatic synthesis) and the product quantity of the following step (e.g., after protein depletion and HPLC).
4.4. Cell Culture
HEK293 cells were cultured in low-glucose Dulbecco’s modified Eagle’s medium (Biowest) containing 10% fetal bovine serum, 2 mM glutamine, non-essential amino acids, 4.5 mg mL−1 glucose, and the antibiotics penicillin and streptomycin. HL-60 cells were cultured in RPMI 1640 w/o l-Glutamine supplemented with 10% fetal bovine serum, 2 mM glutamine, and the antibiotics penicillin and streptomycin. Cells were grown at 37 °C in a humidified atmosphere with 5% CO2.
4.5. XTT Cell Proliferation Assay
Cytotoxicity was measured by the XTT assay at 450 nm as described by the manufacturer (Roche Diagnostics, Basel, Switzerland). Phosphate-buffered saline was used as negative control. For HL-60 and HEK293 cells, 1.2 × 105 cells in growth media were transferred to each well of a 96-well flat-bottom plate. Subsequently, modified nucleosides were added in varying concentrations. XTT assay was performed after 24 h for HL-60 and HEK293 cells, as described previously [35]. At least three replicates for each nucleoside concentration were performed. All experiments were conducted for a minimum of three independent experiments.
4.6. Analytical HPLC
Nucleosides and bases were analyzed by HPLC (λ = 260 nm) using a reversed-phase Kinetex EVO C18 column (Phenomenex, Aschaffenburg, Germany) at 25 °C with a flow rate of 1 mL min−1. Substrates and products were identified by comparing their retention times with those of authentic standards. Quantification was performed using a six-step calibration from a serial dilution in the range from 0 to 1 mM over the peak area.
Gradual elution was performed using eluent A (20 mM ammonium acetate) and eluent B (100% acetonitrile). The following gradient was used: 97% A and 3% B; 10-min linear gradient to 60% A and 40% B. The initial condition (97% A and 3% B) was restored afterwards and maintained for 4 min.
Typical retention times were as follows: 1a: 3.2 min, 4a: 2.4 min, 1b: 4.7 min, 4b: 4 min, 3c: 7.5 min, 3d: 8.2 min, 2b: 7 min, 3a: 8.2 min, 3b: 8.8 min, and 2a: 7.6 min. The percentage of conversion of nucleosides was calculated as described previously (Equation (1)) [36].
4.7. Purification of Nucleoside Analogues by Semi-Preparative HPLC
Nucleoside analogues were purified at room temperature using a KNAUER HPLC system equipped with a Smartline Detector 2600 and an AZURA P 2.1 L pump. A flow rate of 21.24 mL min−1 and a Kinetex® 5 μm EVO C18 250 × 21.2 mm RP column were used. Deionized water and acetonitrile were used as eluents, while the gradient was modified from the analytical method as summarized in Table S4. Fractions containing either the sugar donor or the product were collected.
Collected samples were dried using a Christ Gamma 1–20 freeze-dryer (Osterode am Harz, Germany). Residual water was removed by drying at 50 °C in a drying cabinet (DRY-Line 112 Prime, VWR) for 8 h.
4.8. NMR Analysis
The NMR spectra for 1H, 13C, and 19F were recorded in DMSO-d6 (purchased from Eurisotop) on a Bruker Avance III 500 MHz instrument. Chemical shifts are reported in parts per million (ppm) and are referenced to the residual solvent resonance as the internal standard (1H NMR: δ = 2.50 ppm for DMSO-d5; 13C NMR: δ = 39.52 ppm for DMSO-d6) [37]. 19F NMR spectra are referenced in compliance with the unified scale for NMR chemical shifts as recommended by the IUPAC stating the chemical shift relative to CCl3F [38]. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, mc = centrosymmetric multiplet, br = broad signal), coupling constants (Hz), and integration.
5. Conclusions
The present study demonstrates the value of small-scale experiments combined with thermodynamic equilibrium state calculations to predict conditions for the efficient synthesis of nucleosides at a larger scale. This approach was used to produce 3a–d in an enzymatic transglycosylation reaction. Upscaling of the reaction was successfully performed based on the theoretical predictions. The presented strategy can be transferred to the synthesis of a wide range of nucleoside analogues using nucleoside phosphorylases as biocatalysts.
Supplementary Materials
Table S1. Yields and purity of dihalogenated nucleosides. Yields were determined after each step in the synthesis and purification process. Purity was determined by analytical HPLC. Base concentrations of 5 mM and sugar donor concentrations of 22 to 62.5 mM (Supplementary Table S3) were applied in a reaction volume of 50 mL. Table S2. NMR characteristics of dihalogenated purine nucleosides produced in the present study. Table S3. Conditions used for the synthesis of dihalogenated purine nucleosides with a product yield of 90% or 95%. Reaction temperature was 40 °C. Table S4. HPLC methods applied for the purification of the dihalogenated nucleoside analogues. A flow rate 21.24 mL min−1 was employed. Acetonitrile (ACN) and water were applied as solvents. Figure S1. NMR spectra for 3a (A, C) and 3b (B, D). A, B: 1H NMR (500 MHz, DMSO-d6), C, D: 13C{1H} NMR (126 MHz, DMSO-d6); Figure S2. NMR spectra for 3c (A, C, E) and 3d (B, D, F). A, B: 1H NMR (500 MHz, DMSO-d6); C, D: 13C{1H} NMR (126 MHz, DMSO-d6); E,F: 19F{1H} NMR (471 MHz, DMSO-d6)
Author Contributions
Conceptualization, H.Y., K.P., J.K., and A.W.; methodology, H.Y., S.W., H.F.T.K., R.T.G., F.K., and V.R.; software, F.K. and R.T.G.; validation, F.K., H.Y., S.W., H.F.T.K., and V.R.; formal analysis, H.Y., S.W., V.R., and A.W.; investigation, H.Y., J.K., and A.W.; writing—original draft preparation, H.Y. and A.W; writing—review and editing, H.Y., F.K., A.W, J.K., and P.N.; visualization, A.W.; supervision, J.K., P.N., and A.W.; project administration, P.N., J.K., and A.W.; funding acquisition, R.T.G. All authors have read and agree to the published version of the manuscript.
Funding
We are grateful for the support of R.T.G. by the German Research Foundation (DFG) under Germany’s Excellence Strategy—EXC 2008/1-390540038 (UniSysCat), and by the Einstein Foundation Berlin (ESB)—Einstein Center of Catalysis (EC2).
Acknowledgments
We would like to thank Bernd Krostitz for his support with cell line cultivation, and Erik Wade for revision of the manuscript. We gratefully acknowledge support from the TU Berlin open access funding.
Conflicts of Interest
A.W. is the CEO, P.N. is a board member, and F.K. and S.W. are scientific researchers in the biotech company BioNukleo GmbH. The authors have no other relevant affiliations or financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No conflict of interest is known to the other authors.
References
- Trelles, J.A.; Bentancor, L.; Schoijet, A.; Porro, S.; Lewkowicz, E.S.; Sinisterra, J.V.; Iribarren, A.M. Immobilized Escherichia Coli BL21 as a Catalyst for the Synthesis of Adenine and Hypoxanthine Nucleosides. Chem. Biodivers. 2004, 1, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Rocchietti, S.; Ubiali, D.; Terreni, M.; Albertini, A.M.; Fernandez-Lafuente, R.; Guisan, J.M.; Pregnolato, M. Immobilization and Stabilization of Recombinant Multimeric Uridine and Purine Nucleoside Phosphorylases from Bacillus Subtilis. Biomacromolecules 2004, 5, 2195–2200. [Google Scholar] [CrossRef] [PubMed]
- Rivero, C.W.; Britos, C.N.; Lozano, M.E.; Sinisterra, J.V.; Trelles, J.A. Green Biosynthesis of Floxuridine by Immobilized Microorganisms. FEMS Microbiol. Lett. 2012, 331, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, L.E.; Lewkowicz, E.S.; Medici, R.; Bianchi, P.; Iribarren, A.M. Biocatalytic Approaches Applied to the Synthesis of Nucleoside Prodrugs. Biotechnol. Adv. 2015, 33, 412–434. [Google Scholar] [CrossRef]
- Haki, G.D.; Rakshit, S.K. Developments in Industrially Important Thermostable Enzymes: A Review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Bruins, M.; Janssen, A.; Boom, R. Thermozymes and Their Applications. Appl. Biochem. Biotechnol. 2001, 90, 155–186. [Google Scholar] [CrossRef]
- Quan, D.J.; Peters, M.G. Antiviral Therapy: Nucleotide and Nucleoside Analogs. Clin. Liver Dis. 2004, 8, 371–385. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Older and New Purine Nucleoside Analogs for Patients with Acute Leukemias. Cancer Treat. Rev. 2013, 39, 851–861. [Google Scholar] [CrossRef]
- Khaw, M.; Panosian, C.B. Human Antiprotozoal Therapy: Past, Present, and Future. Clin. Microbiol. Rev. 1995, 8, 427–439. [Google Scholar] [CrossRef]
- Bennett, L., Jr.; Chang, C.-H.; Allan, P.W.; Adamson, D.J.; Rose, L.M.; Brockman, R.W.; Secrist, J.A., III; Shortnacy, A.; Montgomery, J.A. Metabolism and Metabolic Effects of Halopurine Nucleosides in Tumor Cells in Culture. Nucleosides Nucleotides 1985, 4, 107–116. [Google Scholar] [CrossRef]
- Huang, M.-C.; Hatfield, K.; Roetker, A.W.; Montgomery, J.A.; Blakley, R.L. Analogs of 2′-Deoxyadenosine: Facile Enzymatic Preparation and Growth Inhibitory Effects on Human Cell Lines. Biochem. Pharmacol. 1981, 30, 2663–2671. [Google Scholar] [CrossRef]
- Huang, M.C.; Montgomery, J.A.; Thorpe, M.C.; Stewart, E.L.; Secrist, J.A.; Blakley, R.L. Formation of 3-(2′-Deoxyribofuranosyl) and 9-(2′-Deoxyribofuranosyl) Nucleosides of 8-Substituted Purines by Nucleoside Deoxyribosyltransferase. Arch. Biochem. Biophys. 1983, 222, 133–144. [Google Scholar] [CrossRef]
- Kaspar, F.; Giessmann, R.T.; Hellendahl, K.; Neubauer, P.; Wagner, A.; Gimpel, M. General Principles for Yield Optimization of Nucleoside Phosphorylase-catalyzed Transglycosylations. ChemBioChem 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, F.; Giessmann, R.T.; Neubauer, P.; Wagner, A.; Gimpel, M. Thermodynamic Reaction Control of Nucleoside Phosphorolysis. Adv. Synth. Catal. 2020. [Google Scholar] [CrossRef]
- Kaspar, F.; Giessman, R.T. Supplementary Material for “General Principles for Yield Optimization of Nucleoside Phosphorylase-Catalyzed Transglycosylations”. Zenodo 2019. [Google Scholar] [CrossRef]
- Giessmann, R.T. A Python Toolbox to Evaluate Systems under Thermodynamic Reaction Control. Zenodo 2019. [CrossRef]
- Kaspar, F.; Wagner, A. Supplementary Material for “Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations”. Zenodo 2020. [Google Scholar] [CrossRef]
- Stachnik, K.; Grieb, P.; Skierski, J.S. Cytotoxic Effects of Cladribine and Tezacitabine toward HL-60. Acta Biochim. Pol. 2005, 52, 561–565. [Google Scholar] [CrossRef]
- Nishi, R.; Yamauchi, T.; Negoro, E.; Takemura, H.; Ueda, T. Combination of Guanine Arabinoside and Bcl-2 Inhibitor YC137 Overcomes the Cytarabine Resistance in HL-60 Leukemia Cell Line. Cancer Sci. 2013, 104, 502–507. [Google Scholar] [CrossRef]
- Zhou, X.; Szeker, K.; Jiao, L.-Y.; Oestreich, M.; Mikhailopulo, I.A.; Neubauer, P. Synthesis of 2,6-Dihalogenated Purine Nucleosides by Thermostable Nucleoside Phosphorylases. Adv. Synth. Catal. 2015, 357, 1237–1244. [Google Scholar] [CrossRef]
- Zhou, X.; Mikhailopulo, I.A.; Cruz Bournazou, M.N.; Neubauer, P. Immobilization of Thermostable Nucleoside Phosphorylases on MagReSyn® Epoxide Microspheres and Their Application for the Synthesis of 2,6-Dihalogenated Purine Nucleosides. J. Mol. Catal. B Enzym. 2015, 115, 119–127. [Google Scholar] [CrossRef]
- Alexeev, C.S.; Kulikova, I.V.; Gavryushov, S.; Tararov, V.I.; Mikhailov, S.N. Quantitative Prediction of Yield in Transglycosylation Reaction Catalyzed by Nucleoside Phosphorylases. Adv. Synth. Catal. 2018, 360, 3090–3096. [Google Scholar] [CrossRef]
- Faderl, S.; Gandhi, V.; Keating, M.J.; Jeha, S.; Plunkett, W.; Kantarjian, H.M. The Role of Clofarabine in Hematologic and Solid Malignancies—Development of a next-Generation Nucleoside Analog. Cancer 2005, 103, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-C.; Luo, M.-Z.; Mozdziesz, D.E.; Sartorelli, A.C. Synthesis and Biological Evaluation of 2- and 7-Substituted 9-Deazaadenosine Analogues. Nucleosides Nucleotides Nucleic Acids 2005, 24, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, S.; Zhao, M.; Deng, X.-S.; Lee, C.-K.; Yu, X.-D.; Liu, B. Therapeutic Potential of Cladribine in Combination with STAT3 Inhibitor against Multiple Myeloma. BMC Cancer 2011, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Jeha, S.; Kirkpatrick, P. Clofarabine. Nat. Rev. Drug. Discov. 2005, 4, 369–370. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Molina-Arcas, M.; Casado, F.J.; Bellosillo, B.; Colomer, D.; Gil, J. Nucleoside Transporters in Chronic Lymphocytic Leukaemia. Leukemia 2004, 18, 385–393. [Google Scholar] [CrossRef]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and Viral Nucleoside/Nucleotide Kinases Involved in Antiviral Drug Activation: Structural and Catalytic Properties. Antiviral. Res. 2010, 86, 101–120. [Google Scholar] [CrossRef]
- Freyer, C.W.; Gupta, N.; Wetzler, M.; Wang, E.S. Revisiting the Role of Cladribine in Acute Myeloid Leukemia: An Improvement on Past Accomplishments or More Old News? Am. J. Hematol. 2015, 90, 62–72. [Google Scholar] [CrossRef]
- Warzocha, K.; Fabianowska-Majewska, K.; Bloński, J.; Krykowski, E.; Robak, T. 2-Chlorodeoxyadenosine Inhibits Activity of Adenosine Deaminase and S-Adenosylhomocysteine Hydrolase in Patients with Chronic Lymphocytic Leukaemia. Eur. J. Cancer 1997, 33, 170–173. [Google Scholar] [CrossRef]
- Carrera, C.J.; Terai, C.; Lotz, M.; Curd, J.G.; Piro, L.D.; Beutler, E.; Carson, D.A. Potent Toxicity of 2-Chlorodeoxyadenosine toward Human Monocytes in Vitro and in Vivo: A Novel Approach to Immunosuppressive Therapy. J. Clin. Investig. 1990, 86, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis of Nucleosides. In Organic Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 1–630. [Google Scholar]
- Chemo-Enzymatic Preparation Method for Purine Nucleosides and Their Deaza- and Aza-analogues. Available online: https://patents.google.com/patent/WO2016034735A1/en (accessed on 22 January 2020).
- Kaspar, F.; Giessmann, R.T.; Krausch, N.; Neubauer, P.; Wagner, A.; Gimpel, M. A UV/Vis Spectroscopy-Based Assay for Monitoring of Transformations between Nucleosides and Nucleobases. Methods Protoc. 2019, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Hiller, T.; Kissner, M.S.; Qazi, T.H.; Duda, G.N.; Hocke, A.C.; Hippenstiel, S.; Elomaa, L.; Weinhart, M.; Fahrenson, C.; et al. Optimization of Cell-Laden Bioinks for 3D Bioprinting and Efficient Infection with Influenza A Virus. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Szeker, K.; Janocha, B.; Böhme, T.; Albrecht, D.; Mikhailopulo, I.A.; Neubauer, P. Recombinant Purine Nucleoside Phosphorylases from Thermophiles: Preparation, Properties and Activity towards Purine and Pyrimidine Nucleosides. FEBS J. 2013, 280, 1475–1490. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature. Nuclear Spin Properties and Conventions for Chemical Shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds DCR-R, 3b, 3c and 3d are available from the authors. |



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).