
Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments




Abstract
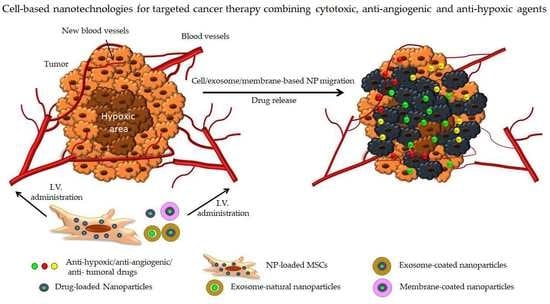
1. Introduction.
2. Anti-Angiogenesis Therapy: A Revealing History
3. Limits of Anti-Angiogenic Therapy
3.1. Tumor Microenvironment Resistance
3.2. Pro-Metastatic Effect of Anti-Angiogenic Therapies
4. New Objective: Targeting Hypoxia
5. The Emergence of Nanomedicine
6. The Limits of Nanomedicine in Clinical Applications
7. Biological Carriers to Deliver NPs
7.1. Mesenchymal Stem Cells as Carriers to Deliver NPs
7.2. Exosomes as Carriers to Deliver NPs
7.3. Cell Membrane-Coated Nanoparticles
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Mattern, J.; Volm, M. Role of oxygenation and vascularization in drug resistance. Cytotechnology 1998, 27, 249–256. [Google Scholar] [CrossRef]
- Rosen, L.S. Clinical Experience with Angiogenesis Signaling Inhibitors: Focus on Vascular Endothelial Growth Factor (VEGF) Blockers. Cancer Control. 2002, 9, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The Vascular Endothelial Growth Factor Family: Identification of a Fourth Molecular Species and Characterization of Alternative Splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Gagner, J.-P.; Law, M.; Fischer, I.; Newcomb, E.W.; Zagzag, D. Angiogenesis in gliomas: Imaging and experimental therapeutics. Brain Pathol. 2005, 15, 342–363. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Ngo, V.C.; Fargnoli, J.; Ayers, M.; Soo, K.C.; Koong, H.N.; Thng, C.H.; Ong, H.S.; Chung, A.; Chow, P.; et al. Brivanib Alaninate, a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Tyrosine Kinases, Induces Growth Inhibition in Mouse Models of Human Hepatocellular Carcinoma. Clin. Cancer Res. 2008, 14, 6146–6153. [Google Scholar] [CrossRef] [PubMed]
- Van Der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, N.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Simpson, D.; Keating, G.M. Sorafenib: In hepatocellular carcinoma. Drugs 2008, 68, 251–258. [Google Scholar] [CrossRef]
- Kontovinis, L.F.; Papazisis, K.T.; Touplikioti, P.; Andreadis, C.; Mouratidou, D.; Kortsaris, A.H. Sunitinib treatment for patients with clear-cell metastatic renal cell carcinoma: Clinical outcomes and plasma angiogenesis markers. BMC Cancer 2009, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Chap, L.I.; Holmes, F.A.; Cobleigh, M.A.; Marcom, P.K.; Fehrenbacher, L.; Dickler, M.; Overmoyer, B.A.; Reimann, J.D.; Sing, A.P.; et al. Randomized Phase III Trial of Capecitabine Compared With Bevacizumab Plus Capecitabine in Patients With Previously Treated Metastatic Breast Cancer. J. Clin. Oncol. 2005, 23, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The Great Escape; the Hallmarks of Resistance to Antiangiogenic Therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef]
- McMillin, U.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour–stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.B.; Meyronet, D.; Hervieu, V.; Frederick, M.J.; Bergsland, E.; Bergers, G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Muñoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef]
- Demir, R.; Naschberger, L.; Demir, I.; Melling, N.; Dimmler, A.; Papadopoulus, T.; Sturzl, M.; Klein, P.; Hohenberger, W. Hypoxia generates a more invasive phenotype of tumour cells: An in vivo experimental setup based on the chorioallantoic membrane. Pathol. Oncol. Res. 2009, 15, 417–422. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef]
- Moeller, B.J.; Richardson, R.A.; Dewhirst, M.W. Hypoxia and radiotherapy: Opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007, 26, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Skarlatos, J.; Corti, L.; Blandamura, S.; Piazza, M.; Gatter, K.C.; Harris, A.L. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res. 2001, 61, 1830–1832. [Google Scholar] [PubMed]
- Sun, X.; Kanwar, J.R.; Leung, E.; Lehnert, K.; Wang, D.; Krissansen, G.W. Gene transfer of antisense hypoxia inducible factor-1 alpha enhances the therapeutic efficacy of cancer immunotherapy. Gene Ther. 2001, 8, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef]
- Siveen, K.S.; Kuttan, G. Role of macrophages in tumour progression. Immunol. Lett. 2009, 123, 97–102. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Huang, H. Recent agents targeting HIF-1alpha for cancer therapy. J. Cell Biochem. 2013, 114, 498–509. [Google Scholar] [CrossRef]
- Shan, K.; Lincoff, A.M.; Young, J.B. Anthracycline-Induced Cardiotoxicity. Ann. Intern. Med. 1996, 125, 47. [Google Scholar] [CrossRef]
- NIH, DCTD Aminoflavone Toxicology Summary. Available online: http://dctd.cancer.gov/FeaturedAgents/pdfs/710464AminoflavoneToxAbstract.pdf (accessed on 12 November 2019).
- Eckardt, J.R. Emerging Role of Weekly Topotecan in Recurrent Small Cell Lung Cancer. Oncology 2004, 9, 25–32. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Deng, Z.; Xu, X.; Li, Z.; Jung, F.; Ma, N.; Lendlein, A.; Wang, Z.D.W. Functional Nanoparticles and their Interactions with Mesenchymal Stem Cells. Curr. Pharm. Des. 2017, 23, 3814–3832. [Google Scholar] [CrossRef]
- Friedman, A.D.; Claypool, S.E.; Liu, R. The smart targeting of nanoparticles. Curr. Pharm. Des. 2013, 19, 6315–6329. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.L.; De La Torre, P.; Cabañas, M.V.; Manzano, M.; Grau, M.; Flores, A.I.; Vallet-Regí, M. Vectorization of ultrasound-responsive nanoparticles in placental mesenchymal stem cells for cancer therapy. Nanoscale 2017, 9, 5528–5537. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, W.; Yu, J.; Zhao, C. Recent Development of pH-Responsive Polymers for Cancer Nanomedicine. Molecules 2018, 24, 4. [Google Scholar] [CrossRef] [PubMed]
- Thambi, T.; Deepagan, V.; Yoon, H.Y.; Han, H.S.; Kim, S.-H.; Son, S.; Jo, N.-G.; Ahn, C.-H.; Suh, Y.D.; Kim, K.; et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials 2014, 35, 1735–1743. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef]
- Meng, H.; Nel, A.E. Use of nano engineered approaches to overcome the stromal barrier in pancreatic cancer. Adv. Drug Deliv. Rev. 2018, 130, 50–57. [Google Scholar] [CrossRef]
- Mohanty, C.; Sahoo, S.K. The in vitro stability and in vivo pharmacokinetics of curcumin prepared as an aqueous nanoparticulate formulation. Biomaterials 2010, 31, 6597–6611. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, M.; Liu, T.; Zhou, J.; Gao, Y.; Wen, Z.; Guan, J.; Zhu, J.; Lin, Z.; He, D. Perfluorocarbon-Encapsulated PLGA-PEG Emulsions as Enhancement Agents for Highly Efficient Reoxygenation to Cell and Organism. ACS Appl. Mater. Interfaces 2015, 7, 18369–18378. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Kim, H.-W.; Lee, J.H.; Oh, S.H. Controlling oxygen release from hollow microparticles for prolonged cell survival under hypoxic environment. Biomaterials 2015, 53, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cheng, H.; Jiang, C.; Qiu, X.; Wang, K.; Huan, W.; Yuan, A.; Wu, J.; Hu, Y. Perfluorocarbon nanoparticles enhance reactive oxygen levels and tumour growth inhibition in photodynamic therapy. Nat. Commun. 2015, 6, 8785. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Liang, C.; Yi, X.; Zhao, Q.; Cheng, L.; Yang, K.; Liu, Z. Perfluorocarbon-Loaded Hollow Bi2Se3Nanoparticles for Timely Supply of Oxygen under Near-Infrared Light to Enhance the Radiotherapy of Cancer. Adv. Mater. 2016, 28, 2716–2723. [Google Scholar] [CrossRef] [PubMed]
- Eisenbrey, J.R.; Albala, L.; Kramer, M.R.; Daroshefski, N.; Brown, D.; Liu, J.B.; Stanczak, M.; O’Kane, P.; Forsberg, F.; Wheatley, M.A. Development of an ultrasound sensitive oxygen carrier for oxygen delivery to hypoxic tissue. Int. J. Pharm. 2015, 478, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, W.-H.; Liu, L.-H.; Qiu, W.-X.; Yu, W.-Y.; Zhang, X.-Z. An O2 Self-Supplementing and Reactive-Oxygen-Species-Circulating Amplified Nanoplatform via H2 O/H2 O2 Splitting for Tumor Imaging and Photodynamic Therapy. Adv. Funct. Mater. 2017, 27, 1700626. [Google Scholar] [CrossRef]
- Zhang, R.; Song, X.; Liang, C.; Yi, X.; Song, G.; Chao, Y.; Yang, Y.; Yang, K.; Feng, L.; Liu, Z. Catalase-loaded cisplatin-prodrug-constructed liposomes to overcome tumor hypoxia for enhanced chemo-radiotherapy of cancer. Biomaterials 2017, 138, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.; Hussain, A.; Vogelzang, N.; Lee, J.; Keam, B.; Rha, S.; Vaishampayan, U.; Harris, W.; Richey, S.; Randall, J.; et al. A randomized phase II trial of CRLX101 in combination with bevacizumab versus standard of care in patients with advanced renal cell carcinoma. Ann. Oncol. 2017, 28, 2754–2760. [Google Scholar] [CrossRef]
- Tardi, P.; Choice, E.; Masin, D.; Redelmeier, T.; Bally, M.; Madden, T.D. Liposomal encapsulation of topotecan enhances anticancer efficacy in murine and human xenograft models. Cancer Res. 2000, 60, 3389–3393. [Google Scholar]
- Zhao, X.; Li, F.; Li, Y.; Wang, H.; Ren, H.; Chen, J.; Nie, G.; Hao, J. Co-delivery of HIF1alpha siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 2015, 46, 13–25. [Google Scholar] [CrossRef]
- Wang, Y.; Saad, M.; Pakunlu, R.I.; Khandare, J.J.; Garbuzenko, O.B.; Vetcher, A.A.; Soldatenkov, V.A.; Pozharov, V.P.; Minko, T. Nonviral nanoscale-based delivery of antisense oligonucleotides targeted to hypoxia-inducible factor 1 alpha enhances the efficacy of chemotherapy in drug-resistant tumor. Clin. Cancer Res. 2008, 14, 3607–3616. [Google Scholar] [CrossRef]
- Greish, K. Enhanced Permeability and Retention (EPR) Effect for Anticancer Nanomedicine Drug Targeting. Adv. Struct. Saf. Stud. 2010, 624, 25–37. [Google Scholar]
- Kong, G.; Braun, R.D.; Dewhirst, M.W. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res. 2001, 61, 3027–3032. [Google Scholar] [PubMed]
- Koning, G.A.; Eggermont, A.M.M.; Lindner, L.H.; Hagen, T.L.M.T. Hyperthermia and Thermosensitive Liposomes for Improved Delivery of Chemotherapeutic Drugs to Solid Tumors. Pharm. Res. 2010, 27, 1750–1754. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Bazan-Peregrino, M.; Rifai, B.; Seymour, L.W.; Coussios, C.C. Cavitation-Enhanced Extravasation for Drug Delivery. Ultrasound Med. Biol. 2011, 37, 1838–1852. [Google Scholar] [CrossRef] [PubMed]
- Mannaris, C.; Bau, L.; Grundy, M.; Gray, M.; Lea-Banks, H.; Seth, A.; Teo, B.; Carlisle, R.; Stride, E.; Coussios, C.C. Microbubbles, Nanodroplets and Gas-Stabilizing Solid Particles for Ultrasound-Mediated Extravasation of Unencapsulated Drugs: An Exposure Parameter Optimization Study. Ultrasound Med. Biol. 2019, 45, 954–967. [Google Scholar] [CrossRef]
- Friedman, A.J.; Han, G.; Navati, M.S.; Chacko, M.; Gunther, L.; Alfieri, A.; Friedman, J.M. Sustained release nitric oxide releasing nanoparticles: Characterization of a novel delivery platform based on nitrite containing hydrogel/glass composites. Nitric Oxide 2008, 19, 12–20. [Google Scholar] [CrossRef]
- Godugu, C.; Patel, A.R.; Doddapaneni, R.; Marepally, S.; Jackson, T.; Singh, M. Inhalation delivery of Telmisartan enhances intratumoral distribution of nanoparticles in lung cancer models. J. Control. Release 2013, 172, 86–95. [Google Scholar] [CrossRef]
- Beik, J.; Abed, Z.; Ghoreishi, F.S.; Hosseini-Nami, S.; Mehrzadi, S.; Shakeri-Zadeh, A.; Kamrava, S.K. Nanotechnology in hyperthermia cancer therapy: From fundamental principles to advanced applications. J. Control. Release 2016, 235, 205–221. [Google Scholar] [CrossRef]
- Saraiva, J.; Marotta-Oliveira, S.S.; Cicillini, S.A.; Eloy, J.D.O.; Marchetti, J.M. Nanocarriers for Nitric Oxide Delivery. J. Drug Deliv. 2011, 2011, 1–16. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popovic, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Yang, W.; Li, W.; Yang, X.-Y.; Liu, S.; Li, X.; Zhao, X.; Ding, H.; Qin, L.; Pan, Y. Conjugation of gold nanoparticles and recombinant human endostatin modulates vascular normalization via interruption of anterior gradient 2–mediated angiogenesis. Tumor Biol. 2017, 39, 1010428317708547. [Google Scholar] [CrossRef]
- Du, S.; Xiong, H.; Xu, C.; Lu, Y.; Yao, J. Attempts to strengthen and simplify the tumor vascular normalization strategy using tumor vessel normalization promoting nanomedicines. Biomater. Sci. 2019, 7, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef]
- Choi, Y.H.; Han, H.-K. Correction to: Nanomedicines: Current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. 2018, 49, 201. [Google Scholar] [CrossRef]
- Paris, J.L.; Baeza, A.; Vallet-Regí, M. Overcoming the stability, toxicity, and biodegradation challenges of tumor stimuli-responsive inorganic nanoparticles for delivery of cancer therapeutics. Expert Opin. Drug Deliv. 2019, 16, 1095–1112. [Google Scholar] [CrossRef]
- Hua, S.; De Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef]
- Park, K. Facing the Truth about Nanotechnology in Drug Delivery. ACS Nano 2013, 7, 7442–7447. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Lee, H.; Hoang, B.; Fonge, H.; Reilly, R.M.; Allen, C. In Vivo Distribution of Polymeric Nanoparticles at the Whole-Body, Tumor, and Cellular Levels. Pharm. Res. 2010, 27, 2343–2355. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Yu, M.; Zheng, J. Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 2018, 3, 358–374. [Google Scholar] [CrossRef]
- Monopoli, M.P.; Walczyk, D.; Campbell, A.; Elia, G.; Lynch, I.; Bombelli, F.B.; Dawson, K.A. Physical-chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Lunova, M.; Smolková, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinová, Š.; Dejneka, A.; Lunov, O. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers 2019, 11, 82. [Google Scholar] [CrossRef]
- Moghimi, S.M. Chemical camouflage of nanospheres with a poorly reactive surface: Towards development of stealth and target-specific nanocarriers. Biochim. Biophys. Acta BBA Bioenerg. 2002, 1590, 131–139. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Andersen, A.; Hashemi, S.; Lettiero, B.; Ahmadvand, D.; Hunter, A.; Andresen, T.L.; Hamad, I.; Szebeni, J. Complement activation cascade triggered by PEG–PL engineered nanomedicines and carbon nanotubes: The challenges ahead. J. Control. Release 2010, 146, 175–181. [Google Scholar] [CrossRef]
- Wang, X.; Ishida, T.; Kiwada, H. Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J. Control. Release 2007, 119, 236–244. [Google Scholar] [CrossRef]
- Mauricio, M.D.; Guerra-Ojeda, S.; Marchio, P.; Valles, S.L.; Aldasoro, M.; Escribano-Lopez, I.; Herance, J.R.; Rocha, M.; Vila, J.M.; Victor, V.M. Nanoparticles in Medicine: A Focus on Vascular Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.; Klopp, A.; Dembinski, J.; Andreeff, M.; Marini, F. Inflammation and tumor microenvironments: Defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008, 15, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Labusca, L.; Herea, D.D.; Mashayekhi, K. Stem cells as delivery vehicles for regenerative medicine-challenges and perspectives. World J. Stem Cells 2018, 10, 43–56. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, P.; Flores, A.I. Nanotechnology and Mesenchymal Stem Cells for Regenerative Medicine. Glob. J. Nanomed. 2017, 1, 555559. [Google Scholar]
- Pittenger, M.F. Mesenchymal Stem Cells from Adult Bone Marrow. Adv. Struct. Saf. Stud. 2008, 449, 27–44. [Google Scholar]
- Miana, V.V.; Gonzalez, E.A.P. Adipose tissue stem cells in regenerative medicine. Ecancermedicalscience 2018, 12, 822. [Google Scholar] [CrossRef]
- Arutyunyan, I.; Elchaninov, A.; Makarov, A.; Fatkhudinov, T. Umbilical Cord as Prospective Source for Mesenchymal Stem Cell-Based Therapy. Stem Cells Int. 2016, 2016, 1–17. [Google Scholar] [CrossRef]
- De Coppi, P.; Bartsch, G., Jr.; Siddiqui, M.M.; Xu, T.; Santos, C.C.; Perin, L.; Mostoslavsky, G.; Serre, A.C.; Snyder, E.Y.; Yoo, J.J.; et al. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 2007, 25, 100–106. [Google Scholar] [CrossRef]
- Macias, M.I.; Grande, J.; Moreno, A.; Domínguez, I.; Bornstein, R.; Flores, A.I. Isolation and characterization of true mesenchymal stem cells derived from human term decidua capable of multilineage differentiation into all 3 embryonic layers. Am. J. Obstet. Gynecol. 2010, 203, e9–e23. [Google Scholar] [CrossRef]
- Vegh, I.; Grau, M.; Gracia, M.; Grande, J.; de la Torre, P.; Flores, A.I. Decidua mesenchymal stem cells migrated toward mammary tumors in vitro and in vivo affecting tumor growth and tumor development. Cancer Gene Ther. 2013, 20, 8–16. [Google Scholar] [CrossRef]
- Droujinine, I.A.; Eckert, M.A.; Zhao, W. To grab the stroma by the horns: From biology to cancer therapy with mesenchymal stem cells. Oncotarget 2013, 4, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Javan, M.R.; Khosrojerdi, A.; Moazzeni, S.M. New Insights Into Implementation of Mesenchymal Stem Cells in Cancer Therapy: Prospects for Anti-angiogenesis Treatment. Front. Oncol. 2019, 9, 840. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, P.; Pérez-Lorenzo, M.J.; Flores, A.I. Human Placenta-Derived Mesenchymal Stromal Cells: A Review from Basic Research to Clinical Applications. In Stromal Cells—Structure, Function, and Therapeutic Implications; InTech Open: London, UK, 2019. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Shah, K. Mesenchymal stem cells engineered for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 739–748. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of Mesenchymal Stem Cells for Therapeutic Agent Delivery in Anti-tumor Treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef]
- Fakiruddin, K.S.; Ghazalli, N.; Lim, M.N.; Zakaria, Z.; Abdullah, S. Mesenchymal Stem Cell Expressing TRAIL as Targeted Therapy against Sensitised Tumour. Int. J. Mol. Sci. 2018, 19, 2188. [Google Scholar] [CrossRef]
- Lu, Y.R.; Yuan, Y.; Wang, X.J.; Wei, L.L.; Chen, Y.N.; Cong, C.; Li, S.F.; Long, D.; Tan, W.D.; Mao, Y.Q.; et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol. Ther. 2008, 7, 245–251. [Google Scholar] [CrossRef]
- Sun, B.; Roh, K.-H.; Park, J.-R.; Lee, S.-R.; Park, S.-B.; Jung, J.-W.; Kang, S.-K.; Lee, Y.-S.; Kang, K.-S. Therapeutic potential of mesenchymal stromal cells in a mouse breast cancer metastasis model. Cytotherapy 2009, 11, 289–298. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.-S. Usage of Human Mesenchymal Stem Cells in Cell-based Therapy: Advantages and Disadvantages. Dev. Reprod. 2017, 21, 1–10. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Natare 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Cheng, S.; Nethi, S.K.; Rathi, S.; Layek, B.; Prabha, S. Engineered Mesenchymal Stem Cells for Targeting Solid Tumors: Therapeutic Potential beyond Regenerative Therapy. J. Pharmacol. Exp. Ther. 2019, 370, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Baxter-Holland, M.; Dass, C.R. Doxorubicin, mesenchymal stem cell toxicity and antitumour activity: Implications for clinical use. J. Pharm. Pharmacol. 2018, 70, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.B.; Kenworthy, R.; Zorio, D.A.; Sang, Q.X. Human mesenchymal stem cells are resistant to Paclitaxel by adopting a non-proliferative fibroblastic state. PLoS ONE 2015, 10, e0128511. [Google Scholar] [CrossRef] [PubMed]
- Pires, M.M.; Emmert, D.; Hrycyna, C.A.; Chmielewski, J. Inhibition of P-glycoprotein-mediated paclitaxel resistance by reversibly linked quinine homodimers. Mol. Pharmacol. 2009, 75, 92–100. [Google Scholar] [CrossRef]
- Kozhukharova, I.; Zemelko, V.; Kovaleva, Z.; Alekseenko, L.; Lyublinskaya, O.; Nikolsky, N. Therapeutic doses of doxorubicin induce premature senescence of human mesenchymal stem cells derived from menstrual blood, bone marrow and adipose tissue. Int. J. Hematol. 2018, 107, 286–296. [Google Scholar] [CrossRef]
- Vallet-Regí, M.; Paris, J.L.; Torre, P.; Cabañas, M.V.; Manzano, M.; Flores, A.I. Mesenchymal Stem Cells from Human Placenta as Nanoparticle Delivery Vectors. Insights Stem. Cells 2018, 4, 1. [Google Scholar]
- Park, J.S.; Suryaprakash, S.; Lao, Y.-H.; Leong, K.W. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods 2015, 84, 3–16. [Google Scholar] [CrossRef]
- Paris, J.L.; De La Torre, P.; Manzano, M.; Cabañas, M.V.; Flores, A.I.; Vallet-Regí, M. Decidua-derived mesenchymal stem cells as carriers of mesoporous silica nanoparticles. In vitro and in vivo evaluation on mammary tumors. Acta Biomater. 2016, 33, 275–282. [Google Scholar] [CrossRef]
- Kim, S.-W.; Lee, Y.K.; Hong, J.H.; Park, J.-Y.; Choi, Y.-A.; Lee, D.U.; Choi, J.; Sym, S.J.; Kim, S.-H.; Khang, D. Mutual Destruction of Deep Lung Tumor Tissues by Nanodrug-Conjugated Stealth Mesenchymal Stem Cells. Adv. Sci. 2018, 5, 1700860. [Google Scholar] [CrossRef]
- Wang, X.; Gao, J.-Q.; Ouyang, X.; Wang, J.; Sun, X.; Lv, Y. Mesenchymal stem cells loaded with paclitaxel-poly(lactic-co-glycolic acid) nanoparticles for glioma-targeting therapy. Int. J. Nanomed. 2018, 13, 5231–5248. [Google Scholar] [CrossRef]
- Bexell, D.; Gunnarsson, S.; Svensson, A.; Tormin, A.; Henriques-Oliveira, C.; Siesjö, P.; Paul, G.; Salford, L.G.; Scheding, S.; Bengzon, J. Rat Multipotent Mesenchymal Stromal Cells Lack Long-Distance Tropism to 3 Different Rat Glioma Models. Neurosurgery 2012, 70, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Sadhukha, T.; O’Brien, T.D.; Prabha, S. Nano-engineered mesenchymal stem cells as targeted therapeutic carriers. J. Control Release 2014, 196, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Layek, B.; Sadhukha, T.; Panyam, J.; Prabha, S. Nano-Engineered Mesenchymal Stem Cells Increase Therapeutic Efficacy of Anticancer Drug Through True Active Tumor Targeting. Mol. Cancer Ther. 2018, 17, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.L.; De La Torre, P.; Cabañas, M.V.; Manzano, M.; Flores, A.I.; Vallet-Regí, M. Suicide-gene transfection of tumor-tropic placental stem cells employing ultrasound-responsive nanoparticles. Acta Biomater. 2019, 83, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. Recent progress toward antiangiogenesis application of nanomedicine in cancer therapy. Futur. Sci. OA 2018, 4, FSO318. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.S.; Jonkman, J.; Tannock, I.F. Quantitative Immunohistochemistry for Evaluating the Distribution of Ki67 and Other Biomarkers in Tumor Sections and Use of the Method to Study Repopulation in Xenografts after Treatment with Paclitaxel. Neoplasia 2012, 14, 324. [Google Scholar] [CrossRef]
- Sun, J.-G.; Jiang, Q.; Zhang, X.-P.; Shan, K.; Liu, B.-H.; Zhao, C.; Yan, B. Mesoporous silica nanoparticles as a delivery system for improving antiangiogenic therapy. Int. J. Nanomed. 2019, 14, 1489–1501. [Google Scholar] [CrossRef]
- Hu, X.; Wei, L.; Taylor, T.M.; Wei, J.; Zhou, X.; Wang, J.-A.; Yu, S.P. Hypoxic preconditioning enhances bone marrow mesenchymal stem cell migration via Kv2.1 channel and FAK activation. Am. J. Physiol. Physiol. 2011, 301, C362–C372. [Google Scholar] [CrossRef]
- Hsu, F.-T.; Wei, Z.-H.; Hsuan, Y.C.-Y.; Lin, W.; Su, Y.-C.; Liao, C.-H.; Hsieh, C.-L. MRI tracking of polyethylene glycol-coated superparamagnetic iron oxide-labelled placenta-derived mesenchymal stem cells toward glioblastoma stem-like cells in a mouse model. Artif. Cells Nanomed. Biotechnol. 2018, 46, S448–S459. [Google Scholar] [CrossRef]
- Menon, L.G.; Picinich, S.; Koneru, R.; Gao, H.; Lin, S.Y.; Koneru, M.; Mayer-Kuckuk, P.; Glod, J.; Banerjee, D. Differential Gene Expression Associated with Migration of Mesenchymal Stem Cells to Conditioned Medium from Tumor Cells or Bone Marrow Cells. Stem Cells 2007, 25, 520–528. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, C.; Fitch, S.; Yang, F. Targeting Tumor Hypoxia Using Nanoparticle-engineered CXCR4-overexpressing Adipose-derived Stem Cells. Theranostics 2018, 8, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.-H.; Hsiao, J.-K.; Hsu, S.-C.; Yao, M.; Chen, Y.-C.; Wang, S.-W.; Kuo, M.Y.-P.; Yang, C.-S.; Huang, D.-M. Iron Oxide Nanoparticle-Induced Epidermal Growth Factor Receptor Expression in Human Stem Cells for Tumor Therapy. ACS Nano 2011, 5, 9807–9816. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, Z.; Lv, H.; Wu, L.; Cui, Y.; Yao, H.; Li, J.; Zhang, H.; Yang, B.; Jiang, J. Iron oxide nanoparticles promote the migration of mesenchymal stem cells to injury sites. Int. J. Nanomed. 2019, 14, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Momin, E.; Choi, J.; Yuan, K.; Zaidi, H.; Kim, J.; Park, M.; Lee, N.; McMahon, M.T.; Quiñones-Hinojosa, A.; et al. Mesoporous Silica-Coated Hollow Manganese Oxide Nanoparticles as PositiveT1Contrast Agents for Labeling and MRI Tracking of Adipose-Derived Mesenchymal Stem Cells. J. Am. Chem. Soc. 2011, 133, 2955–2961. [Google Scholar] [CrossRef]
- Meir, R.; Betzer, O.; Motiei, M.; Kronfeld, N.; Brodie, C.; Popovtzer, R. Design principles for noninvasive, longitudinal and quantitative cell tracking with nanoparticle-based CT imaging. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 421–429. [Google Scholar] [CrossRef]
- Wu, X.; Hu, J.; Zhou, L.; Mao, Y.; Yang, B.; Gao, L.; Xie, R.; Xu, F.; Zhang, D.; Liu, J.; et al. In vivo tracking of superparamagnetic iron oxide nanoparticle–labeled mesenchymal stem cell tropism to malignant gliomas using magnetic resonance imaging. J. Neurosurg. 2008, 108, 320–329. [Google Scholar] [CrossRef]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of nanoparticles for diagnosis and therapy of cancer. Br. J. Radiol. 2015, 88, 20150207. [Google Scholar] [CrossRef]
- Vella, L.J.; Sharples, R.A.; Nisbet, R.M.; Cappai, R.; Hill, A.F. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 2008, 37, 323–332. [Google Scholar] [CrossRef]
- Halkein, J.; Tabruyn, S.P.; Ricke-Hoch, M.; Haghikia, A.; Nguyen, N.-Q.-N.; Scherr, M.; Castermans, K.; Malvaux, L.; Lambert, V.; Thiry, M.; et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Investig. 2013, 123, 2143–2154. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Masyuk, T.V.; LaRusso, N.F. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J. Hepatol. 2013, 59, 621–625. [Google Scholar] [CrossRef]
- Hannafon, B.N.; Ding, W.-Q. Intercellular Communication by Exosome-Derived microRNAs in Cancer. Int. J. Mol. Sci. 2013, 14, 14240–14269. [Google Scholar] [CrossRef] [PubMed]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Larssen, P.; Wik, L.; Czarnewski, P.; Eldh, M.; Lof, L.; Ronquist, K.G.; Dubois, L.; Freyhult, E.; Gallant, C.J.; Oelrich, J.; et al. Tracing Cellular Origin of Human Exosomes Using Multiplex Proximity Extension Assays. Mol. Cell Proteom. 2017, 16, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Rezvani, K.; Shpall, E. Mesenchymal stem cell-derived exosomes for clinical use. Bone Marrow Transplant. 2019, 54, 789–792. [Google Scholar] [CrossRef]
- You, B.; Xu, W.; Zhang, B. Engineering exosomes: A new direction for anticancer treatment. Am. J. Cancer Res. 2018, 8, 1332–1342. [Google Scholar]
- Sancho-Albero, M.; Encabo-Berzosa, M.D.M.; Beltran-Visiedo, M.; Fernandez-Messina, L.; Sebastian, V.; Sanchez-Madrid, F.; Arruebo, M.; Santamaria, J.; Martin-Duque, P. Efficient encapsulation of theranostic nanoparticles in cell-derived exosomes: Leveraging the exosomal biogenesis pathway to obtain hollow gold nanoparticle-hybrids. Nanoscale 2019, 11, 18825–18836. [Google Scholar] [CrossRef]
- Illes, B.; Hirschle, P.; Barnert, S.; Cauda, V.; Wuttke, S.; Engelke, H. Exosome-Coated Metal–Organic Framework Nanoparticles: An Efficient Drug Delivery Platform. Chem. Mater. 2017, 29, 8042–8046. [Google Scholar] [CrossRef]
- Dumontel, B.; Susa, F.; Limongi, T.; Canta, M.; Racca, L.; Chiodoni, A.; Garino, N.; Chiabotto, G.; Centomo, M.L.; Pignochino, Y.; et al. ZnO nanocrystals shuttled by extracellular vesicles as effective Trojan nano-horses against cancer cells. Nanomedicine 2019, 14, 2815–2833. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Shi, J.-B.; Li, C. Small extracellular vesicle loading systems in cancer therapy: Current status and the way forward. Cytotherapy 2019, 21, 1122–1136. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, K.; Wu, S.; Cui, M.; Xu, T. Focus on Mesenchymal Stem Cell-Derived Exosomes: Opportunities and Challenges in Cell-Free Therapy. Stem Cells Int. 2017, 2017, 6305295. [Google Scholar] [CrossRef] [PubMed]
- Betzer, O.; Barnoy, E.; Sadan, T.; Elbaz, I.; Braverman, C.; Liu, Z.; Popovtzer, R. Advances in imaging strategies for in vivo tracking of exosomes. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 1594, e1594. [Google Scholar] [CrossRef] [PubMed]
- Gangadaran, P.; Hong, C.M.; Ahn, B.-C. Current Perspectives on In Vivo Noninvasive Tracking of Extracellular Vesicles with Molecular Imaging. BioMed. Res. Int. 2017, 2017, 9158319. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chhour, P.; Hsu, J.; Litt, H.I.; Ferrari, V.A.; Popovtzer, R.; Cormode, D.P. Use of Nanoparticle Contrast Agents for Cell Tracking with Computed Tomography. Bioconjug. Chem. 2017, 28, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Yeo, R.W.Y.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Naseri, Z.; Oskuee, R.K.; Jaafari, M.R.; Forouzandeh-Moghadam, M. Exosome-mediated delivery of functionally active miRNA-142-3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo. Int. J. Nanomed. 2018, 13, 7727–7747. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.O.; Jo, H.; Yu, J.H.; Gambhir, S.S.; Pratx, G. Development and MPI tracking of novel hypoxia-targeted theranostic exosomes. Biomaterials 2018, 177, 139–148. [Google Scholar] [CrossRef]
- Gonzalez-King, H.; Garcia, N.A.; Ontoria-Oviedo, I.; Ciria, M.; Montero, J.A.; Sepulveda, P. Hypoxia Inducible Factor-1alpha Potentiates Jagged 1-Mediated Angiogenesis by Mesenchymal Stem Cell-Derived Exosomes. Stem. Cells 2017, 35, 1747–1759. [Google Scholar] [CrossRef]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef]
- Reza, A.M.M.T.; Choi, Y.-J.; Yasuda, H.; Kim, J.-H. Human adipose mesenchymal stem cell-derived exosomal-miRNAs are critical factors for inducing anti-proliferation signalling to A2780 and SKOV-3 ovarian cancer cells. Sci. Rep. 2016, 6, 38498. [Google Scholar] [CrossRef]
- Ko, S.-F.; Yip, H.-K.; Zhen, Y.-Y.; Lee, C.-C.; Lee, C.-C.; Huang, C.-C.; Ng, S.-H.; Lin, J.-W. Adipose-Derived Mesenchymal Stem Cell Exosomes Suppress Hepatocellular Carcinoma Growth in a Rat Model: Apparent Diffusion Coefficient, Natural Killer T-Cell Responses, and Histopathological Features. Stem Cells Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhang, L. Coating nanoparticles with cell membranes for targeted drug delivery. J. Drug Target. 2015, 23, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Lv, P.; Chen, Z.; Ni, D.; Zhang, L.; Yue, H.; Yue, Z.; Wei, W.; Ma, G. Programmed co-delivery of paclitaxel and doxorubicin boosted by camouflaging with erythrocyte membrane. Nanoscale 2015, 7, 4020–4030. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Quattrocchi, N.; van de Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, K.; Li, C.; Guo, Q.; Chen, Q.; He, X.; Liu, L.; Zhang, Y.; Lu, Y.; Chen, X.; et al. Macrophage-Membrane-Coated Nanoparticles for Tumor-Targeted Chemotherapy. Nano Lett. 2018, 18, 1908–1915. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Qian, C.; Wang, C.; Bomba, H.N.; Gu, Z. Anticancer Platelet-Mimicking Nanovehicles. Adv. Mater. 2015, 27, 7043–7050. [Google Scholar] [CrossRef]
- Gao, C.; Lin, Z.; Jurado-Sánchez, B.; Lin, X.; Wu, Z.; He, Q. Stem Cell Membrane-Coated Nanogels for Highly Efficient In Vivo Tumor Targeted Drug Delivery. Small 2016, 12, 4056–4062. [Google Scholar] [CrossRef]
- Gao, W.; Fang, R.H.; Thamphiwatana, S.; Luk, B.T.; Li, J.; Angsantikul, P.; Zhang, Q.; Hu, C.-M.J.; Zhang, L. Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles. Nano Lett. 2015, 15, 1403–1409. [Google Scholar] [CrossRef]
- Rao, L.; Bu, L.-L.; Cai, B.; Xu, J.-H.; Li, A.; Zhang, W.-F.; Sun, Z.-J.; Guo, S.-S.; Liu, W.; Wang, T.-H.; et al. Cancer Cell Membrane-Coated Upconversion Nanoprobes for Highly Specific Tumor Imaging. Adv. Mater. 2016, 28, 3460–3466. [Google Scholar] [CrossRef]
- Vijayan, V.; Uthaman, S.; Park, I.-K. Cell Membrane-Camouflaged Nanoparticles: A Promising Biomimetic Strategy for Cancer Theragnostics. Polym. 2018, 10, 983. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; He, Y.; Zhang, S.; Qin, J.; Wang, J. Cell membrane-based nanoparticles: A new biomimetic platform for tumor diagnosis and treatment. Acta Pharm. Sin. B 2018, 8, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Yu, W.; Ji, B.; Chen, C.; Yang, H.; Du, Y.; Song, M.; Cai, H.; Yan, F.; Su, R. Saikosaponin D loaded macrophage membrane-biomimetic nanoparticles target angiogenic signaling for breast cancer therapy. Appl. Mater. Today 2020, 18, 100505. [Google Scholar] [CrossRef]
- Su, J.; Sun, H.; Meng, Q.; Yin, Q.; Tang, S.; Zhang, P.; Chen, Y.; Yu, H.; Li, Y. Long Circulation Red-Blood-Cell-Mimetic Nanoparticles with Peptide-Enhanced Tumor Penetration for Simultaneously Inhibiting Growth and Lung Metastasis of Breast Cancer. Adv. Funct. Mater. 2016, 26, 1243–1252. [Google Scholar] [CrossRef]
- Zuo, H.; Tao, J.; Shi, H.; He, J.; Zhou, Z.; Zhang, C. Platelet-mimicking nanoparticles co-loaded with W18O49 and metformin alleviate tumor hypoxia for enhanced photodynamic therapy and photothermal therapy. Acta Biomater. 2018, 80, 296–307. [Google Scholar] [CrossRef]
- Amo, L.; Tamayo-Orbegozo, E.; Maruri, N.; Eguizabal, C.; Zenarruzabeitia, O.; Rinon, M.; Arrieta, A.; Santos, S.; Monge, J.; Vesga, M.A.; et al. Involvement of platelet-tumor cell interaction in immune evasion. Potential role of podocalyxin-like protein 1. Front. Oncol. 2014, 4, 245. [Google Scholar] [CrossRef]
- Aviram, I.; Shaklai, N. The association of human erythrocyte catalase with the cell membrane. Arch. Biochem. Biophys. 1981, 212, 329–337. [Google Scholar] [CrossRef]
- Bidkar, A.P.; Sanpui, P.; Ghosh, S.S. Red Blood Cell-Membrane-Coated Poly(Lactic-co-glycolic Acid) Nanoparticles for Enhanced Chemo- and Hypoxia-Activated Therapy. ACS Appl. Bio Mater. 2019, 2, 4077–4086. [Google Scholar] [CrossRef]
- Gao, M.; Liang, C.; Song, X.; Chen, Q.; Jin, Q.; Wang, C.; Liu, Z. Erythrocyte-Membrane-Enveloped Perfluorocarbon as Nanoscale Artificial Red Blood Cells to Relieve Tumor Hypoxia and Enhance Cancer Radiotherapy. Adv. Mater. 2017, 29, 1701429. [Google Scholar] [CrossRef]
- Li, C.; Yang, X.-Q.; An, J.; Cheng, K.; Hou, X.-L.; Zhang, X.-S.; Hu, Y.-G.; Liu, B.; Zhao, Y.-D. Red blood cell membrane-enveloped O2 self-supplementing biomimetic nanoparticles for tumor imaging-guided enhanced sonodynamic therapy. Theranostics 2020, 10, 867–879. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Categories | Cargo | Type of Nanoparticle | Mode of Action | Ref. |
|---|---|---|---|---|
| O2 carriers | ||||
| Perfluorocarbonand derivatives | Perfluorocarbon Perfluorooctane Perfluorohexane Perfluorocarbon | PLGA-PEG emulsion Hollow microparticles Liposomes Hollow Bi2Se3 NPs | Rapid release of O2 by hydrolysis | [41] [42] [43] [44] |
| Ultrasound-based carrier | Oxygen | Microbubbles | Ultrasound controlled release and imaging by ultrasonography | [45] |
| H2O2 catalysis platforms | ||||
| MnO2 Catalase | UPCNPs Liposomes | Decomposition of H2O2 into O2 and H2O | [46] [47] | |
| Inhibitors of HIF-1 signaling | ||||
| Camptothecin Topotecan HIF-1 siRNA HIF-1 ASO | Cyclodextrin-based polymer Liposomes PEGylated ε-polylysine copolymer Liposomes | Topoisomerase I inhibition Topoisomerase I inhibition Reduction of HIF-1 levels Reduction of HIF-1 levels | [48] [49] [50] [51] | |
| Cell-Based Strategy | Advantages | Disadvantages |
|---|---|---|
| MSCs | Easy isolation from accessible sources Easy culture in vitro No immunogenicity Tissue regeneration capacity Tumor tropism Migration ability into the site of damage Homing capacity | Uncertain tumorigenic effect High retention in lungs after systemic administration Possible occlusion of microvessels after systemic administration Repeated injection may result in production of alloantibodies |
| EXOSOMES | High stability in physiological and pathological conditions Unlikely to be immunogenic Small and relatively homogeneous size Intracellular delivery of cargo by fusion of membranes Able to cross natural barriers such as blood–brain barrier Autologous use allowing personalized medicine | Need of standardized protocols for isolation and purification Need of adequate characterization of cell of origin Undesired effects due to exosome components themselves Lack of standardized mass production protocols |
| PLASMA MEMBRANE-COATING | Provide biocompatibility to nanoparticles Immune escape and longer circulation lifetime High versatility Easy functionalization | Need of techniques for large-scale cell culture Need of high-yield methods for membrane derivation Lack of knowledge about all membrane components |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Torre, P.; Pérez-Lorenzo, M.J.; Alcázar-Garrido, Á.; Flores, A.I. Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments. Molecules 2020, 25, 715. https://doi.org/10.3390/molecules25030715
de la Torre P, Pérez-Lorenzo MJ, Alcázar-Garrido Á, Flores AI. Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments. Molecules. 2020; 25(3):715. https://doi.org/10.3390/molecules25030715
Chicago/Turabian Stylede la Torre, Paz, María Jesús Pérez-Lorenzo, Álvaro Alcázar-Garrido, and Ana I. Flores. 2020. "Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments" Molecules 25, no. 3: 715. https://doi.org/10.3390/molecules25030715
APA Stylede la Torre, P., Pérez-Lorenzo, M. J., Alcázar-Garrido, Á., & Flores, A. I. (2020). Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments. Molecules, 25(3), 715. https://doi.org/10.3390/molecules25030715