
Tumor Targeted Nanocarriers for Immunotherapy

Abstract
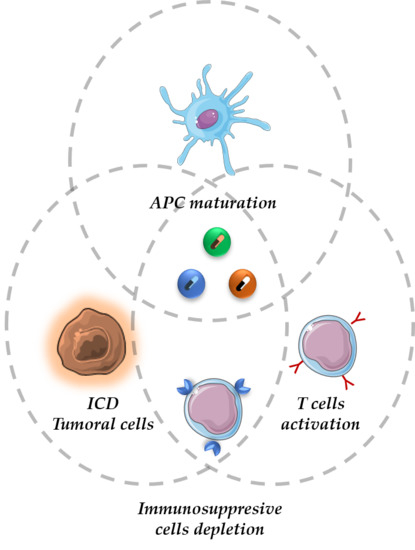
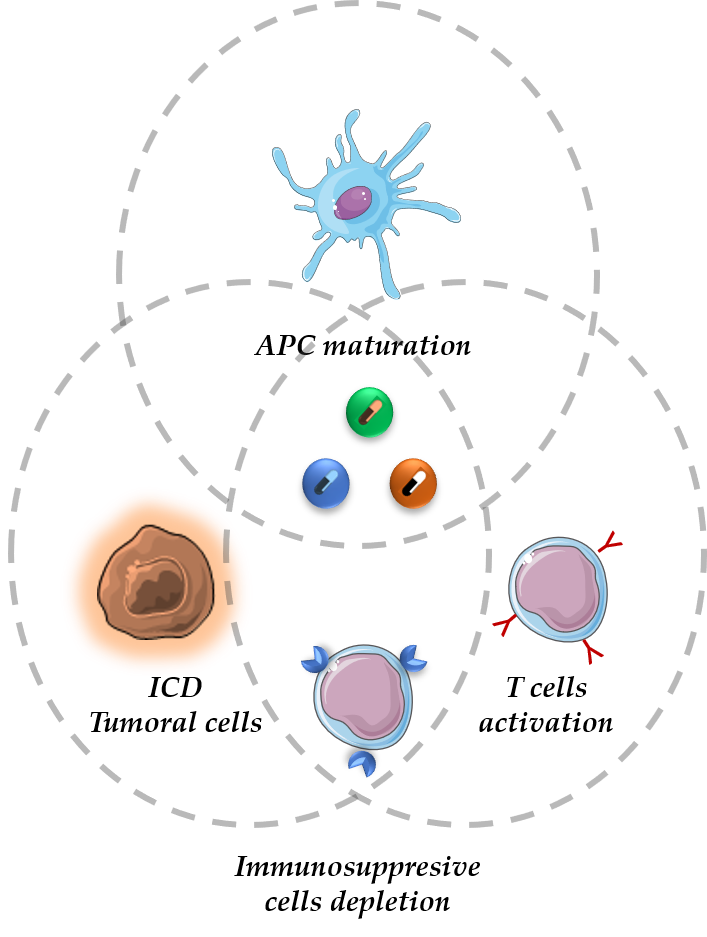
1. Introduction
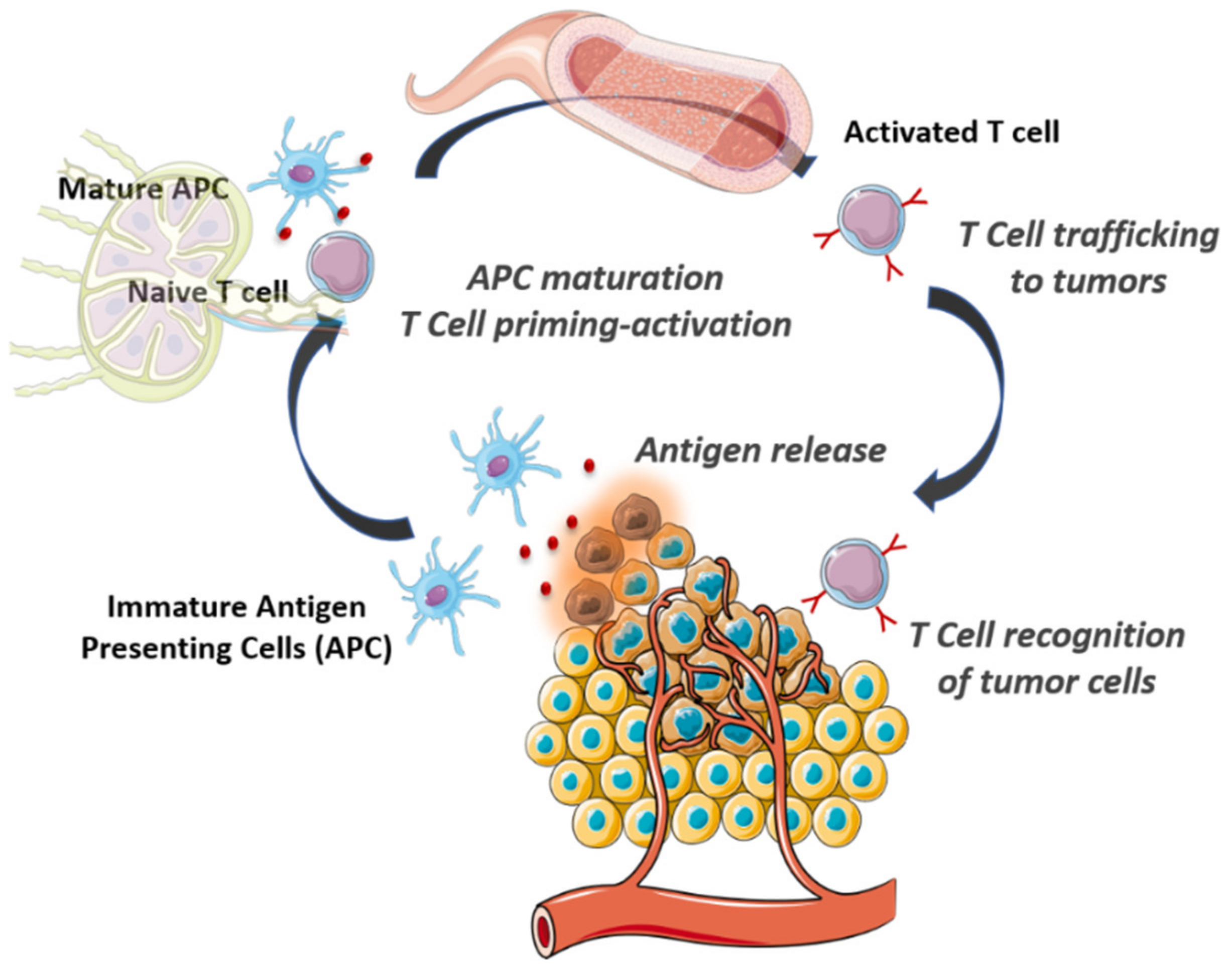
2. The Cancer-Immunity Cycle
3. Nanoparticles to Enhance the Antitumoral Action of Innate Immune Cells
4. Nanoparticles to Enhance the Antitumoral Action of Adaptive Immune Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kaasgaard, T.; Andresen, T.L. Liposomal cancer therapy: Exploiting tumor characteristics. Expert Opin. Drug Deliv. 2010, 7, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2014, 12, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Cao-Milán, R.; Liz-Marzán, L.M. Gold nanoparticle conjugates: Recent advances toward clinical applications. Expert Opin. Drug Deliv. 2014, 11, 741–752. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P.; Patrick, C. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Caster, J.M.; Patel, A.N.; Zhang, T.; Wang, A.Z. Investigational nanomedicines in 2016: A review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 9, e1416. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Florence, A.T. “Targeting” nanoparticles: The constraints of physical laws and physical barriers. J. Control. Release 2012, 164, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Villegas, M.R.; Baeza, A.; Noureddine, A.; Durfee, P.; Butler, K.; Agola, J.O.; Brinker, C.J.; Vallet-Regí, M. Multifunctional Protocells for Enhanced Penetration in 3D Extracellular Tumoral Matrices. Chem. Mater. 2017, 30, 112–120. [Google Scholar] [CrossRef]
- Wang, T.-Y.; Choe, J.W.; Pu, K.; Devulapally, R.; Bachawal, S.; Machtaler, S.; Chowdhury, S.M.; Luong, R.; Tian, L.; Khuri-Yakub, B.; et al. Ultrasound-guided delivery of microRNA loaded nanoparticles into cancer. J. Control. Release 2015, 203, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer active targeting by nanoparticles: A comprehensive review of literature. J. Cancer Res. Clin. Oncol. 2014, 141, 769–784. [Google Scholar] [CrossRef]
- Bahrami, B.; Farsangi, M.H.; Mohammadi, H.; Anvari, E.; Ghalamfarsa, G.; Yousefi, M.; Jadidi-Niaragh, F. Nanoparticles and targeted drug delivery in cancer therapy. Immunol. Lett. 2017, 190, 64–83. [Google Scholar] [CrossRef]
- Arranja, A.G.; Pathak, V.; Lammers, T.; Shi, Y. Tumor-targeted nanomedicines for cancer theranostics. Pharmacol. Res. 2017, 115, 87–95. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Villaverde, G.; Nairi, V.; Baeza, A.; Vallet-Regí, M. Double Sequential Encrypted Targeting Sequence: A New Concept for Bone Cancer Treatment. Chem. Eur. J. 2017, 23, 7174–7179. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Zhang, Y.; Li, Z.; Hou, X.; Feng, N. Red blood cell membrane-camouflaged nanoparticles: A novel drug delivery system for antitumor application. Acta Pharm. Sin. B 2019, 9, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; He, Y.; Zhang, S.; Qin, J.; Wang, J. Cell membrane-based nanoparticles: A new biomimetic platform for tumor diagnosis and treatment. Acta Pharm. Sin. B 2018, 8, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Björnmalm, M.; Thurecht, K.J.; Michael, M.; Scott, A.M.; Caruso, F.; Björnmalm, A.M.H. Bridging Bio–Nano Science and Cancer Nanomedicine. ACS Nano 2017, 11, 9594–9613. [Google Scholar] [CrossRef]
- Fan, Y.; Moon, J. Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy. Vaccines 2015, 3, 662–685. [Google Scholar] [CrossRef]
- Kroll, A.V.; Jiang, Y.; Zhou, J.; Holay, M.; Fang, R.H.; Zhang, L. Biomimetic Nanoparticle Vaccines for Cancer Therapy. Adv. Biosyst. 2018, 3, 1800219. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immun. 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Riley, J. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Loeb, K.R.; Loeb, L.A. Genetic Instability and the Mutator Phenotype. Am. J. Pathol. 1999, 154, 1621–1626. [Google Scholar] [CrossRef]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Limon, P.L.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ng, S.S.; Huo, L.-F.; Chow, B.K.C.; Shen, Z.; Yang, M.; Sze, J.; Ko, O.; Li, M.; Yue, A.; et al. Effective Melanoma Immunotherapy with Interleukin-2 Delivered by a Novel Polymeric Nanoparticle. Mol. Cancer Ther. 2011, 10, 1082–1092. [Google Scholar] [CrossRef]
- Deng, G.; Sun, Z.; Li, S.; Peng, X.; Li, W.; Zhou, L.; Ma, Y.; Gong, P.; Cai, L. Cell-Membrane Immunotherapy Based on Natural Killer Cell Membrane Coated Nanoparticles for the Effective Inhibition of Primary and Abscopal Tumor Growth. ACS Nano 2018, 12, 12096–12108. [Google Scholar] [CrossRef]
- Pitchaimani, A.; Nguyen, T.D.T.; Marasini, R.; Eliyapura, A.; Azizi, T.; Jaberi-Douraki, M.; Aryal, S. Biomimetic Natural Killer Membrane Camouflaged Polymeric Nanoparticle for Targeted Bioimaging. Adv. Funct. Mater. 2018, 29, 1806817. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019, 20, 7–24. [Google Scholar] [CrossRef]
- Fang, R.H.; Kroll, A.V.; Zhang, L. Nanoparticle-Based Manipulation of Antigen-Presenting Cells for Cancer Immunotherapy. Small 2015, 11, 5483–5496. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Goldberg, M.S. Targeting myeloid cells using nanoparticles to improve cancer immunotherapy. Adv. Drug Deliv. Rev. 2015, 91, 38–51. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Wang, Z.; Cai, H.; Lu, Z.; Li, Y.; Du, M.; Huang, G.; Wang, C.; Chen, X.; et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 648–654. [Google Scholar] [CrossRef]
- Park, S.; Kang, S.; Chen, X.; Kim, E.J.; Kim, J.; Kim, N.; Kim, J.; Jin, M.M. Tumor suppression via paclitaxel-loaded drug carriers that target inflammation marker upregulated in tumor vasculature and macrophages. Biomaterials 2013, 34, 598–605. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Van Der Schoot, J.M.S.; Rueda, F.; Torensma, R.; Figdor, C.G. ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation. Molecules 2019, 24, 1825. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, K.; Zhao, R.; Ji, T.; Wang, X.; Yang, X.; Zhang, Y.; Cheng, K.; Liu, S.; Hao, J.-H.; et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials 2016, 102, 187–197. [Google Scholar] [CrossRef] [PubMed]
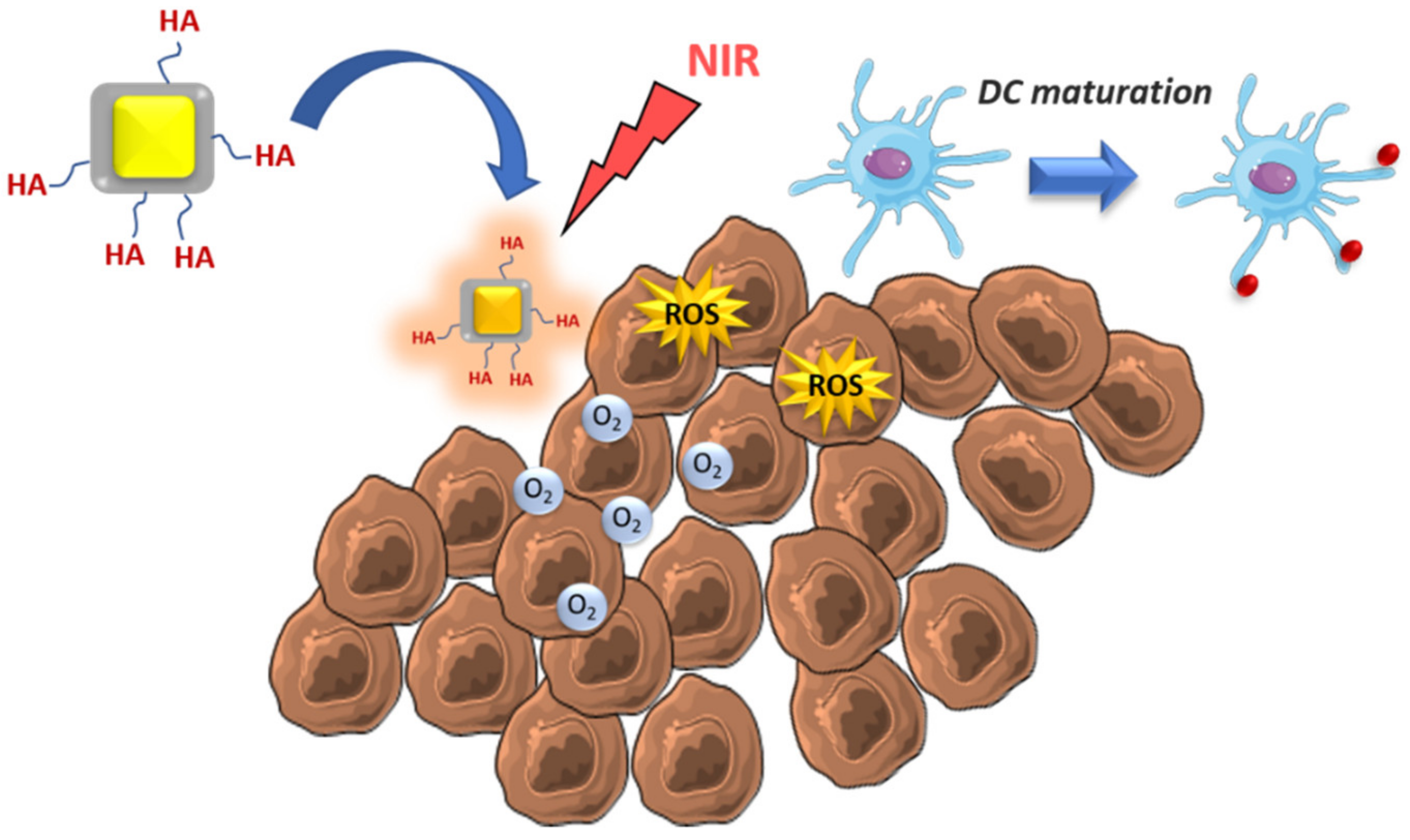
- He, H.; Liu, L.; Liang, R.; Zhou, H.; Pan, H.; Zhang, S.; Cai, L. Tumor-targeted nanoplatform for in situ oxygenation-boosted immunogenic phototherapy of colorectal cancer. Acta Biomater. 2020, 104, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ito, A.; Kobayashi, T.; Kawamura, T.; Shimada, S.; Matsumoto, K.; Saida, T.; Honda, H. Heat immunotherapy using magnetic nanoparticles and dendritic cells for T-lymphoma. J. Biosci. Bioeng. 2005, 100, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Jin, H.; Cho, M.-H. Mannosylated chitosan nanoparticle-based cytokine gene therapy suppressed cancer growth in BALB/c mice bearing CT-26 carcinoma cells. Mol. Cancer Ther. 2006, 5, 1723–1732. [Google Scholar] [CrossRef]
- Kwong, B.; Liu, H.; Irvine, D.J. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 2011, 32, 5134–5147. [Google Scholar] [CrossRef]
- An, M.; Yu, C.; Xi, J.; Reyes, J.; Mao, G.; Wei, W.-Z.; Liu, H. Induction of necrotic cell death and activation of STING in the tumor microenvironment via cationic silica nanoparticles leading to enhanced antitumor immunity. Nanoscale 2018, 10, 9311–9319. [Google Scholar] [CrossRef]
- Min, Y.; Roche, K.C.; Tian, S.; Eblan, M.J.; McKinnon, K.P.; Caster, J.M.; Chai, S.; Herring, L.E.; Zhang, L.; Zhang, T.; et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 877–882. [Google Scholar] [CrossRef]
- Tran, T.-H.; Mattheolabakis, G.; Aldawsari, H.; Amiji, M. Exosomes as nanocarriers for immunotherapy of cancer and inflammatory diseases. Clin. Immunol. 2015, 160, 46–58. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sasso, M.S.; Lollo, G.; Pitorre, M.; Solito, S.; Pinton, L.; Valpione, S.; Bastiat, G.; Mandruzzato, S.; Bronte, V.; Marigo, I.; et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials 2016, 96, 47–62. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liang, P.; Guo, G.; Huang, Z.; Niu, Y.; Dong, L.; Wang, C.; Zhang, J. Re-polarizing Myeloid-derived Suppressor Cells (MDSCs) with Cationic Polymers for Cancer Immunotherapy. Sci. Rep. 2016, 6, 24506. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Muroski, M.E.; Miska, J.; Lee-Chang, C.; Shen, Y.; Rashidi, A.; Zhang, P.; Xiao, T.; Han, Y.; Lopez-Rosas, A.; et al. Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment. Nanomed. Nanotechnol. Boil. Med. 2019, 16, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Mai, J.; Shen, J.; Wolfram, J.; Li, Z.; Zhang, G.; Xu, R.; Li, Y.; Mu, C.; Zu, Y.; et al. A Novel DNA Aptamer for Dual Targeting of Polymorphonuclear Myeloid-derived Suppressor Cells and Tumor Cells. Theranostics 2018, 8, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Yang, Y.; Liu, Y.; Cun, X.; Li, M.; Xu, S.; Zhao, W.; Xiang, Y.; Qiu, Y.; Yu, Q.; et al. Sequential Depletion of Myeloid-Derived Suppressor Cells and Tumor Cells with a Dual-PH-Sensitive Conjugated Micelle System for Cancer Chemoimmunotherapy. J. Control. Release 2020, 317, 43–56. [Google Scholar] [CrossRef]
- Yu, G.-T.; Rao, L.; Wu, H.; Yang, L.-L.; Bu, L.-L.; Deng, W.-W.; Wu, L.; Nan, X.; Zhang, W.-F.; Zhao, X.-Z.; et al. Myeloid-Derived Suppressor Cell Membrane-Coated Magnetic Nanoparticles for Cancer Theranostics by Inducing Macrophage Polarization and Synergizing Immunogenic Cell Death. Adv. Funct. Mater. 2018, 28, 1801389. [Google Scholar] [CrossRef]
- Long, Y.; Lu, Z.; Xu, S.; Li, M.; Wang, X.; Zhang, Z.; He, Q. Self-Delivery Micellar Nanoparticles Prevent Premetastatic Niche Formation by Interfering with the Early Recruitment and Vascular Destruction of Granulocytic Myeloid-Derived Suppressor Cells. Nano Lett. 2019. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Condeelis, J.S. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Poh, A.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin Encapsuled Nanoparticles “Re-Educate” Tumor-Associated Macrophages and Exhibit Anti-Tumor Effects on Breast Cancer Following STAT3 Suppression. PLoS ONE 2013, 8, e65896. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Z.; Jiang, Y.; Zhang, D.; Chen, J.; Dong, L.; Zhang, J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J. Control. Release 2012, 158, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Niu, M.; O’Mary, H.; Cui, Z. Targeting of Tumor-Associated Macrophages Made Possible by PEG-Sheddable, Mannose-Modified Nanoparticles. Mol. Pharm. 2013, 10, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Tsoi, K.M.; Ouyang, B.; Ma, X.-Z.; Manuel, J.; Fawaz, A.; Ostrowski, M.; Alman, B.A.; Zilman, A.; Chan, W.C.W.; et al. Phenotype Determines Nanoparticle Uptake by Human Macrophages from Liver and Blood. ACS Nano 2017, 11, 2428–2443. [Google Scholar] [CrossRef]
- Song, M.; Liu, T.; Shi, C.; Zhang, X.; Chen, X. Bioconjugated Manganese Dioxide Nanoparticles Enhance Chemotherapy Response by Priming Tumor-Associated Macrophages toward M1-like Phenotype and Attenuating Tumor Hypoxia. ACS Nano 2015, 10, 633–647. [Google Scholar] [CrossRef]
- Shi, C.; Liu, T.; Guo, Z.; Zhuang, R.; Zhang, X.; Chen, X.S. Reprogramming Tumor-Associated Macrophages by Nanoparticle-Based Reactive Oxygen Species Photogeneration. Nano Lett. 2018, 18, 7330–7342. [Google Scholar] [CrossRef]
- Tian, L.; Yi, X.; Dong, Z.; Xu, J.; Liang, C.; Chao, Y.; Wang, Y.; Yang, K.; Liu, Z. Calcium Bisphosphonate Nanoparticles with Chelator-Free Radiolabeling to Deplete Tumor-Associated Macrophages for Enhanced Cancer Radioisotope Therapy. ACS Nano 2018, 12, 11541–11551. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Wu, A.; Drake, V.; Huang, A.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. OncoImmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-Delivered Transforming Growth Factor-β siRNA Enhances Vaccination against Advanced Melanoma by Modifying Tumor Microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, Z.; Xu, Z.; Chen, X.; Zhu, G. A Cisplatin-Loaded Immunochemotherapeutic Nanohybrid Bearing Immune Checkpoint Inhibitors for Enhanced Cervical Cancer Therapy. Angew. Chem. Int. Ed. 2018, 57, 3426–3430. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhou, F.; Hou, B.; Wang, D.; Wang, T.; Fu, Y.; Ma, Y.; Yu, H.; Yu, H. Binary Cooperative Prodrug Nanoparticles Improve Immunotherapy by Synergistically Modulating Immune Tumor Microenvironment. Adv. Mater. 2018, 30, 1803001. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; He, C.; Guo, N.; Chan, C.; Ni, K.; Lan, G.; Tang, H.; Pelizzari, C.; Fu, Y.-X.; Spiotto, M.T.; et al. Low-dose X-ray radiotherapy-radiodynamic therapy via nanoscale metal-organic frameworks enhances checkpoint blockade immunotherapy. Nat. Biomed. Eng. 2018, 2, 600–610. [Google Scholar] [CrossRef]
- Cheng, K.; Ding, Y.; Zhao, Y.; Ye, S.; Zhao, X.; Zhang, Y.; Ji, T.; Wu, H.; Wang, B.; Anderson, G.J.; et al. Sequentially Responsive Therapeutic Peptide Assembling Nanoparticles for Dual-Targeted Cancer Immunotherapy. Nano Lett. 2018, 18, 3250–3258. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. The PD-1–PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, E.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, W.; Zhou, M.; Huang, Q.; Wen, X.; Zhao, J.; Shi, S.; Geng, K.; Li, F.; Hatakeyama, H.; et al. Induction of Antitumor Immunity in Mice by the Combination of Nanoparticle-Based Photothermolysis and Anti-PD-1 Checkpoint Inhibition. Nanomed. Nanotechnol. Biol. Med. 2020, 25, 102169. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Y.; Zhu, J.; Jin, L.; Liu, B.; Xia, K.; Wang, J.; Gao, J.; Liang, C.; Tao, H. Autophagy inhibitor enhance ZnPc/BSA nanoparticle induced photodynamic therapy by suppressing PD-L1 expression in osteosarcoma immunotherapy. Biomaterials 2019, 192, 128–139. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Han, W.; Guo, N.; Weichselbaum, R.R.; Lin, W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat. Commun. 2019, 10, 1899. [Google Scholar] [CrossRef] [PubMed]
- Phung, C.D.; Nguyen, H.T.; Choi, J.Y.; Pham, T.T.; Acharya, S.; Timilshina, M.; Chang, J.-H.; Kim, J.-H.; Jeong, J.-H.; Ku, S.K.; et al. Reprogramming the T cell response to cancer by simultaneous, nanoparticle-mediated PD-L1 inhibition and immunogenic cell death. J. Control. Release 2019, 315, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-W.; Hsu, F.-F.; Qiu, J.T.; Chern, G.-J.; Lee, Y.-A.; Chang, C.-C.; Huang, Y.-T.; Sung, Y.-C.; Chiang, C.-C.; Huang, R.-L.; et al. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci. Adv. 2020, 6, eaax5032. [Google Scholar] [CrossRef] [PubMed]
- Kosmides, A.K.; Sidhom, J.-W.; Fraser, A.K.; Bessell, C.A.; Schneck, J.P. Dual targeting nanoparticle stimulates the immune system to inhibit tumor growth. ACS Nano 2017, 11, 5417–5429. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Yu, Y. Janus Nanoparticles for T Cell Activation: Clustering Ligands to Enhance Stimulation. J. Mater. Chem. B 2017, 5, 4410–4415. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Van Hooren, L.; Sandin, L.; Moskalev, I.; Ellmark, P.; Dimberg, A.; Black, P.; Tötterman, T.H.T.H.; Mangsbo, S. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur. J. Immunol. 2016, 47, 385–393. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 2016, 7, 13193. [Google Scholar] [CrossRef]
- Xu, J.; Xu, L.; Wang, C.; Yang, R.; Zhuang, Q.; Han, X.; Dong, Z.; Zhu, W.; Peng, R.; Liu, Z. Near-Infrared-Triggered Photodynamic Therapy with Multitasking Upconversion Nanoparticles in Combination with Checkpoint Blockade for Immunotherapy of Colorectal Cancer. ACS Nano 2017, 11, 4463–4474. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Kohrt, H.; Sagiv-barfi, I.; Ajami, B.; Axtell, R.C.; Zhou, G.; Rajapaksa, R.; Green, M.R.; Torchia, J.; Brody, J.; et al. Depleting Tumor-Specific Tregs at a Single Site Eradicates Disseminated Tumors. J. Clin. Investig. 2013, 123, 2447–2463. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, N.; Janikashvili, N.; Lacasse, C.J.; Larmonier, C.B.; Cantrell, J.; Situ, E.; Lundeen, T.; Bonnotte, B.; Katsanis, E. Imatinib mesylate inhibits CD4+CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABLnegative tumors. J. Immunol. 2008, 181, 6955–6963. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Thapa, R.K.; Jiang, L.; Soe, Z.C.; Gautam, M.; Chang, J.-H.; Jeong, J.-H.; Ku, S.K.; Choi, H.-G.; Yong, C.S.; et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J. Control. Release 2018, 281, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Luan, X.; Paholak, H.J.; Burnett, J.P.; Stevers, N.O.; Sansanaphongpricha, K.; He, M.; Chang, A.; Li, Q.; Sun, D. Depleting tumor-associated Tregs via nanoparticle-mediated hyperthermia to enhance anti-CTLA-4 immunotherapy. Nanomedicine 2020, 15, 77–92. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, B.; Zai, W.; Kang, L.; Yuan, A.; Hu, Y.; Wu, J. Perfluorocarbon nanoparticle-mediated platelet inhibition promotes intratumoral infiltration of T cells and boosts immunotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 11972–11977. [Google Scholar] [CrossRef]
- Rao, L.; Bu, L.-L.; Meng, Q.-F.; Cai, B.; Deng, W.-W.; Li, A.; Li, K.; Guo, S.-S.; Zhang, W.-F.; Liu, W.; et al. Antitumor Platelet-Mimicking Magnetic Nanoparticles. Adv. Funct. Mater. 2017, 27, 1604774. [Google Scholar] [CrossRef]
- Park, K. The beginning of the end of the nanomedicine hype. J. Control. Release 2019, 305, 221–222. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action – Immune Cell Target | Nanoparticle Type | Payload | Tumor Model | Ref |
|---|---|---|---|---|
| Enhance NK population | Liposomal-polymer core-shell | IL-2 and TGF-β inhibitors | Melanoma | [33] |
| Recruitment and activation NK and T cells | Polyethylenimine-β-cyclodextrin | IL-2 gene | Melanoma | [34] |
| Production of DAMPs – NK and APC activation | PLGA | NIR photosensitizers | Breast cancer | [35] |
| ICD of tumoral cells – DC activation and high T cell infiltration | PLGA-PEG | oxaliplatin | Pancreatic cancer | [43] |
| ROS generation by NIR exposition – DC activation | Core-shell Au@MnO2 | - | Colorectal cancer | [44] |
| DC activation | Chitosan | IL-12 | Colon adeno-carcinoma | [46] |
| DC activation | PEGylated -liposomes | anti-CD40 and CpG | Melanoma | [47] |
| Capture of tumor antigens after radiotherapy – DC activation | PLGA | Amino- and maleimide groups | Melanoma | [49] |
| MSDC depletion | Liposomes | gemcitabine | Melanoma | [52] |
| Tumoral cell elimination by ROS and MSDC repolarization | Zinc-doped iron oxide-PEI | - | Glioblastoma | [54] |
| MSDC depletion | Aptamer-liposomes | Dox | Breast cancer | [55] |
| Tumoral cell elimination and MSDC depletion | pH-sensitive micelles | RGX-104 and PTX | Breast cancer | [56] |
| Reduction of MSDC recruitment after tumor surgery | self-assembled micelles | Dox, αGC and TOS | Lung metastasis | [58] |
| TAM repolarization M2-M1 by SAT3 inhibition | Liposomes | Hidrazinocurcumin | Breast cancer | [61] |
| TAM repolarization M2-M1 | Dextran PEG-histidine-modified alginate | CpG, anti-IL-10 and anti-IL-10RA | Hepatoma | [62] |
| TAM repolarization M2-M1 | Core-shell manganese dioxide@HA | Dox (coadministration) | Breast cancer | [65] |
| TAM repolarization M2-M1 by ROS generation under NIR | PLGA | ICG, TiO2 and NH4HCO3 | Breast cancer | [66] |
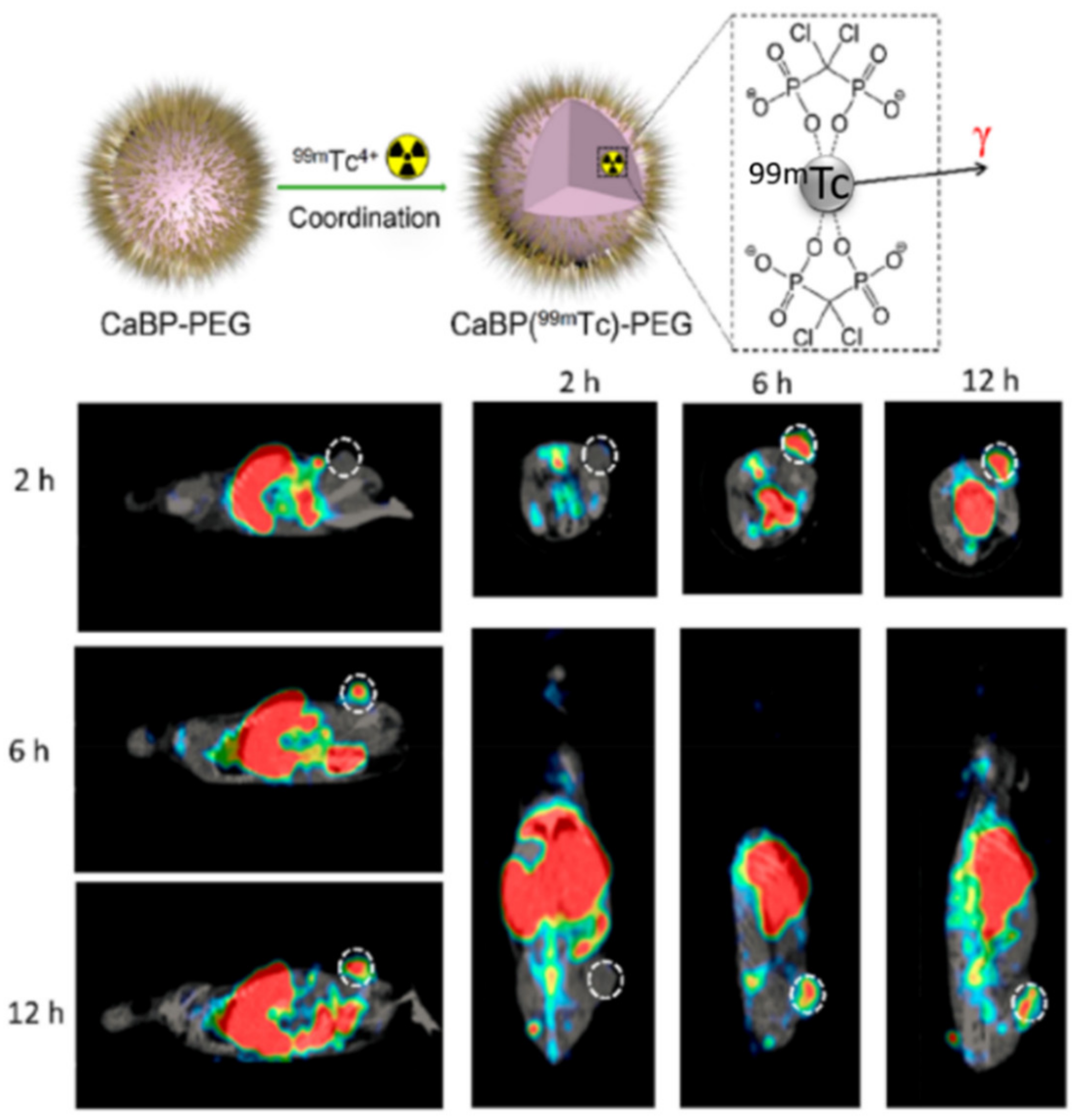
| TAM depletion by calcium bisphosphonate dissolution and tumor imaging by SPECT | calcium bisphosphonate | 99mTc and 32P radioisotopes | Breast cancer | [67] |
| Mechanism of Action – Immune Cell Target | Nanoparticle Type | Payload | Tumor Model | Ref |
|---|---|---|---|---|
| Elimination of immunosuppressive environment - higher infiltration CD8+ T cells | liposome-protamine-hyaluronic acid and lipid-calcium-phosphate | siRNA to silence TGF-β and tumor antigens/CpG | Melanoma | [71] |
| Tumoral cell elimination and IDO inhibition - Higher infiltration of T cells | Layered double hydroxide Mg/Al | IDO inhibitors and disuccinatocisplatin | Cervical cancer | [72] |
| IDO inhibition and ROS generation under X-rays - T cell activation, abscopal effect and tumor rechallenge resistance | Hafnium (Hf)-based MOF | IDO inhibitors and porphyrins | Several tumor models | [74] |
| Enhanced survival of T cells | Self-assembled amphiphilic peptide | IDO inhibitors and PD-L1 antagonist | Melanoma | [75] |
| Higher expression of CRT, TNF-α, INF-γ and IL-6 - higher APC maturation and higher T cell activation and infiltration | Core-shell coordination polymers | Photosensitizer/oxalilplatin and anti-PD-L1 | Colorectal cancer | [78] |
| ROS generation which enhances expression CRT - T cell activation, memory effect | coordination polymer | Dihydroartemisinin and anti-PD-L1 | Colorectal cancer | [81] |
| ICD tumoral cells, higher T cell activation and infiltration | PLGA-PEI-PEG | Dox and microRNA that silence PD-L1 | Colon adenocarcinoma | [82] |
| Improve CD8+ T cell infiltration | Lipid-dendrimer-calcium-phosphate | siRNA that silence PD-L1 and plasmid encoding IL-2 | Lung metastasis | [83] |
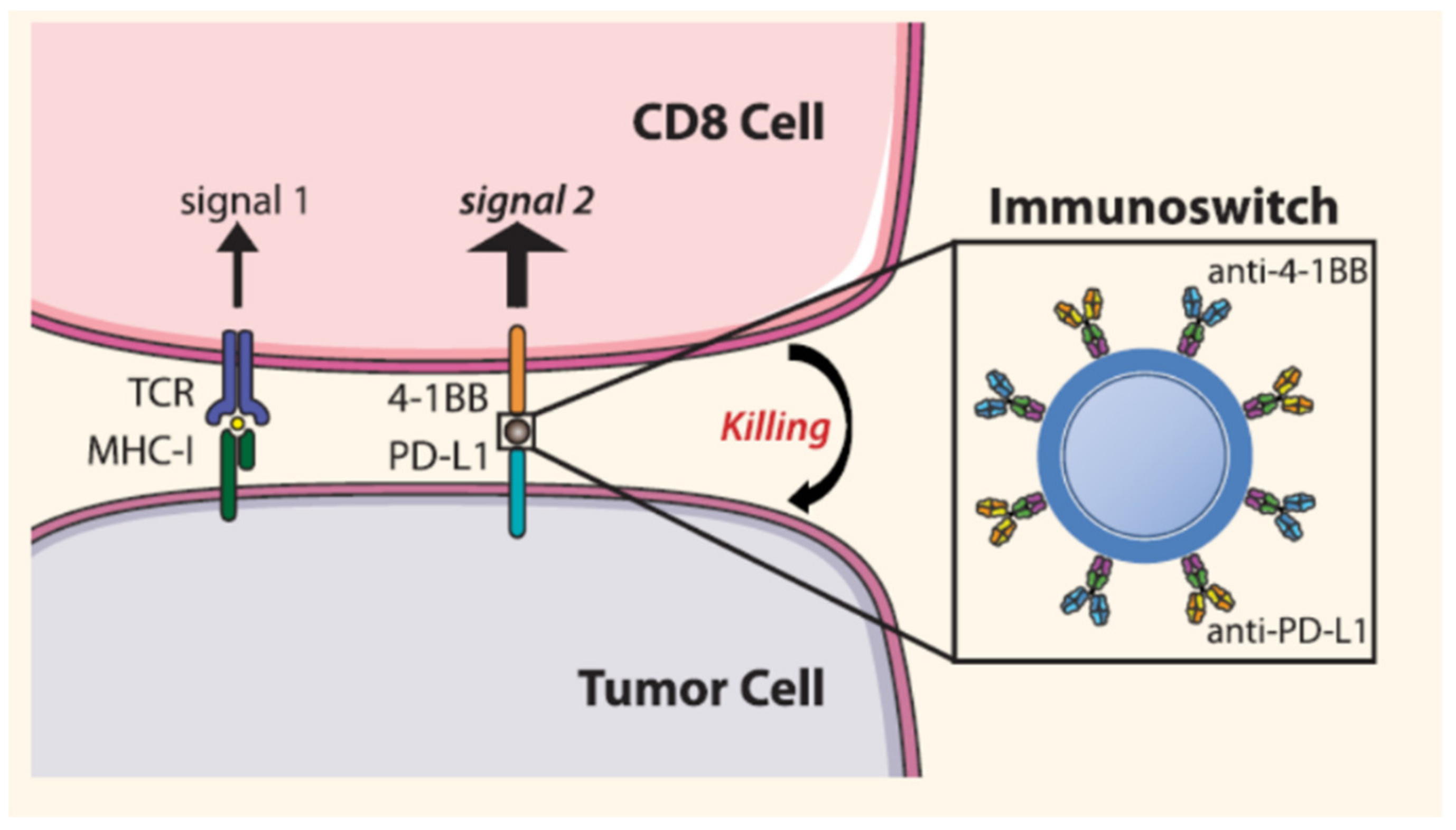
| Blocks PD-L1 and stimulate T cell with anti-4-1BB | Iron oxide | Anti-PD-L1 and anti-4-1BB | Melanoma | [84] |
| Thermal ablation under NIR -DC stimulation; T cell activation; abscopal effect | PLGA | Photosensitizer/imiquimod and anti-CTLA-4 | Breast and colon cancer | [88] |
| Selective depletion Treg by targeting neuropilin-1 receptor with tLyp1 | PLGA@lipid decorated with tLyp1 | Imatinib combined with anti-CTLA-4 | Melanoma | [94] |
| PTT by NIR to destroy Treg | Polymer-coated iron oxide | Combined with anti-CTLA-4 | Breast cancer | [95] |
| Platelet depletion; increase blood vessel permeability of T cells | perfluorotributylamine (PFTBA)-albumin | Combined with anti-PD-L1 | Colorectal and melanoma | [97] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baeza, A. Tumor Targeted Nanocarriers for Immunotherapy. Molecules 2020, 25, 1508. https://doi.org/10.3390/molecules25071508
Baeza A. Tumor Targeted Nanocarriers for Immunotherapy. Molecules. 2020; 25(7):1508. https://doi.org/10.3390/molecules25071508
Chicago/Turabian StyleBaeza, Alejandro. 2020. "Tumor Targeted Nanocarriers for Immunotherapy" Molecules 25, no. 7: 1508. https://doi.org/10.3390/molecules25071508
APA StyleBaeza, A. (2020). Tumor Targeted Nanocarriers for Immunotherapy. Molecules, 25(7), 1508. https://doi.org/10.3390/molecules25071508