Silica Coated Iron/Iron Oxide Nanoparticles as a Nano-Platform for T2 Weighted Magnetic Resonance Imaging



Abstract
:1. Introduction
2. Results and Discussion
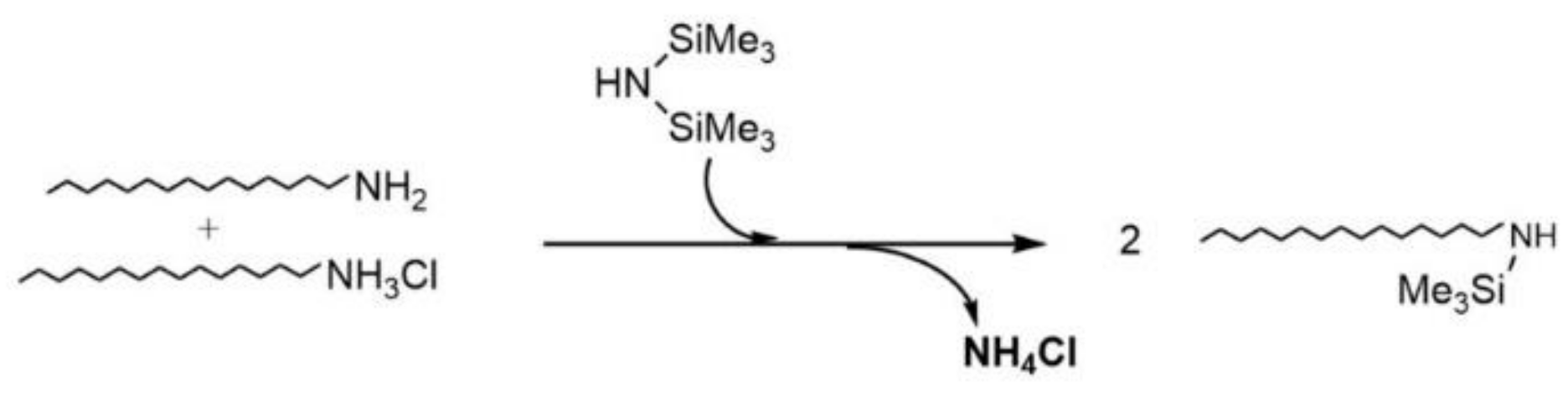
2.1. Iron NPs
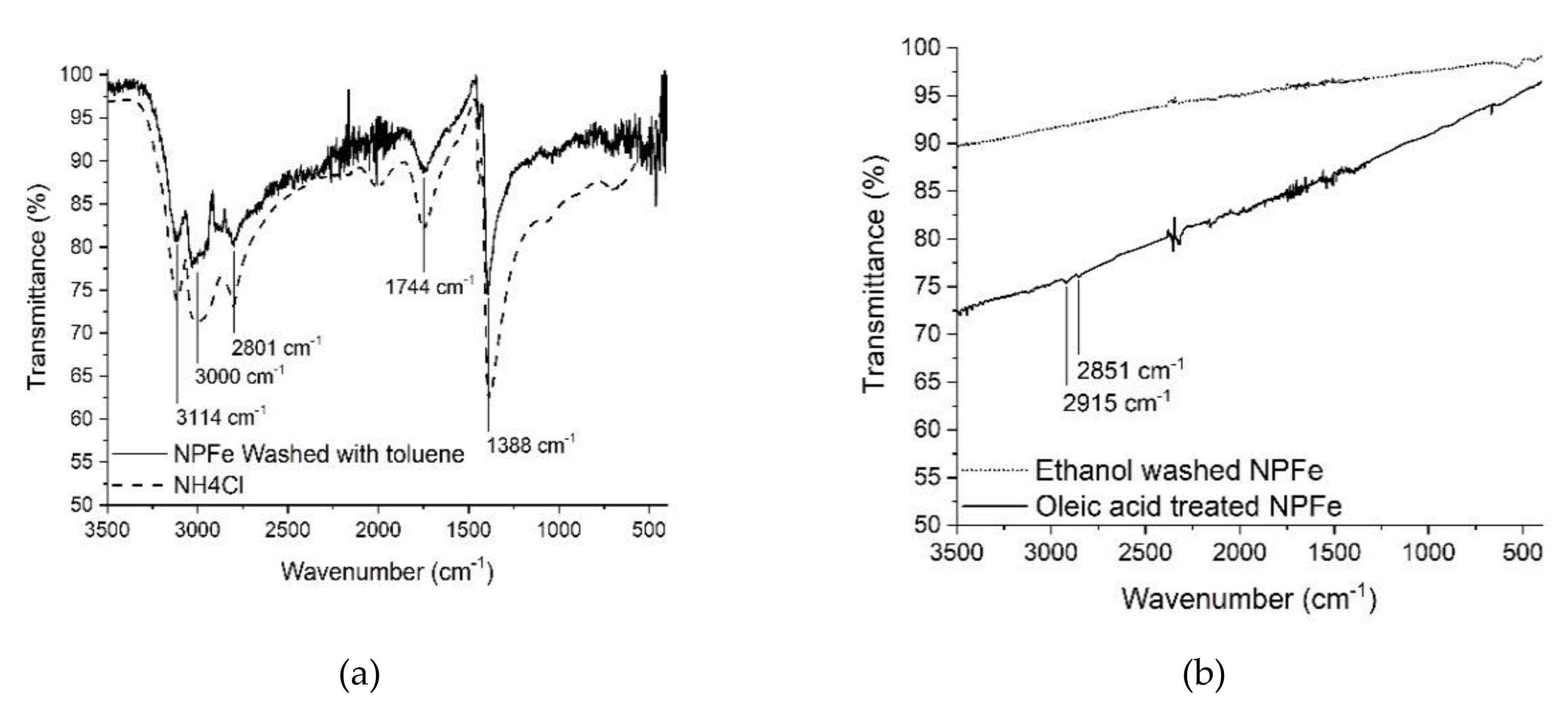
2.2. Silica Coated Fe@FeOx NPs
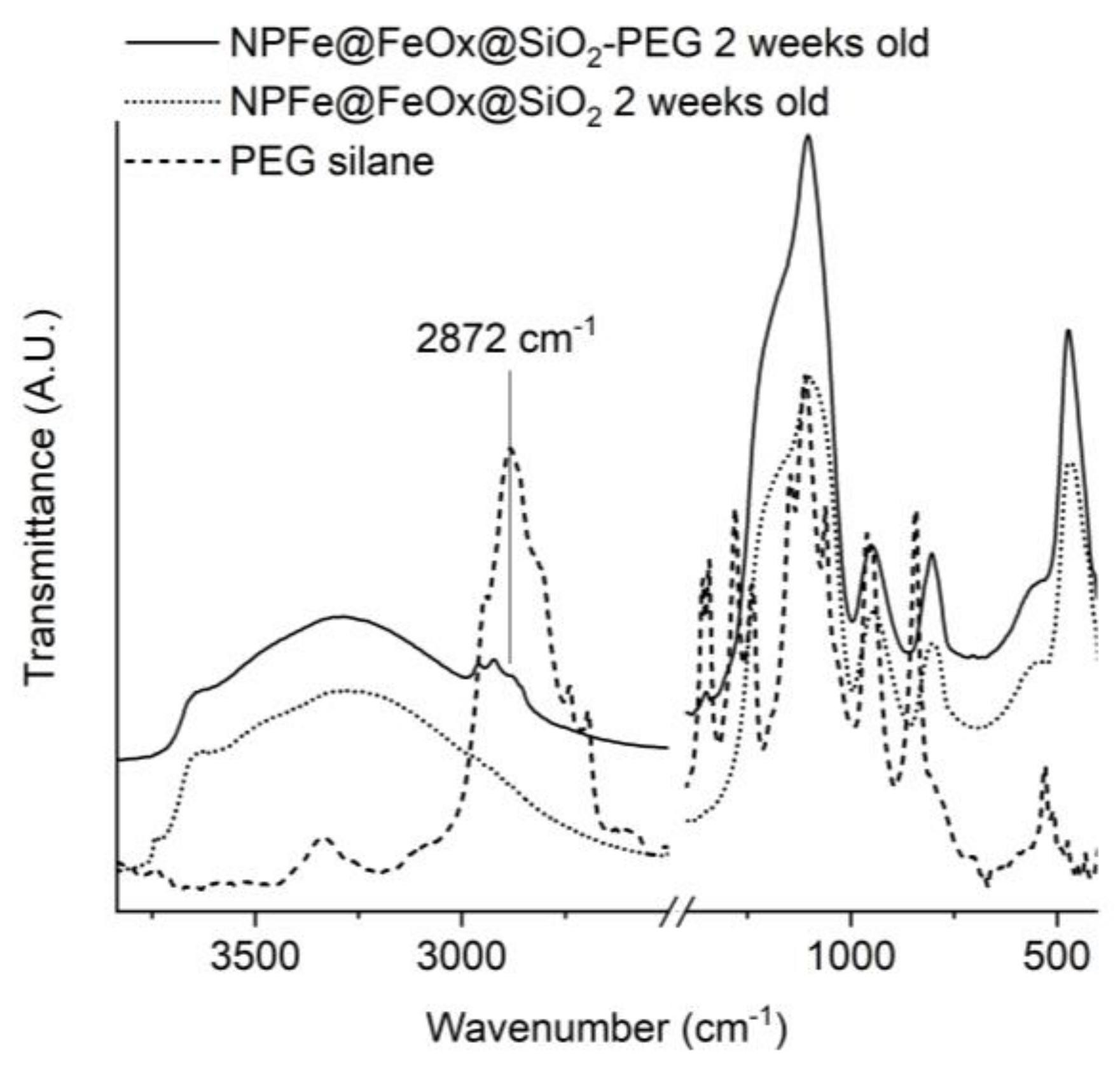
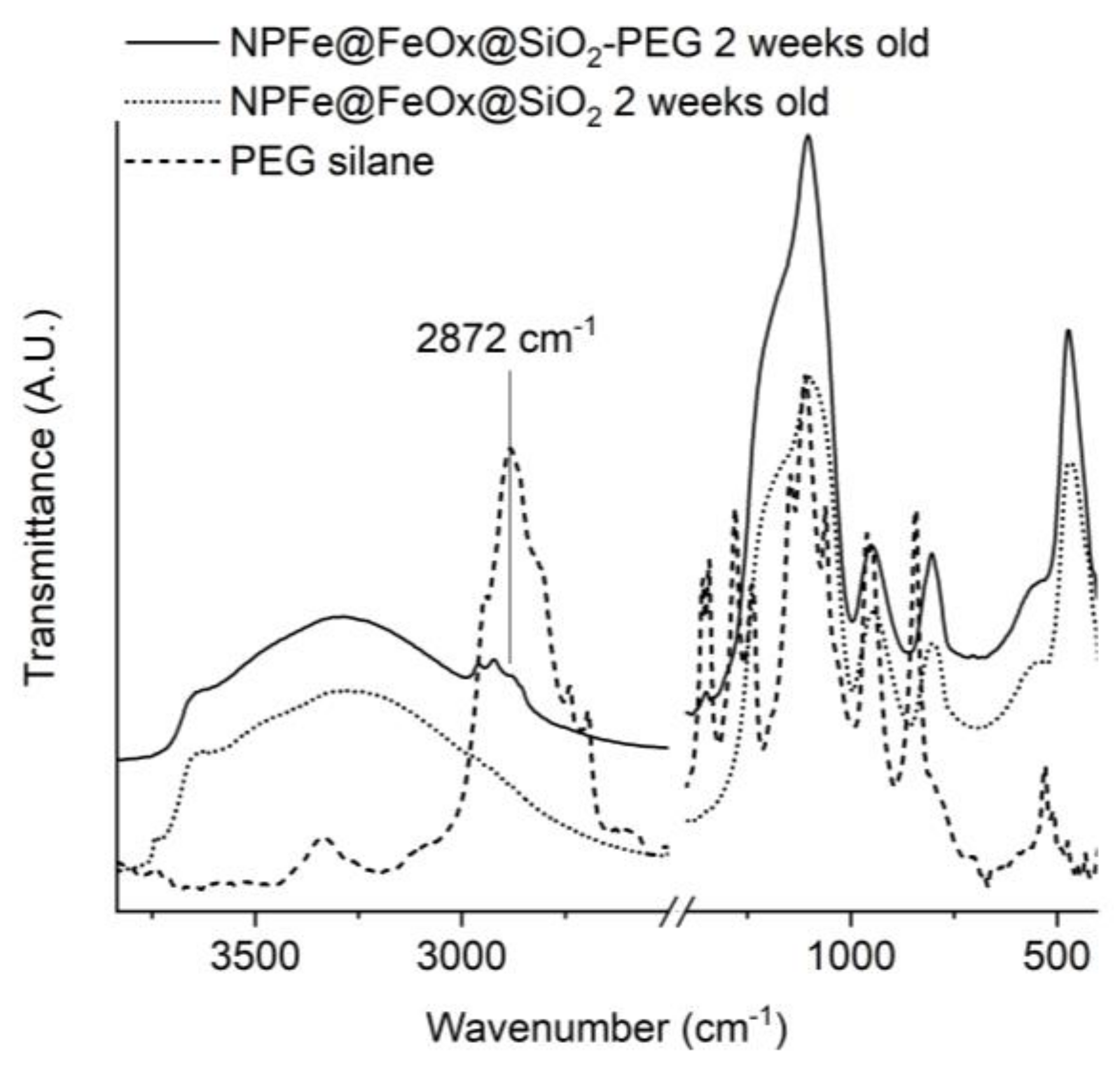
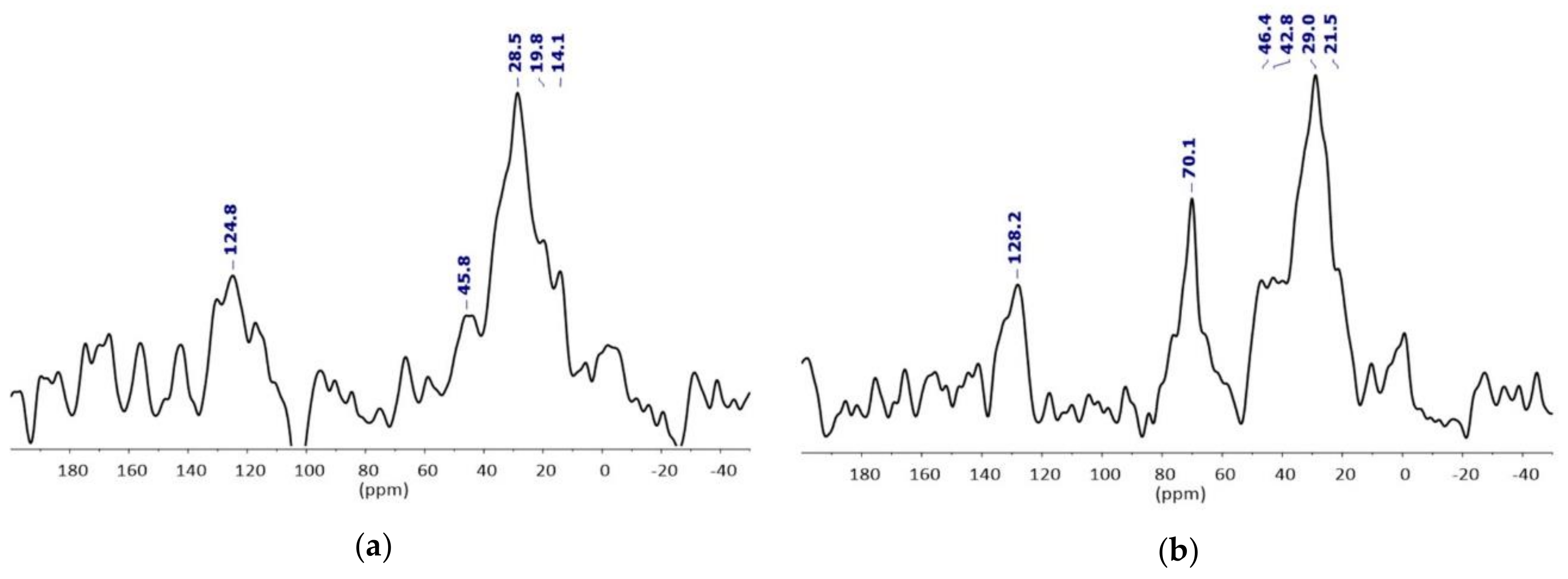
2.3. Surface Functionnalization
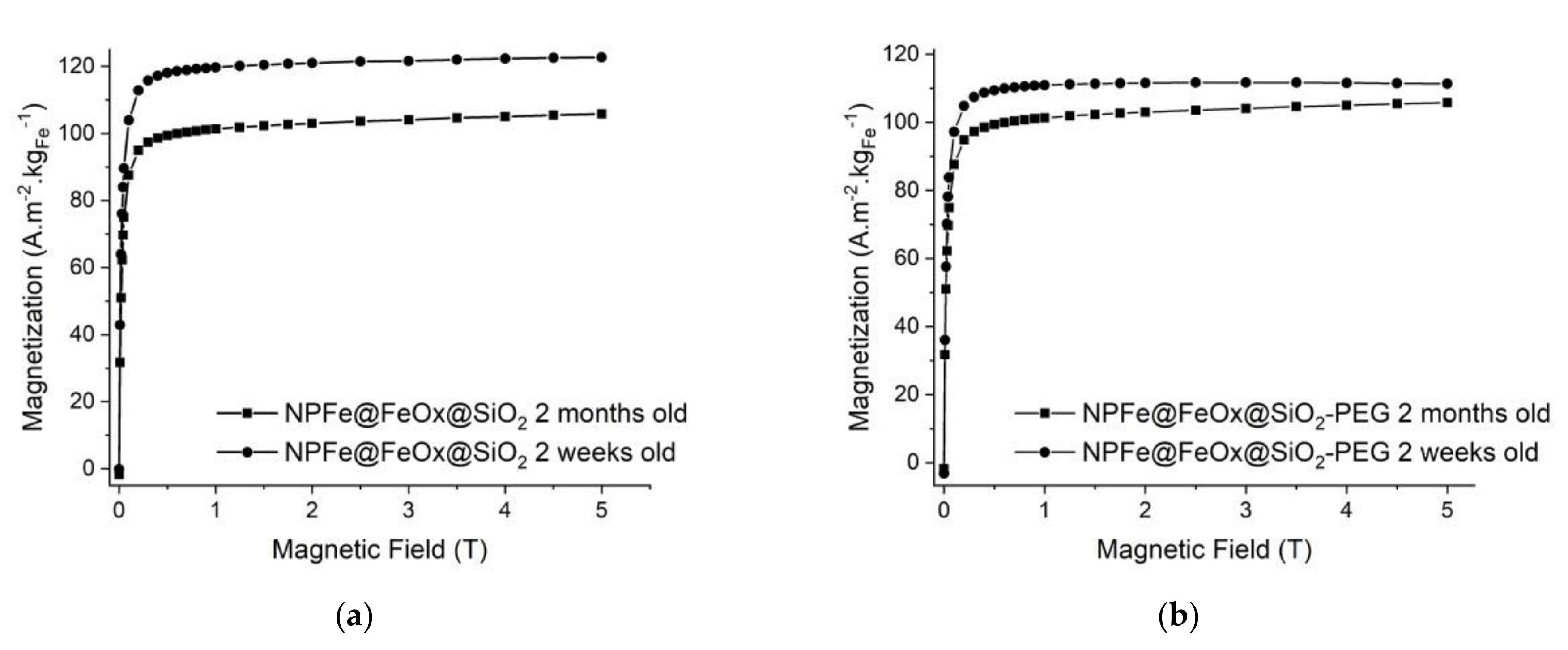
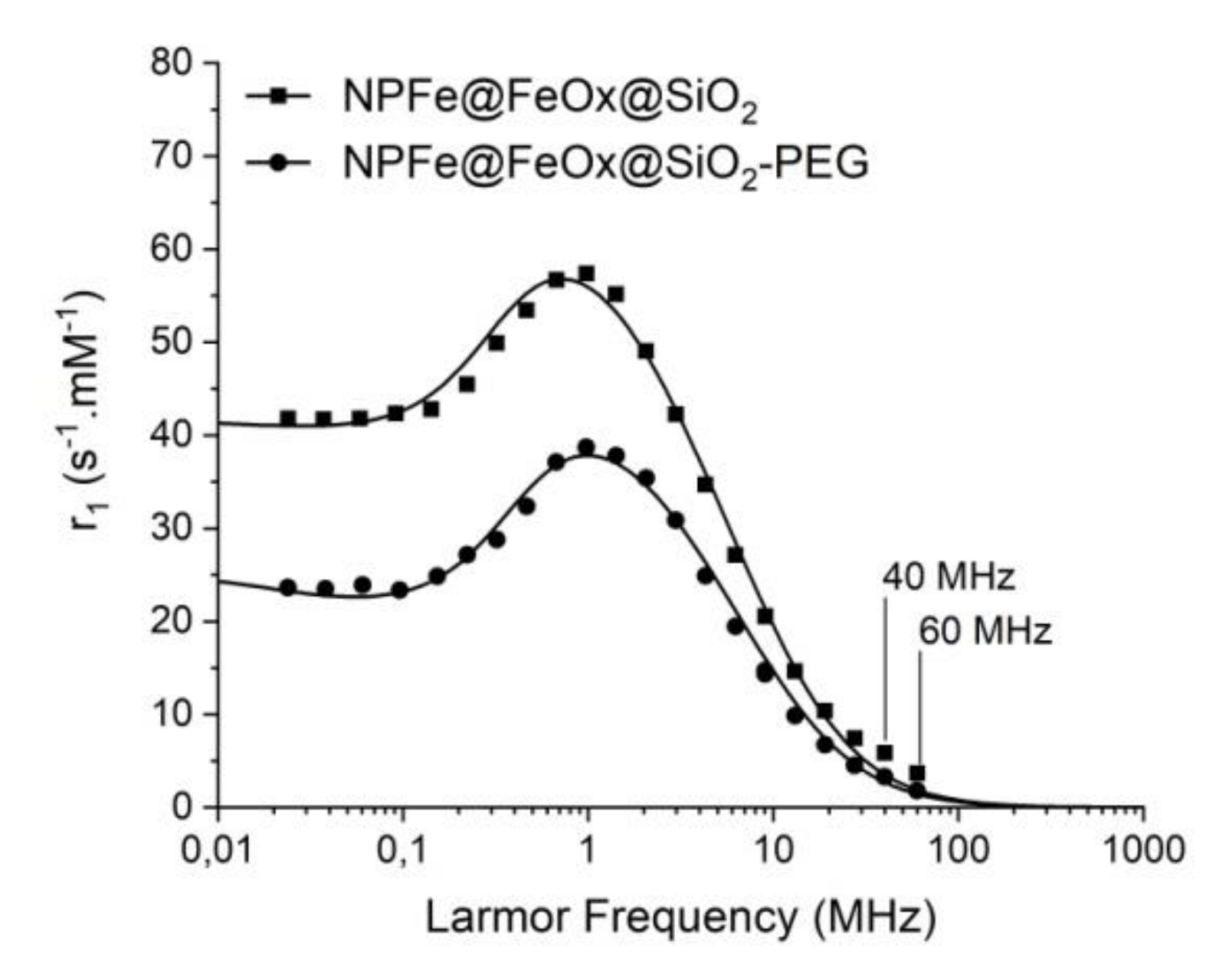
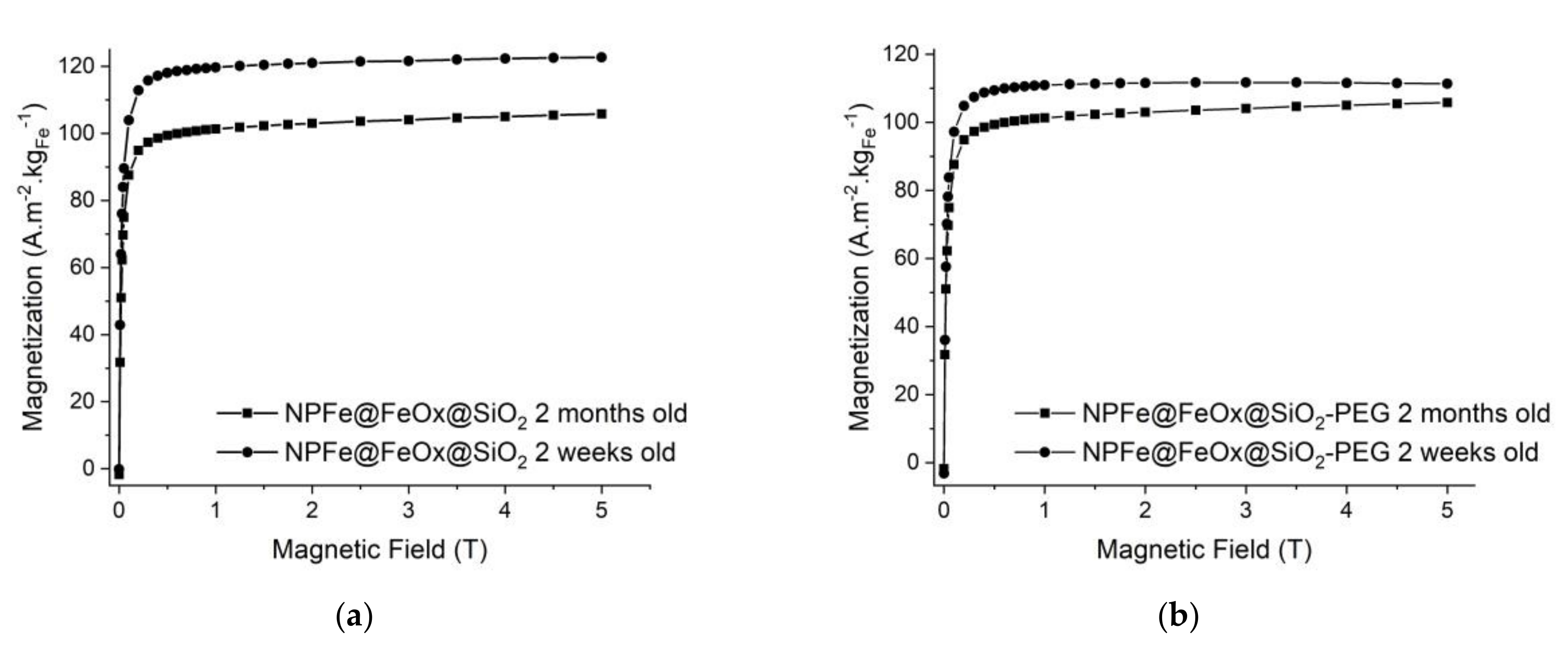
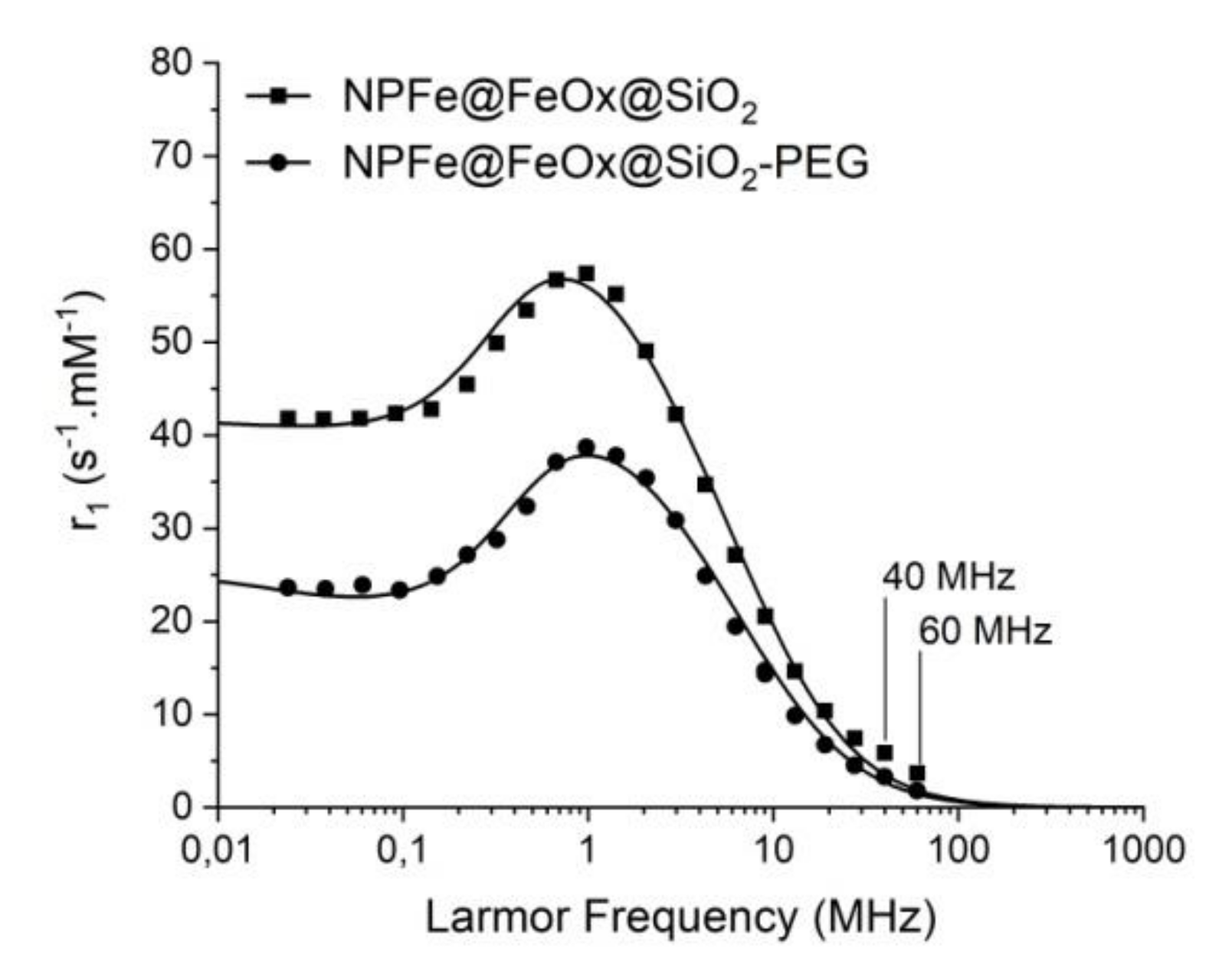
2.4. Magnetic and Relaxometric Properties
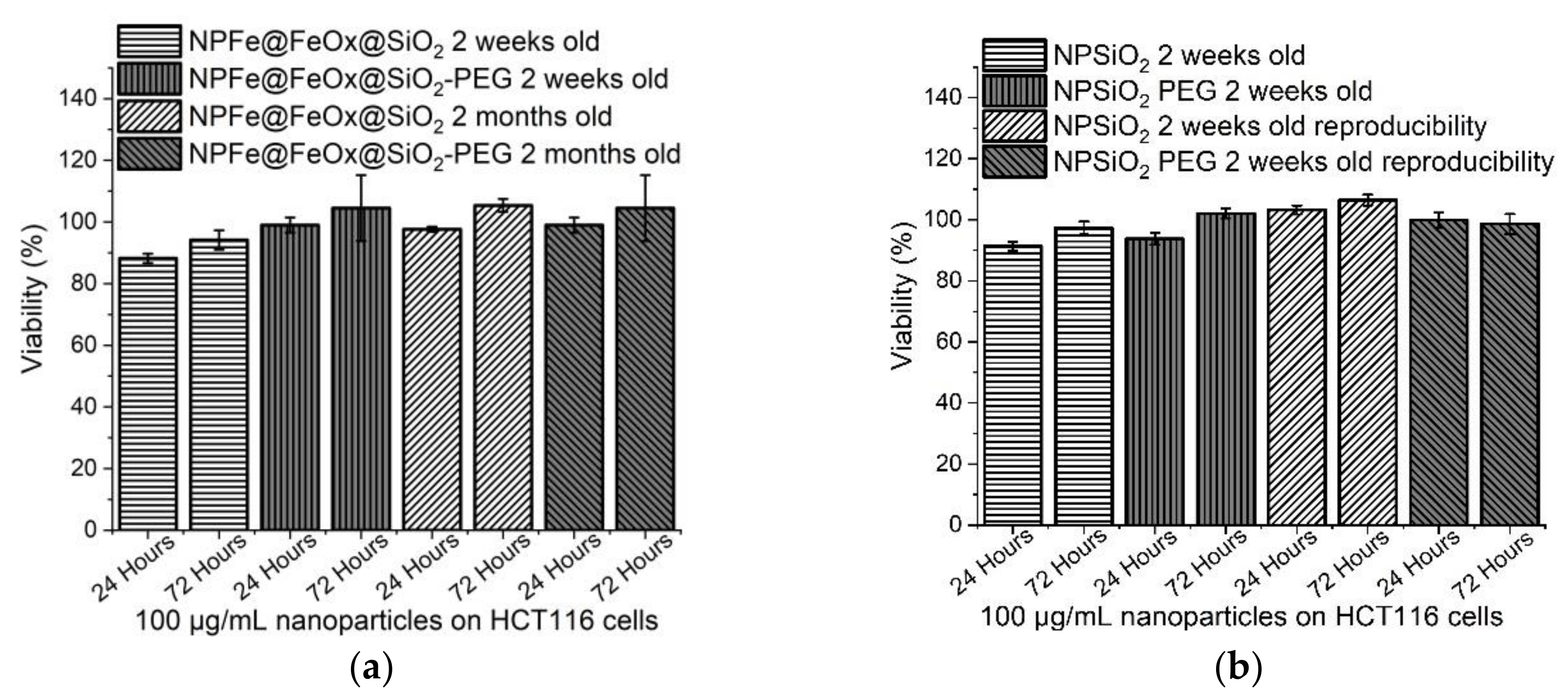
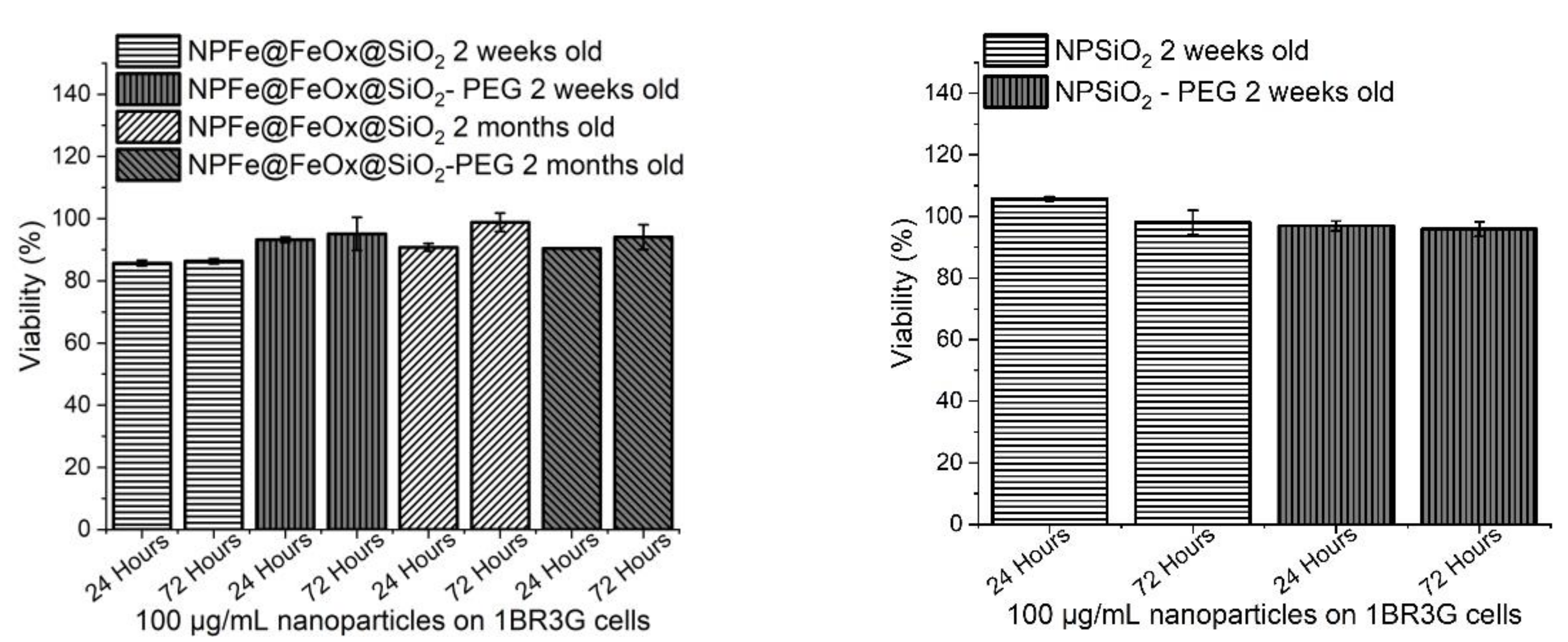
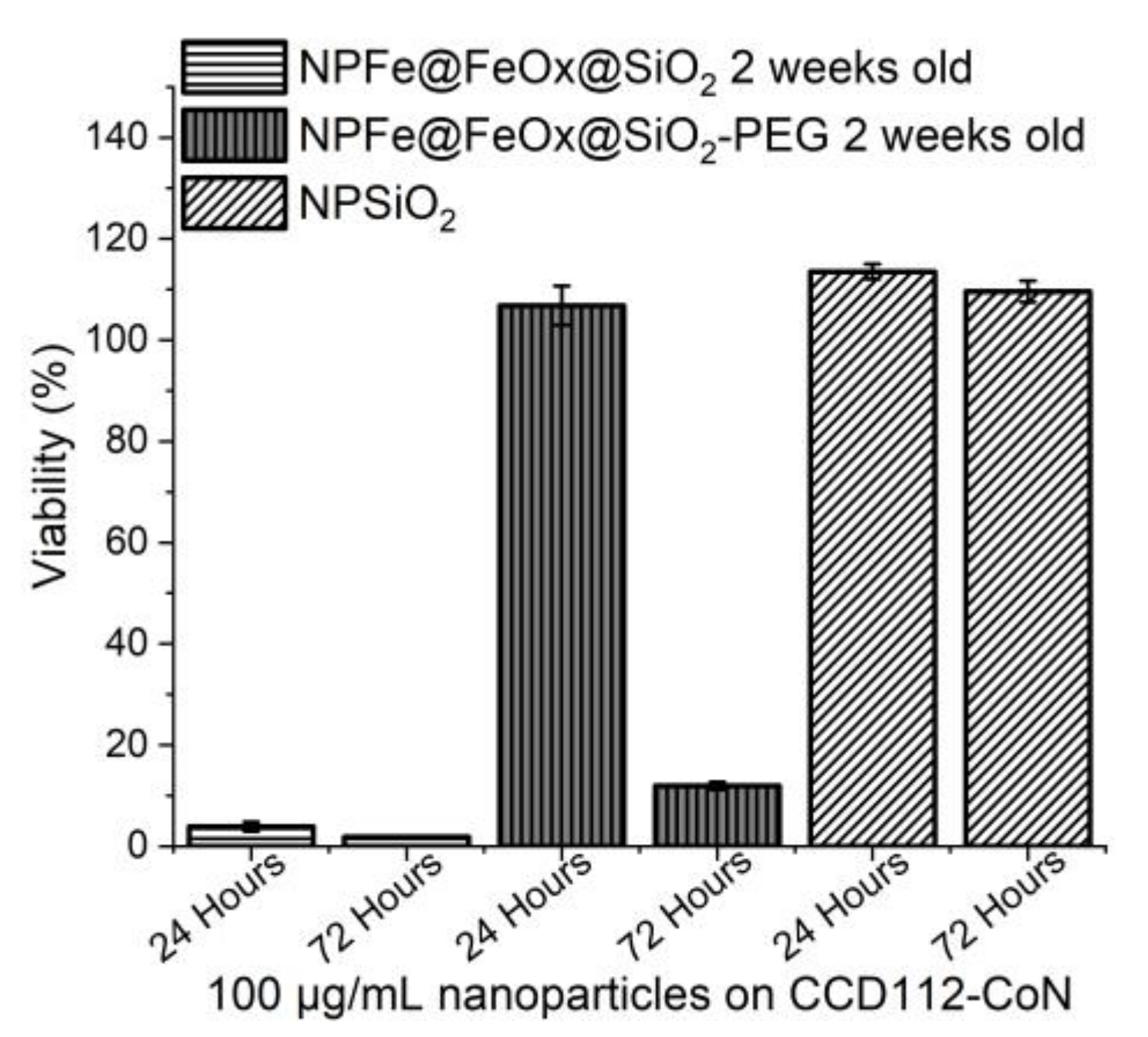
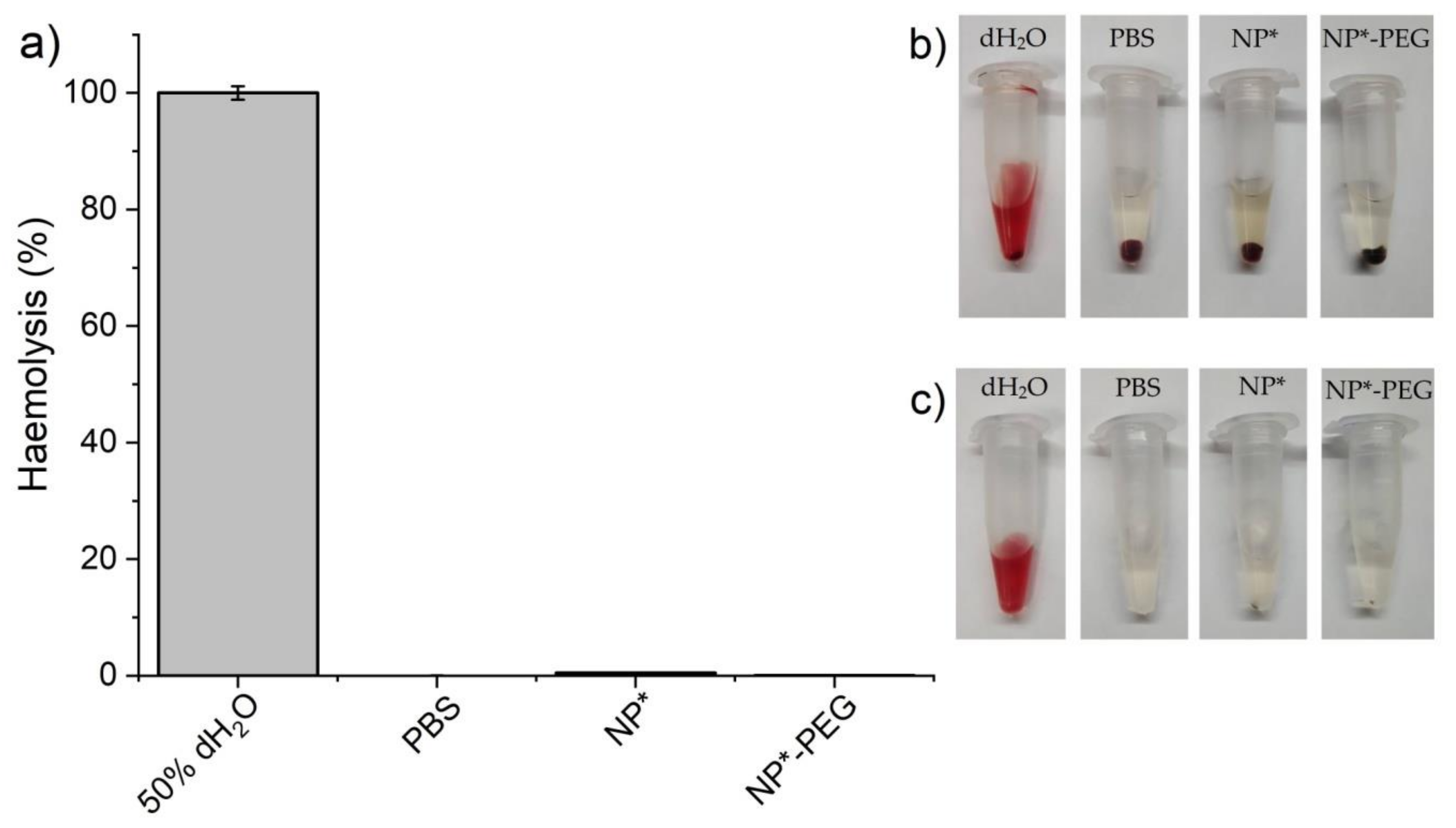
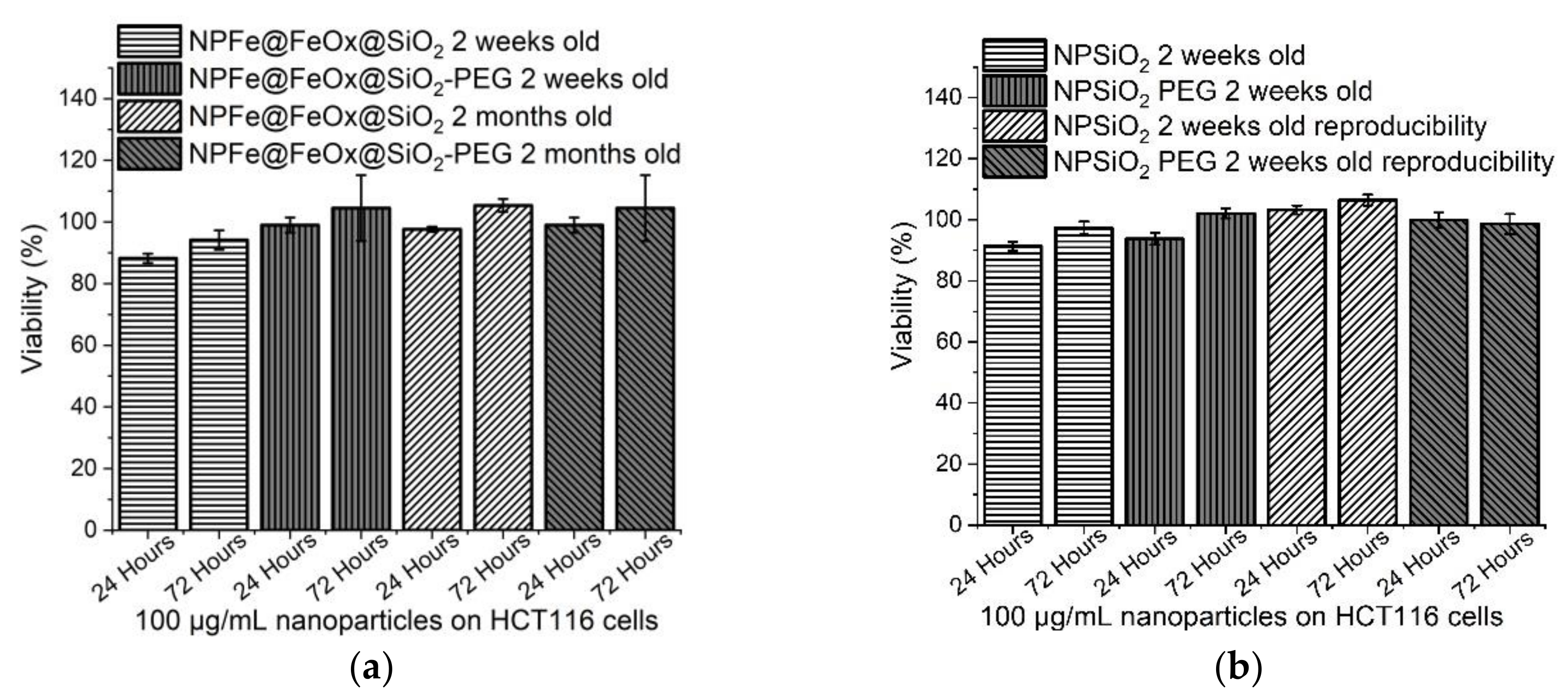
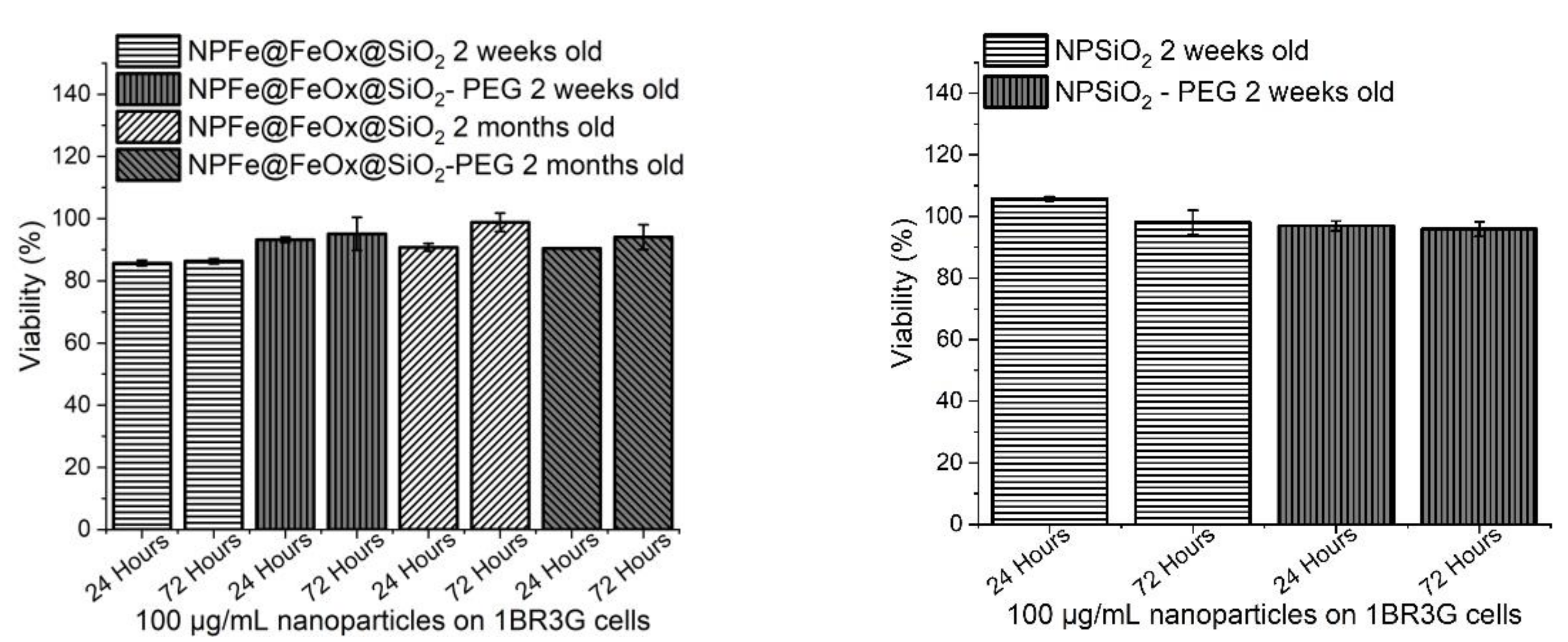
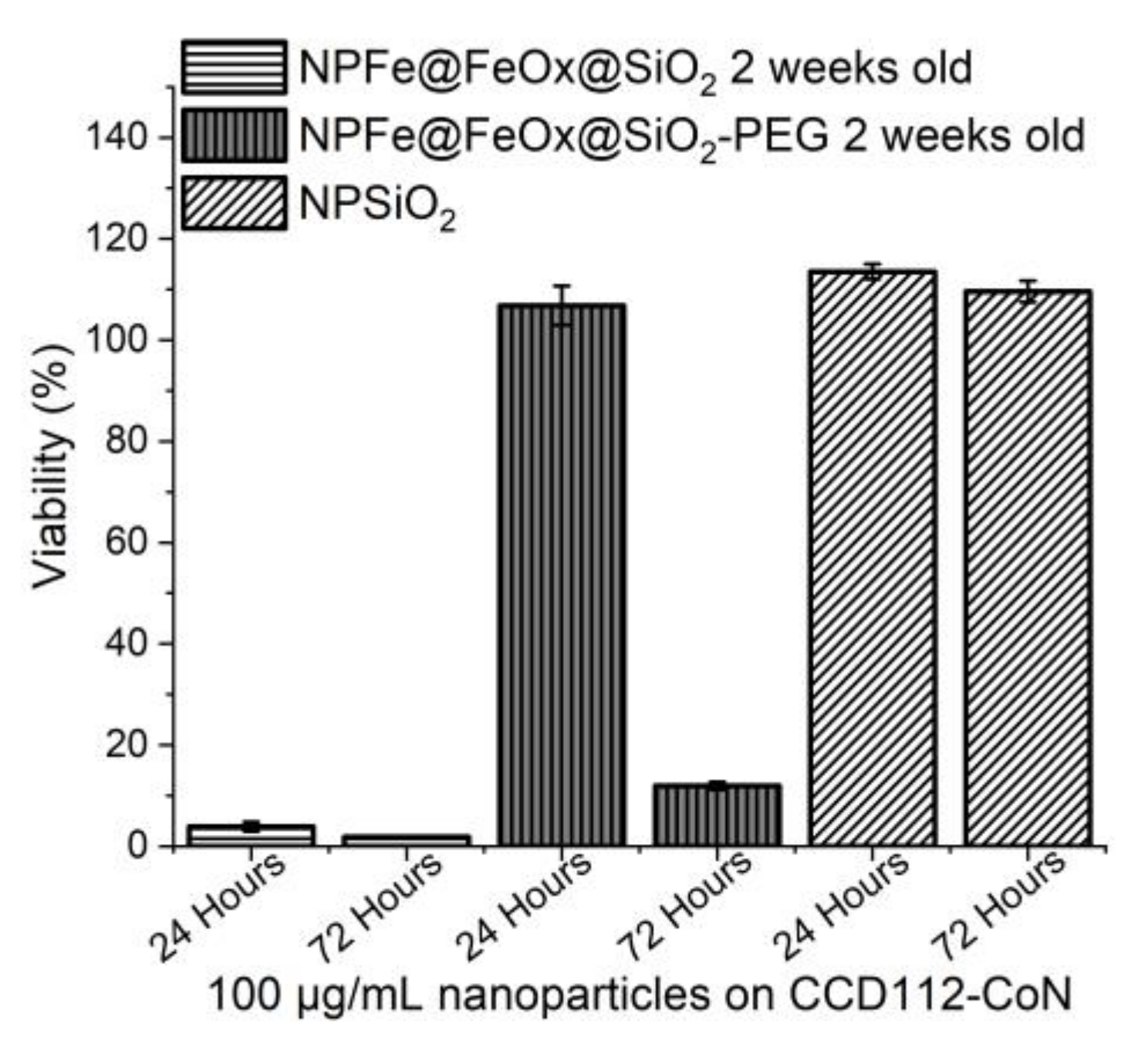
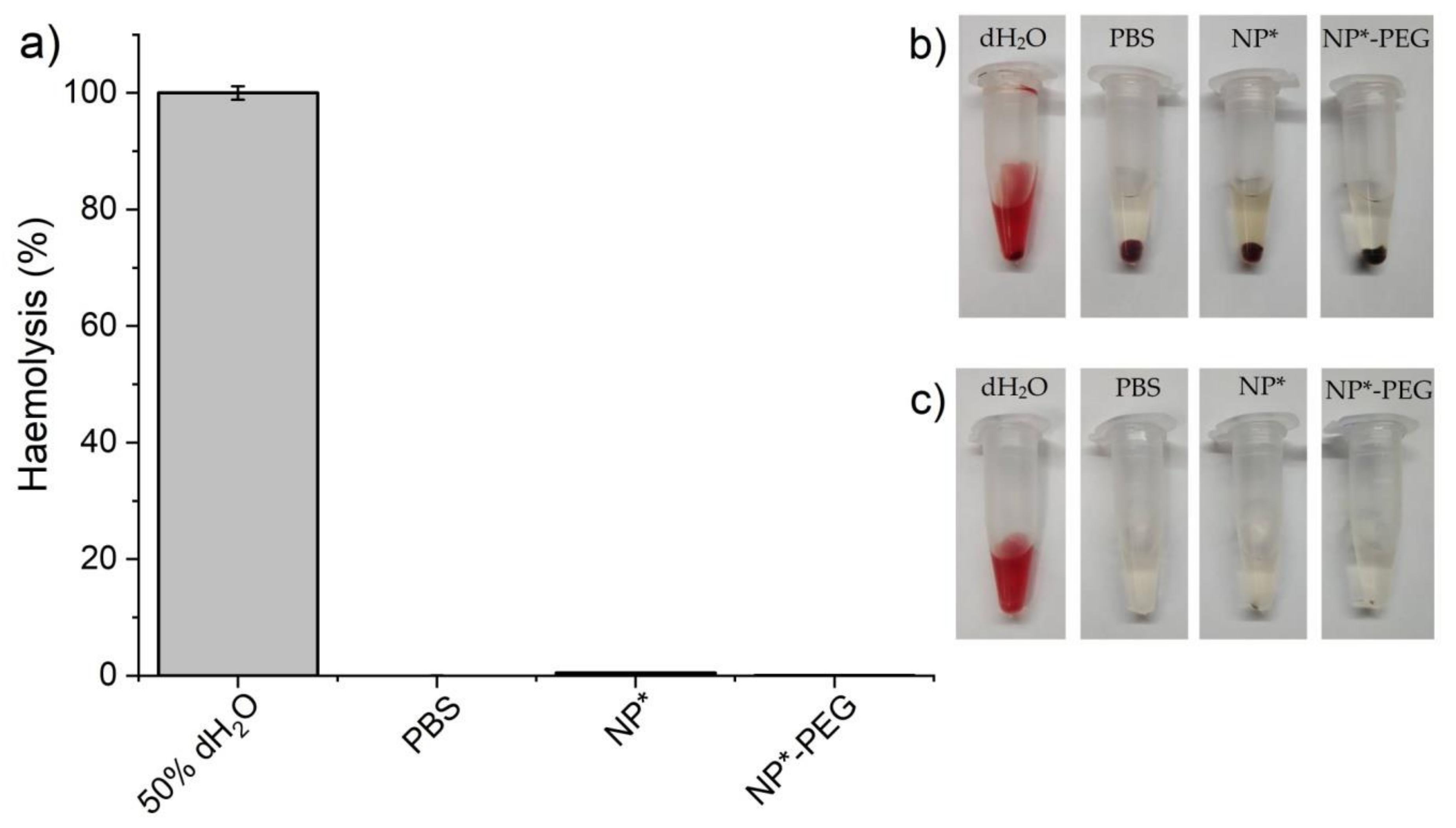
2.5. Cytotoxicity Assessment
3. Materials and Methods
3.1. Synthesis of Iron NPs (NPFe)
3.2. Oleic Acid Treatment of the NPFe
3.3. Coating of the Iron NPs with Silica (NPFe@FeOx@SiO2)
3.4. Synthesis of PEG-Si(OCH2CH3)3
3.5. PEGylation of NPFe@FeOx@SiO2 (NPFe@FeOx@SiO2-PEG)
3.6. Synthesis of Silica NPs (NPSiO2) and PEGylated-NPSiO2 (NPSiO2-PEG)
3.7. Characterizations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and notes
- Laurent, S.; Henoumont, C.; Stanicki, D.; Boutry, S.; Lipani, E.; Belaid, S.; Muller, R.N.; Vander Elst, L. MRI Contrast Agents: From Molecules to Particles; SpringerBriefs in Applied Sciences and Technology; Springer: Singapore, 2017. [Google Scholar]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Bu, W.; Ehlerding, E.B.; Cai, W.; Shi, J. Engineering of inorganic nanoparticles as magnetic resonance imaging contrast agents. Chem. Soc. Rev. 2017, 46, 7438–7468. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wu, A.; Chen, X. Iron Oxide Nanoparticle Based Contrast Agents for Magnetic Resonance Imaging. Mol. Pharm. 2017, 14, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Vallabani, N.V.S.; Singh, S. Recent advances and future prospects of iron oxide nanoparticles in biomedicine and diagnostics. 3 Biotech 2018, 8, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-García, S.; Arias-Ramos, N.; Frias, C.; Candiota, A.P.; Arús, C.; Lorenzo, J.; Ruiz-Molina, D.; Novio, F. Dual T1/T2 Nanoscale Coordination Polymers as Novel Contrast Agents for MRI: A Preclinical Study for Brain Tumor. ACS Appl. Mater. Interfaces 2018, 10, 38819–38832. [Google Scholar] [CrossRef] [PubMed]
- Tse, B.W.-C.; Cowin, G.J.; Soekmadji, C.; Jovanovic, L.; Vasireddy, R.S.; Ling, M.-T.; Khatri, A.; Liu, T.; Thierry, B.; Russell, P.J. PSMA-targeting iron oxide magnetic nanoparticles enhance MRI of preclinical prostate cancer. NanoMed 2015, 10, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Jafari, A.; Salouti, M.; Shayesteh, S.F.; Heidari, Z.; Rajabi, A.B.; Boustani, K.; Nahardani, A. Synthesis and characterization of Bombesin-superparamagnetic iron oxide nanoparticles as a targeted contrast agent for imaging of breast cancer using MRI. Nanotechnology 2015, 26, 075101. [Google Scholar] [CrossRef]
- Hayashi, K.; Nakamura, M.; Sakamoto, W.; Yogo, T.; Miki, H.; Ozaki, S.; Abe, M.; Matsumoto, T.; Ishimura, K. Superparamagnetic Nanoparticle Clusters for Cancer Theranostics Combining Magnetic Resonance Imaging and Hyperthermia Treatment. Theranostics 2013, 3, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Zhang, D.; Zeng, Y.; Wu, L.; Liu, X.; Liu, J. Nanocluster of superparamagnetic iron oxide nanoparticles coated with poly (dopamine) for magnetic field-targeting, highly sensitive MRI and photothermal cancer therapy. Nanotechnology 2015, 26, 115102. [Google Scholar] [CrossRef]
- Revia, R.A.; Zhang, M. Magnetite nanoparticles for cancer diagnosis, treatment, and treatment monitoring: Recent advances. Mater. Today 2016, 19, 157–168. [Google Scholar] [CrossRef]
- Glaria, A.; Soulé, S.; Hallali, N.; Ojo, W.S.; Mirjolet, M.; Fuks, G.; Cornejo, A.; Allouche, J.; Dupin, J.C.; Martinez, H.; et al. Silica coated iron nanoparticles: Synthesis, interface control, magnetic and hyperthermia properties. 2018, 8, 32146–32156. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.M.; Baer, D.R.; Thomas, L.E.; Amonette, J.E.; Antony, J.; Qiang, Y.; Duscher, G. Void formation during early stages of passivation: Initial oxidation of iron nanoparticles at room temperature. J. Appl. Phys. 2005, 98, 094308. [Google Scholar] [CrossRef]
- Miguel, O.B.; Gossuin, Y.; Morales, M.P.; Gillis, P.; Muller, R.N.; Veintemillas-Verdaguer, S. Comparative analysis of the 1H NMR relaxation enhancement produced by iron oxide and core-shell iron–iron oxide nanoparticles. Magn. Reson. Imaging 2007, 25, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Cheong, S.; Ferguson, P.; Feindel, K.W.; Hermans, I.F.; Callaghan, P.T.; Meyer, C.; Slocombe, A.; Su, C.-H.; Cheng, F.-Y.; Yeh, C.-S.; et al. Simple Synthesis and Functionalization of Iron Nanoparticles for Magnetic Resonance Imaging. Angew. Chem. Int. Ed. 2011, 50, 4206–4209. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, L.-M.; Frey Huls, N.; Ho, D.; Sun, X.; Cheng, K.; Sun, S. Stable Single-Crystalline Body Centered Cubic Fe Nanoparticles. Nano Lett. 2011, 11, 1641–1645. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Bonder, M.J.; Balakrishnan, S.; Wang, X.; Mao, H.; Hadjipanayis, G.C. Metallic Iron Nanoparticles for MRI Contrast Enhancement and Local Hyperthermia. Small 2008, 4, 1925–1929. [Google Scholar] [CrossRef]
- Ferguson, P.M.; Feindel, K.W.; Slocombe, A.; MacKay, M.; Wignall, T.; Delahunt, B.; Tilley, R.D.; Hermans, I.F. Strongly Magnetic Iron Nanoparticles Improve the Diagnosis of Small Tumours in the Reticuloendothelial System by Magnetic Resonance Imaging. PLoS ONE 2013, 8, e56572. [Google Scholar] [CrossRef] [Green Version]
- Yoon, T.-J.; Lee, H.; Shao, H.; Weissleder, R. Highly Magnetic Core-Shell Nanoparticles with a Unique Magnetization Mechanism. Angew. Chem. Int. Ed. 2011, 50, 4663–4666. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Sun, Y.; Shen, J.; Wei, J.; Yu, C.; Kong, B.; Liu, W.; Yang, H.; Yang, S.; Wang, W. Iron/iron oxide core/shell nanoparticles for magnetic targeting MRI and near-infrared photothermal therapy. Biomaterials 2014, 35, 7470–7478. [Google Scholar] [CrossRef]
- Masoudi, A.; Madaah Hosseini, H.R.; Seyed Reyhani, S.M.; Shokrgozar, M.A.; Oghabian, M.A.; Ahmadi, R. Long-term investigation on the phase stability, magnetic behavior, toxicity, and MRI characteristics of superparamagnetic Fe/Fe-oxide core/shell nanoparticles. Int. J. Pharm. 2012, 439, 28–40. [Google Scholar] [CrossRef]
- Dumestre, F.; Chaudret, B.; Amiens, C.; Renaud, P.; Fejes, P. Superlattices of Iron Nanocubes Synthesized from Fe[N(SiMe3)2]2. Science 2004, 303, 821–823. [Google Scholar] [CrossRef] [Green Version]
- Margeat, O.; Respaud, M.; Amiens, C.; Lecante, P.; Chaudret, B. Ultrafine metallic Fe nanoparticles: Synthesis, structure and magnetism. Beilstein J. Nanotechnol. 2010, 1, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, L.-M.; Lachaize, S.; Falqui, A.; Blon, T.; Carrey, J.; Respaud, M.; Dumestre, F.; Amiens, C.; Margeat, O.; Chaudret, B.; et al. Ultrasmall iron nanoparticles: Effect of size reduction on anisotropy and magnetization. J. Appl. Phys. 2008, 103, 07D521. [Google Scholar] [CrossRef]
- Lacroix, L.-M.; Lachaize, S.; Falqui, A.; Respaud, M.; Chaudret, B. Iron Nanoparticle Growth in Organic Superstructures. J. Am. Chem. Soc. 2009, 131, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Margeat, O.; Dumestre, F.; Amiens, C.; Chaudret, B.; Lecante, P.; Respaud, M. Synthesis of iron nanoparticles: Size effects, shape control and organisation. Prog. Solid State Chem. 2005, 33, 71–79. [Google Scholar] [CrossRef]
- Meffre, A.; Lachaize, S.; Gatel, C.; Respaud, M.; Chaudret, B. Use of long chain amine as a reducing agent for the synthesis of high quality monodisperse iron(0) nanoparticles. J. Mater. Chem. 2011, 21, 13464–13469. [Google Scholar] [CrossRef]
- Caltagirone, C.; Bettoschi, A.; Garau, A.; Montis, R. Silica-based nanoparticles: A versatile tool for the development of efficient imaging agents. Chem. Soc. Rev. 2015, 44, 4645–4671. [Google Scholar] [CrossRef]
- Chen, F.; Hableel, G.; Zhao, E.R.; Jokerst, J.V. Multifunctional nanomedicine with silica: Role of silica in nanoparticles for theranostic, imaging, and drug monitoring. J. Colloid Interface Sci. 2018, 521, 261–279. [Google Scholar] [CrossRef]
- Ding, H.L.; Zhang, Y.X.; Wang, S.; Xu, J.M.; Xu, S.C.; Li, G.H. Fe3O4@SiO2 Core/Shell Nanoparticles: The Silica Coating Regulations with a Single Core for Different Core Sizes and Shell Thicknesses. Chem. Mater. 2012, 24, 4572–4580. [Google Scholar] [CrossRef]
- Chen, F.; Bu, W.; Chen, Y.; Fan, Y.; He, Q.; Zhu, M.; Liu, X.; Zhou, L.; Zhang, S.; Peng, W.; et al. A Sub-50-nm Monosized Superparamagnetic Fe3O4@SiO2 T2-Weighted MRI Contrast Agent: Highly Reproducible Synthesis of Uniform Single-Loaded Core-Shell Nanostructures. Chem. Asian J. 2009, 4, 1809–1816. [Google Scholar] [CrossRef]
- Glorani, G.; Marin, R.; Canton, P.; Pinto, M.; Conti, G.; Fracasso, G.; Riello, P. Pegylated silica nanoparticles: Cytotoxicity and macrophage uptake. J. Nanoparticle Res. 2017, 19, 294. [Google Scholar] [CrossRef]
- Karakoti, A.S.; Das, S.; Thevuthasan, S.; Seal, S. PEGylated Inorganic Nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 1980–1994. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.A.J.; Ferguson, P.; Cheong, S.; Hermans, I.F.; Ruck, B.J.; Allan, K.M.; Prabakar, S.; Spencer, J.L.; Lendrum, C.D.; Tilley Richard, D. Hot-injection synthesis of iron/iron oxide core/shell nanoparticles for T2 contrast enhancement in magnetic resonance imaging. Chem. Commun. 2011, 47, 9221. [Google Scholar] [CrossRef] [PubMed]
- Schumaker, N.E.; Garland, C.W. Infrared Investigation of Structural and Ordering Changes in Ammonium Chloride and Bromide. J. Chem. Phys. 1970, 53, 392–407. [Google Scholar] [CrossRef]
- Fredrickson, L.R.; Decius, J.C. The Raman spectrum of the ordered phase of NH4Cl and ND4Cl: Dipole and polarizability derivatives. J. Chem. Phys. 1977, 66, 2297–2305. [Google Scholar] [CrossRef]
- Gharbi, K. Elaboration de Nanoparticules d’or et de fer pour des Applications Biomédicales. Ph.D. Thesis, Université Toulouse III Paul Sabatier, Toulouse, France, 2017. [Google Scholar]
- Accordingly, the IR spectrum of the dried ethanol supernatant showed the presence of NH4Cl (3114 cm−1, 3003 cm−1, 2816 cm−1, 1390 cm−1), and aliphatic chains (2916 cm−1 and 2849 cm−1) attributed to residual HAD·HCl (Figure SI 3b).
- Trunova, A.V.; Meckenstock, R.; Barsukov, I.; Hassel, C.; Margeat, O.; Spasova, M.; Lindner, J.; Farle, M. Magnetic characterization of iron nanocubes. J. Appl. Phys. 2008, 104, 093904. [Google Scholar] [CrossRef]
- Branca, M.; Marciello, M.; Ciuculescu-Pradines, D.; Respaud, M.; Morales, M.d.P.; Serra, R.; Casanove, M.-J.; Amiens, C. Towards MRI T2 contrast agents of increased efficiency. J. Magn. Magn. Mater. 2015, 377, 348–353. [Google Scholar] [CrossRef]
- Toney, M.; Davenport, A.; Oblonsky, L.; Ryan, M.; Vitus, C. Atomic Structure of the Passive Oxide Film Formed on Iron. Phys. Rev. Lett. 1997, 79, 4282–4285. [Google Scholar] [CrossRef]
- Wang, C.; Baer, D.R.; Amonette, J.E.; Engelhard, M.H.; Antony, J.; Qiang, Y. Morphology and Electronic Structure of the Oxide Shell on the Surface of Iron Nanoparticles. J. Am. Chem. Soc. 2009, 131, 8824–8832. [Google Scholar] [CrossRef]
- Signorini, L.; Pasquini, L.; Savini, L.; Carboni, R.; Boscherini, F.; Bonetti, E.; Giglia, A.; Pedio, M.; Mahne, N.; Nannarone, S. Size-dependent oxidation in iron/iron oxide core-shell nanoparticles. Phys. Rev. B 2003, 68. [Google Scholar] [CrossRef]
- Liz-Marzán, L.M.; Giersig, M.; Mulvaney, P. Synthesis of Nanosized Gold−Silica Core−Shell Particles. Langmuir 1996, 12, 4329–4335. [Google Scholar] [CrossRef]
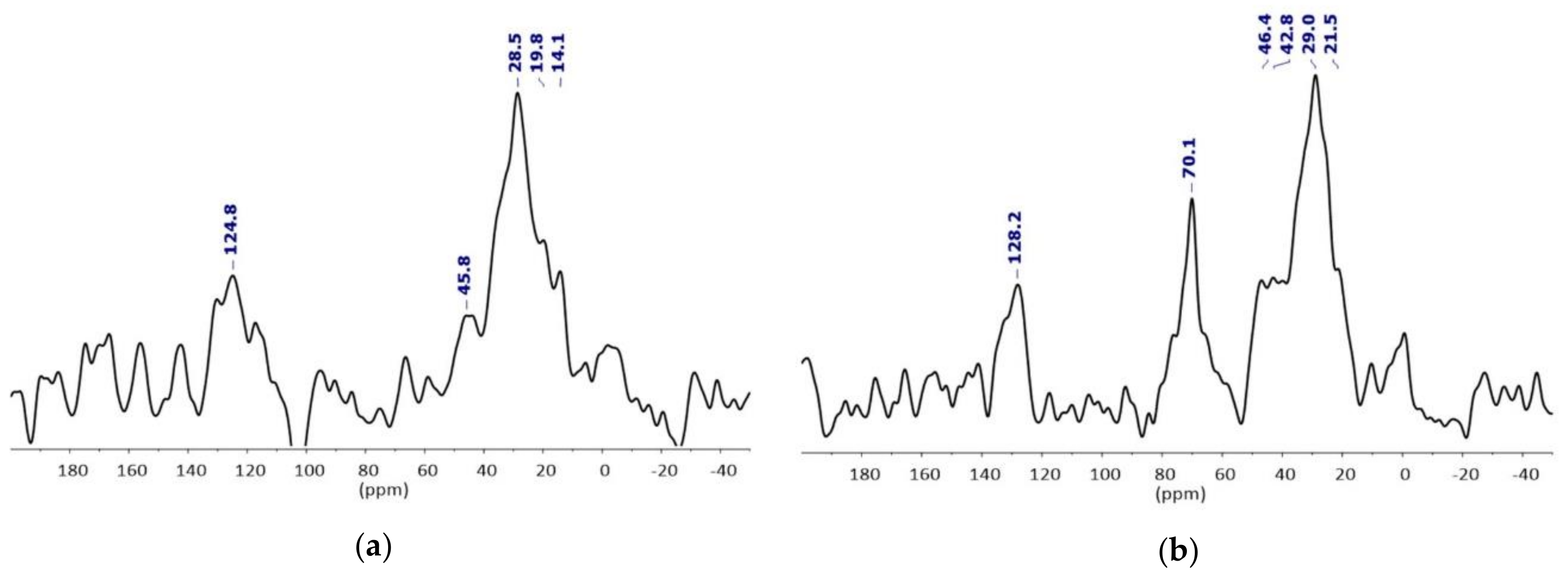
- EDX study confirmed the silica shell / iron rich core architecture (Figure S6). We assume that TEOS is not fully condensed at this stage, based on 29Si MAS NMR studies carried out on NPSiO2 taken as a reference (see supplementary information for details).
- Joshi, H.M.; De, M.; Richter, F.; He, J.; Prasad, P.V.; Dravid, V.P. Effect of silica shell thickness of Fe3O4 –SiOx core–shell nanostructures on MRI contrast. J. Nanoparticle Res. 2013, 15, 1448. [Google Scholar] [CrossRef]
- Perry, J.L.; Reuter, K.G.; Kai, M.P.; Herlihy, K.P.; Jones, S.W.; Luft, J.C.; Napier, M.; Bear, J.E.; DeSimone, J.M. PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett. 2012, 12, 5304–5310. [Google Scholar] [CrossRef]
- Lee, N.; Hyeon, T. Designed synthesis of uniformly sized iron oxide nanoparticles for efficient magnetic resonance imaging contrast agents. Chem. Soc. Rev. 2012, 41, 2575–2589. [Google Scholar] [CrossRef]
- Koenig, S.H.; Kellar, K.E. Theory of 1/T1 and 1/T2 NMRD profiles of solutions of magnetic nanoparticles. Magn. Reson. Med. 1995, 34, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.L.C.; Laurent, S.; Rocha, J.; Roch, A.; Delville, M.-H.; Mornet, S.; Carlos, L.D.; Vander Elst, L.; Muller, R.N.; Geraldes, C.F.G.C. Relaxometric Studies of γ-Fe2O3@SiO2 Core Shell Nanoparticles: When the Coating Matters. J. Phys. Chem. C 2012, 116, 2285–2291. [Google Scholar] [CrossRef]
- Khurshid, H.; Hadjipanayis, C.G.; Chen, H.; Li, W.; Mao, H.; Machaidze, R.; Tzitzios, V.; Hadjipanayis, G.C. Core/shell structured iron/iron-oxide nanoparticles as excellent MRI contrast enhancement agents. J. Magn. Magn. Mater. 2013, 331, 17–20. [Google Scholar] [CrossRef]
- Harisinghani, M.G.; Barentsz, J.; Hahn, P.F.; Deserno, W.M.; Tabatabaei, S.; van de Kaa, C.H.; de la Rosette, J.; Weissleder, R. Noninvasive Detection of Clinically Occult Lymph-Node Metastases in Prostate Cancer. N. Engl. J. Med. 2003, 348, 2491–2499. [Google Scholar] [CrossRef] [Green Version]
- McGrath, A.J.; Dolan, C.; Cheong, S.; Herman, D.A.J.; Naysmith, B.; Zong, F.; Galvosas, P.; Farrand, K.J.; Hermans, I.F.; Brimble, M.; et al. Stability of polyelectrolyte-coated iron nanoparticles for T2-weighted magnetic resonance imaging. J. Magn. Magn. Mater. 2017, 439, 251–258. [Google Scholar] [CrossRef]
- He, Q.; Zhang, J.; Shi, J.; Zhu, Z.; Zhang, L.; Bu, W.; Guo, L.; Chen, Y. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials 2010, 31, 1085–1092. [Google Scholar] [CrossRef]
- Roch, A.; Muller, R.N.; Gillis, P. Theory of proton relaxation induced by superparamagnetic particles. J. Chem. Phys. 1999, 110, 5403–5411. [Google Scholar] [CrossRef]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic Iron Oxide Nanoparticles: Synthesis, Stabilization, Vectorization, Physicochemical Characterizations, and Biological Applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; McCanna, D.J.; Sivak, J.G. Use of the viability reagent PrestoBlue in comparison with alamarBlue and MTT to assess the viability of human corneal epithelial cells. J. Pharmacol. Toxicol. Methods 2015, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Chen, C. Fate and Toxicity of Metallic and Metal-Containing Nanoparticles for Biomedical Applications. Small 2011, 7, 2965–2980. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | NPFe@FeOx@SiO2 | NPFe@FeOx@SiO2-PEG | ||
|---|---|---|---|---|
| Aging | 2 weeks | 2 months | 2 weeks | 2 months |
| Z average | 99 nm | 172 nm | 111 nm | 202 nm |
| PdI | ±16% | ±30% | ±20% | ±21% |
| NPFe@FeOx@SiO2 | NPFe@FeOx@SiO2-PEG | |
|---|---|---|
| Ms (A.m². kgMM) | 23.20 | 20.20 |
| Radius (nm) | 13.7 | 13.5 |
| D (cm². s−1)* | 3.01 e-5 | 3.01 e−5 |
| τNéel (ns) | 40.1 | 24.3 |
| Frequency. | Relaxivity (mM−1.s−1) | NPs* 2 weeks Old | NPs*-PEG 2 weeks Old | NPs* 2 months Old | NPs*-PEG 2 months Old | RESOVIST® Experimental | DOTAREM® Experimental |
|---|---|---|---|---|---|---|---|
| 20 MHz | r1 | 9.5 ± 0.5 | 9.8 ± 0.5 | 8.9 ± 0.4 | 3.6 ± 0.2 | 25.0 ± 1.3 | 4.1 ± 0.2 |
| r2 | 165.5 ± 8.3 | 186.5 ± 9.3 | 163.1 ± 8.2 | 181.7 ± 9.1 | 161.8 ± 8.1 | 4.9 ± 0.2 | |
| r2/r1 | 17.5 | 19 | 18.3 | 50 | 6.5 | 1.2 | |
| 60 MHz | r1 | 3.1 ± 0.2 | 2.5 ± 0.1 | 3.5 ± 0.2 | 1.30 ± 0.07 | 11.0 ± 0.6 | 3.7 ± 0.2 |
| r2 | 166.0 ± 8.3 | 188.0 ± 9.4 | 175.0 ± 8.8 | 185.1 ± 9.3 | 157.8 ± 7.9 | 4.0 ± 0.2 | |
| r2/r1 | 55.3 | 75.2 | 50 | 142 | 14.3 | 1.08 | |
| 400 MHz | r2 | 208.0 ± 10.4/237.0 ± 11.9 | 254.0 ± 12.7 | 224.0 ±11.2 | 239.0 ± 12.0 | 232.0 ± 11.6 | 4.7 ± 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathieu, P.; Coppel, Y.; Respaud, M.; Nguyen, Q.T.; Boutry, S.; Laurent, S.; Stanicki, D.; Henoumont, C.; Novio, F.; Lorenzo, J.; et al. Silica Coated Iron/Iron Oxide Nanoparticles as a Nano-Platform for T2 Weighted Magnetic Resonance Imaging. Molecules 2019, 24, 4629. https://doi.org/10.3390/molecules24244629
Mathieu P, Coppel Y, Respaud M, Nguyen QT, Boutry S, Laurent S, Stanicki D, Henoumont C, Novio F, Lorenzo J, et al. Silica Coated Iron/Iron Oxide Nanoparticles as a Nano-Platform for T2 Weighted Magnetic Resonance Imaging. Molecules. 2019; 24(24):4629. https://doi.org/10.3390/molecules24244629
Chicago/Turabian StyleMathieu, Paul, Yannick Coppel, Marc Respaud, Quyen T. Nguyen, Sébastien Boutry, Sophie Laurent, Dimitri Stanicki, Céline Henoumont, Fernando Novio, Julia Lorenzo, and et al. 2019. "Silica Coated Iron/Iron Oxide Nanoparticles as a Nano-Platform for T2 Weighted Magnetic Resonance Imaging" Molecules 24, no. 24: 4629. https://doi.org/10.3390/molecules24244629
APA StyleMathieu, P., Coppel, Y., Respaud, M., Nguyen, Q. T., Boutry, S., Laurent, S., Stanicki, D., Henoumont, C., Novio, F., Lorenzo, J., Montpeyó, D., & Amiens, C. (2019). Silica Coated Iron/Iron Oxide Nanoparticles as a Nano-Platform for T2 Weighted Magnetic Resonance Imaging. Molecules, 24(24), 4629. https://doi.org/10.3390/molecules24244629