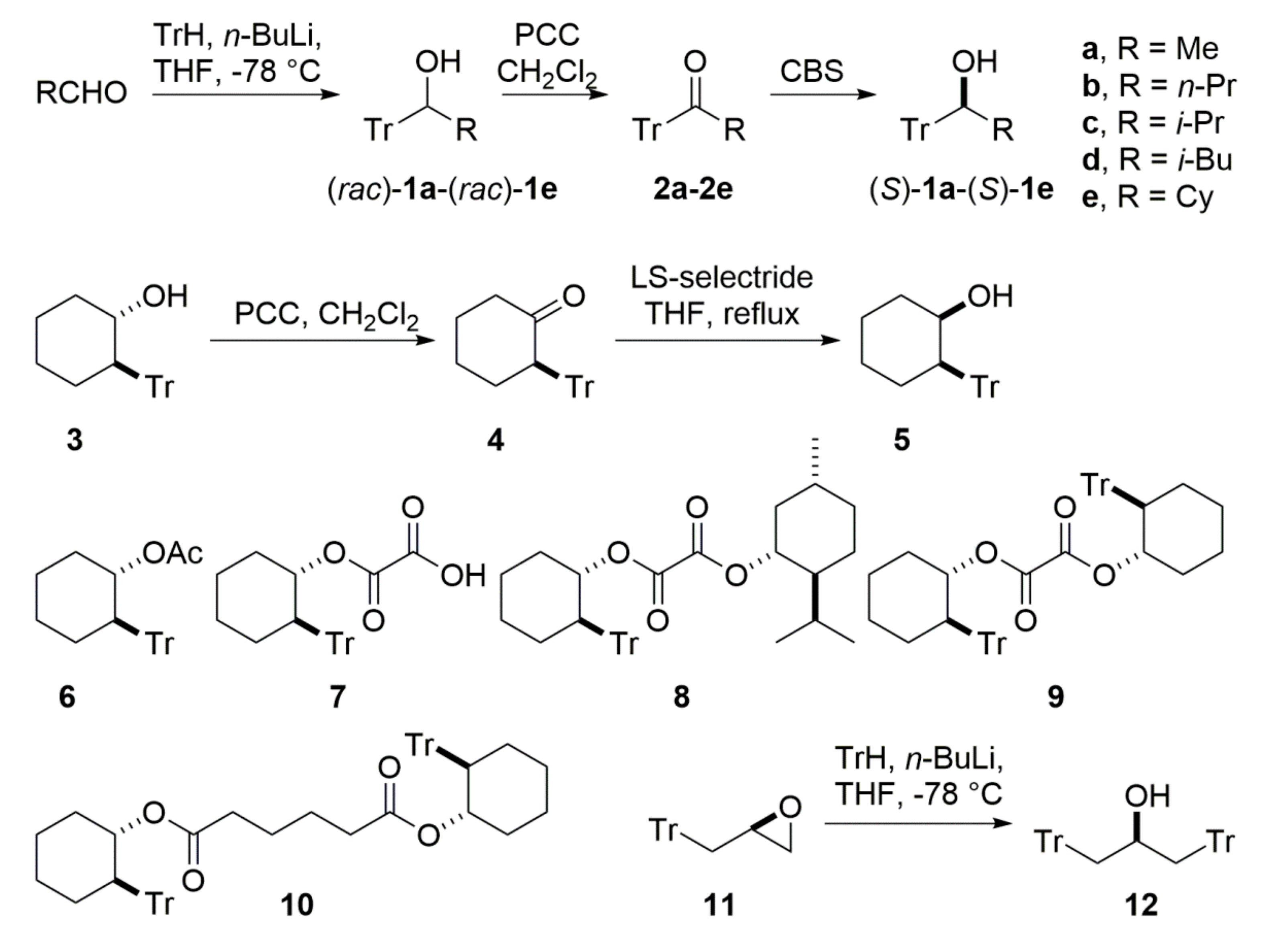
2.1. Synthesis
In these studies, we have focused on derivatives
1–
12 (
Scheme 1) which, in principle, may be conveniently obtained by addition of a carbon nucleophile to a suitable electrophile and then further functionalized. However, the initially tested stereoselective addition of aliphatic
C-nucleophiles to the triphenylacetaldehyde did not yield alcohols
1a–
1e. Thus, we have taken advantage of a facile generation and stability of trityl anion obtained through a direct reaction between triphenylmethane and
n-buthyllithium. We have selected compounds
1,
3,
11, and
12 as the basic structures, which might be obtained through the addition/substitution of the trityl anion to the respective electrophile (see
Scheme 1).
With the exception of
11, the applied synthetic method did not allow us to obtain the desired compounds in the optically pure form. In the case of
1a–
1e, we were forced to extend the synthetic procedure to the step of oxidation of racemic alcohols and asymmetric Corey–Bakshi–Shibata (CBS) reduction of the resulting ketones
2a–
2e, which, in turn, provided the enantiomerically enriched
1a–
1e [
26,
27,
28]. The obtained enantiomeric excesses of
1a–
1e did not exceed 85%, but were sufficient to allow us to proceed with the study.
The racemic
trans-2-tritylcyclohexanol (
rac–
3) was obtained from cyclohexene oxide through opening the epoxide ring with a trityl anion. The optically active
3, in both enantiomeric forms, has been obtained from the racemate via oxalate
8 and by employing the procedure previously described by Hazmi et al [
29]. The reactions provided both diasteroisomers of
8, differing in the absolute configurations of the
trans-2-tritylcyclohexanol moiety, denoted here as
8a ((1
S,2
R)) and
8b ((1
R,2
S)), respectively.
Oxidation of racemic and optically enriched (1S,2R)–3 by PCC led to the ketone 4, respectively, as the racemate and as the optically active form ((R)-5, ee = 80%). The ketone rac- or (R)-5 may be further diastereoselectively reduced to the cis-2-tritylcyclohexanol (rac-6 or (1R,2R)-6, if the enantiomerically enriched 5 was used), with the use of LS-Selectride. To complete the reduction reaction, a prolongated heating was required, and (1R,2R)-5 was obtained with moderate yield (60%) and enantioselectivity (ee = 49%).
To enhance the steric bulkiness at the neighboring stereogenic center, and to show the influence of an external chiral unit on the possible chirality transfer, the optically pure 3 was acylated with various acid chlorides. While simple acetylation of 3 with acetyl chloride proceeded smoothly and gave acetate 6, the initial attempt at synthesis of the diester 9, which consists of two trans-2-tritylcyclohexanol units linked by the oxalyl acid moiety, failed. As the only product, we have obtained the monoester 7, having one free carboxyl group. It is worth noting that the ESI-MS mass spectrum of 7 does not show peaks that might be ascribed to structure of isolated 7. Instead, we observed a sodiated dimeric structure of the general formula [2 × 7 + Na]+. Surprisingly, the replacement of methylene chloride by freshly distilled THF and by performing the reaction at elevated temperature within 20 h led to the diester 9 with a yield of 41% after column chromatography. Despite the fact that the starting 3 was not optically pure (ee = 45%), the reaction was stereoselective and provided only chiral products. For the purpose of this study, we obtained both enantiomers 9 and ent-9. For no obvious reason, ent-9, which consists of two units characterized by the 1R,2S absolute configuration of each trans-2-tritylcyclohexanol moiety, crystallized better than 9 and provided crystals of a sufficient quality for X-ray structure determination.
The oxirane
11 was obtained according the procedure published by Zhang and coworkers [
30]. In principle, oxirane
11 was to be the starting point for the further synthesis of an achiral stereodynamic reporter system consisting of two trityl fragments and an anchoring group enabling covalent or non-covalent binding of an inducer molecule. However, while the opening of the oxirane by the trityl anion led to an achiral
meso-
12 with good yield, the further modification of the hydroxyl group turned out not to be possible, probably due to the steric congestion around the OH group.
2.2. Structure and Induced Circular Dichroism of 1, 3–11
The complementary use of experimental CD measurements and DFT calculations allows a study of the structure/chiroptical properties and mechanism of chirality transmission within the selected compounds. To achieve this goal, ECD spectra measurements are required in order to compare them to the results of DFT calculations, carried out at a sufficiently high level of theory (calculation details can be found in
Supplementary Information). In this study, we have decided to discuss experimental and theoretical results together to preserve the logical stream.
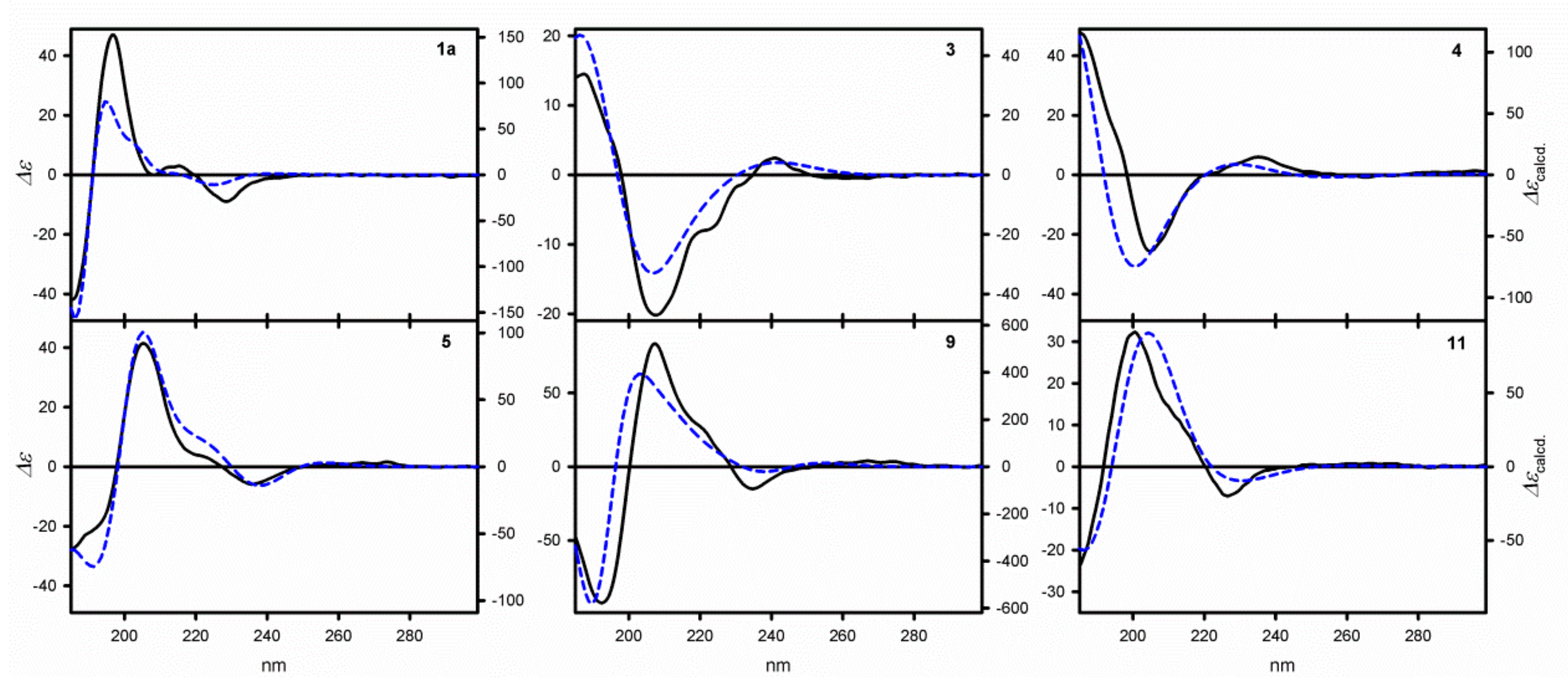
The ECD data for
1–
11 are juxtaposed in
Table 1, whereas the exemplary ECD spectra measured in non-polar cyclohexane for
1a,
3–
5,
9, and
11 are shown in
Figure 1. The structure of the compounds under study can be described by providing the sets of torsion angles strictly determining the structure of the particular species. The angle
α (X-C(=O)-C(H
2)-C) characterizes the conformation of the carbon chain. The angles
β1 –
β3 ((O=C)-C(H
2)-C-C
ipso) describes the conformation of the phenyl groups in the relation to the inductor carbon chain. The sign, and to the same extent, the magnitude of observed CE’s depends on the helicity of the trityl group. The angles
γ1 –
γ3 (C-C-C
ipso-C
ortho) values may be used quantitatively for a precise description of the trityl helicity. However, for the needs of this work, it seems more convenient to use a qualitive descriptor, namely helicity of the blades:
M (−90° <
γ < 0°),
P (0° <
γ < 90°) or
0 (for
γ angles deviating from zero by less than |5°|). Since, for the majority of the cases, the chiroptical properties are dominated by the most abundant conformer, we have limited our structural discussion to the
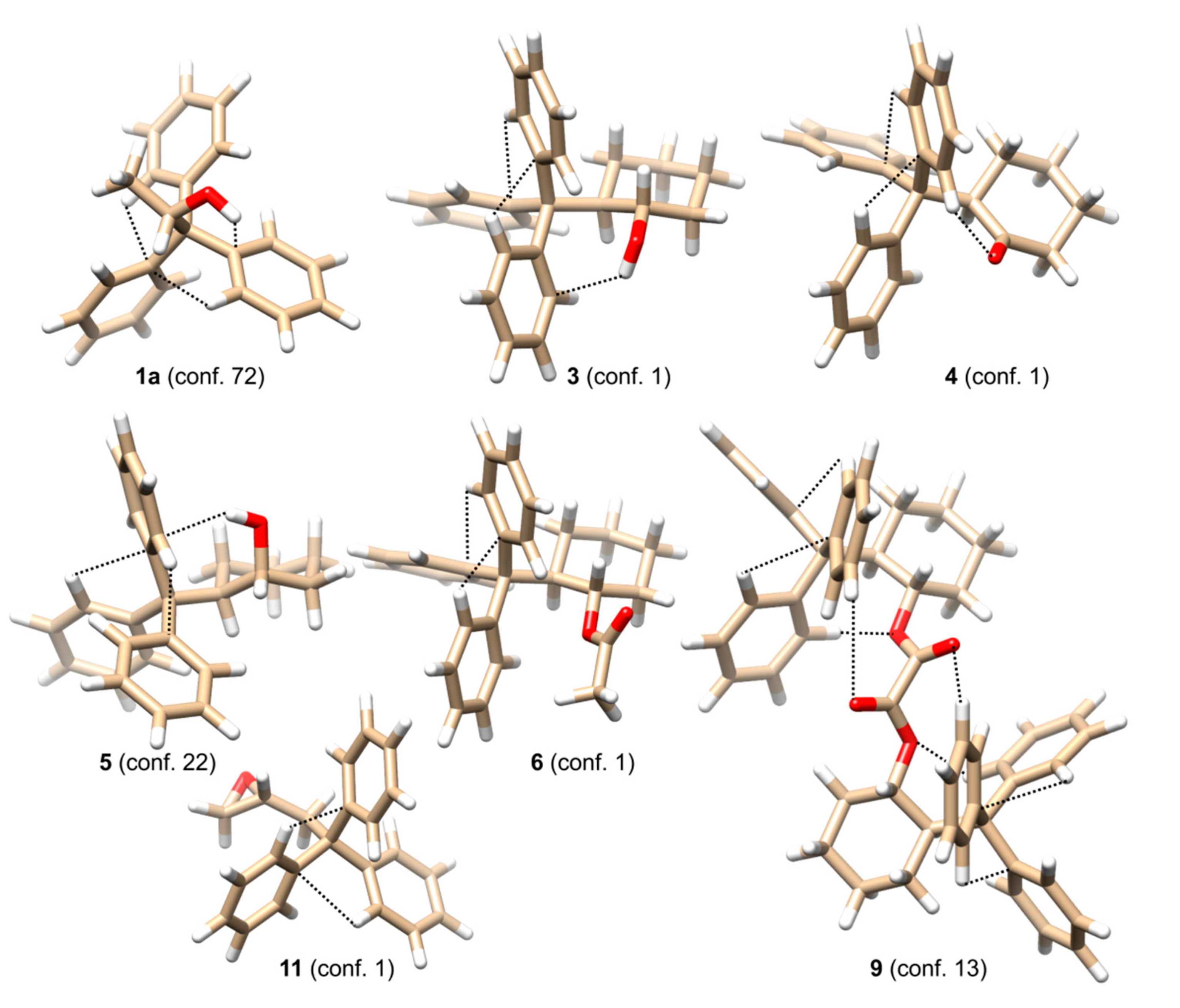
ΔΔG-based the lowest energy species only. The structures of the lowest energy conformers of selected compounds
1a,
3–
6,
9, and
11 are shown in
Figure 2, whereas
Table 2 collects the essential information extracted from the theoretical results (the remaining data can be found in
Supplementary Information).
The data listed in
Table 1 clearly indicates large chiroptical response of the trityl chromophore in the presence of chirality element in its neighborhood. The measured ECD spectra are characterized by the presence of at least three well-defined absorption bands typical of trityl chromophore. The low-energy absorption band appearing at around 220 nm, is preceded by two bands with much higher intensity, which appear around 200 and 185 nm. In the case of the monoester
7, the presence of an oxalyl acid fragment makes the ECD spectrum more complex.
Generally, the –COOR ester group is recognized as the weak UV active chromphore of the π-π* transition separated from the strong
1B band (195–210 nm) in phenyl rings. However, in the case of
7, we are dealing not only with the presence of two different directly connected groups (COOH and COOR), but also with the possibility to form various associates [
17].
As expected, the direct connection between the chirality element and the stereodynamic part of the molecule for
1a–
1e leads to the CEs of the highest intensity within the whole series. In tritylcarbinols
1a–
1e, the amplitudes (
A) that are estimated for the higher-energy CEs reach values between 94 to 195. In fact, these
A values are the highest for all the monotrityl derivatives studied so far. The signal patterns observed in all the spectra are identical: Negative/positive/negative (−/+/−), which suggests the same absolute configuration at the stereogenic centers. To confirm this hypothesis, for all of the alcohols
1a–
1e, DFT calculations (B3LYP/6-311++G(d,p)) were carried out [
31]. Note that the choice of a calculation method was based on our previous studies. We have previously shown that the use of hybrid functionals, B3LYP for geometry and CAM-B3LYP for excited states calculations and for compounds containing trityl groups, gives results closest to the experimental ones [
17,
18]. Hence, we have decided to use these functionals in conjunction with the enhanced 6-311++G(d,p) basis set (or 6-311G(d,p) for larger systems) for calculations. The empirical correction for dispersion interactions does not improve the quality of the results obtained. In other words, the trityl–trityl interactions are usually overestimated, which ultimately leads to large discrepancies between the experiment and calculations [
20].
The use of B3LYP hybrid functional is not recommended for excited state calculations, which is in accordance with our observations [
17,
18,
20,
32]. We received comparable results (let us say—of the same quality) to those obtained with the use of CAM-B3LYP functional, when the Thrular’s M06–2X hybrid functional was employed. In this work, only the results obtained with the use of TD-CAM-B3LYP hybrid functional were discussed.
In the simplest case of 1a, only two thermally accessible conformers have been found. However, the number of thermally accessible conformers increases in alcohols with longer or more flexible alkyl backbones. Despite the structure of the alkyl substituent flanking the stereogenic center, two general factors can be distinguished as responsible for inducing helicity and, thus, the optical activity in the trityl chromophore. The first of them are attractive interactions between an oxygen atom from the hydroxyl group and a hydrogen atom in ortho position from one of the phenyl rings. The proton from the hydroxyl group is directed towards the plane of the second phenyl group. The O-H∙∙∙π interactions together with C-H∙∙∙π interactions stabilize the conformation of this particular phenyl ring. The third phenyl ring adjusts its conformation to the conformation of the remaining two.
The −/+/− signal pattern of CEs, observed for 1a–1e, are due to the predominance of PPP or PP0 helical conformers in conformational equilibria. This experimental/theoretical analysis provides an additional reward—the absolute configuration, determined for 1a–1e, at the stereogenic centers is uniformly S for all of these compounds.
(1S,2R)-trans-2-Tritylcyclohexanol (3), its diastereoisomer (1R,2R)-cis-2-Tritylcyclohexanol (5), and intermediate ketone (R)-2-tritylcyclohexanone (4) constitute interesting examples showing the influence of a neighboring group (hydroxyl group or carbonyl oxygen atom) on trityl conformation. The lowest-energy conformations of 3 and 4 are stabilized mostly by Cortho-H∙∙∙O or O-H∙∙∙π attractive interactions and to a lesser extent by Cortho-H∙∙∙π and steric interactions between axial proton(s) attached to C1 and C3 carbon atoms in cyclohexane ring. The importance of Cortho-H∙∙∙O interactions is particularly seen for the lowest-energy conformer of 4. The calculated (Cortho-)H∙∙∙O(=C) distance is 2.210 Å. The case of 5 is different. The axial position of hydroxyl group prevents direct Cortho-H∙∙∙O interactions. Therefore, the primordial factor that determines the conformation of 5 seems to be O-H∙∙∙π interactions.
As mentioned previously, the process of optical induction in the trityl chromophore is cascading. The electrostatic Cortho-H∙∙∙O (if applicable) interactions constitute the primary factor that fixes the conformation of the phenyl rings that faces the axial protons at C1 and C3 and enforces a (+)-synclinal ((+)-sc) conformation of associated angle β. Then a second ring is positioned in such a way as to allow O-H∙∙∙π interactions with the hydroxyl proton, and Cortho-H∙∙∙π interactions with the first phenyl ring. Finally, the third ring adjusts its conformation to the first two in a way to maximize Cortho-H∙∙∙π interactions. The opposite absolute configuration reached by the change in the position of hydroxyl group from equatorial to axial is reflected in the change of CE sequence. For 3 and 4, the CE sequence which appears in the region of trityl absorption is +/−/+, whereas for 5 it is reversed (−/+/−). These sequences correspond to the preferred helicities of the trityl group, namely MPM and PPM for the most abundant conformers of 3 and 5, respectively. It is worth mentioning that the presence of the trityl, which acts as a protective umbrella, is responsible for the non-reactivity of the axial hydroxyl group, particularly in reactions requiring OH activation prior nucleophilic substitution.
Having compared the ECD spectra of 3 and 6, we may conclude that the acetylation of the equatorial hydroxyl group does not have any significant effect on the amplitude and the sequence of the respective CEs. In both cases, the same +/−/+ sequence is observed. However, the mechanism of optical activity induction, deducted from our theoretical results, is slightly different from that established for the free alcohol. In the case of 6, the Cortho-H∙∙∙O attractive interactions lose their importance in favor of sterical interactions between the acetyl group and the adjacent phenyl ring of trityl.
Due to the unrestricted rotation around the C-C bond in the linker, the adipinic acid derivative 10 is characterized by a great conformational flexibility. While the shape of the ECD spectrum of 10 resembles that of acetate 6 and of the free alcohol 3, the magnitudes of its CE doubles. This is not surprising, given that the observed effects are a linear combination of contributions from individual (non-interacting) trityl chromophores.
Compounds 7–9 represent systems whose common denominator is the presence of an oxalyl moiety. Despite structural differences, these compounds display the same −/+/− sequence of CE observed in the region of the trityl UV absorption. This is apparently due to the dominant influence of conformers of PPM helicity of the trityl group(s) on the overall ECD spectra.
Among oxalates 7–9, the most interesting one is represented by the symmetrical bis((1S,2R)-2-tritylcyclohexyl) oxalate (9). Conformation of the C2-symmetrical, lowest-energy conformer of 9 is stabilized by a set of weak Cortho-H∙∙∙O interactions that involve all C=O and C-O oxygen atoms present in the linker. Moreover, the Cortho-H∙∙∙O=C interactions involve the phenyl group characterized by the (+)-sc conformation of the β angle and a more distant oxygen atom, while Cortho-H∙∙∙O-C interactions take place between the phenyl group of (−)-sc conformation for the β angle and with a closer oxygen atom. The third phenyl group of (+)-antiperiplanar ((+)-ap) conformation for the β angle adjusts its position in a way to minimize steric repulsion while allowing some Cortho-H∙∙∙π interactions.
The C
ortho-H∙∙∙O interactions not only stabilize the conformation of the trityl groups, but also cause a bending of the oxalate linker. In the lowest-energy conformer of
9, both COOR fragments are arranged almost perpendicularly (
ϕ = 91.4°, where
ϕ is the O=C-C=O angle), whereas the higher energy conformer number 26 retains the (−)-
ap orientation (
ϕ = −162.9°). The non-planarity of the
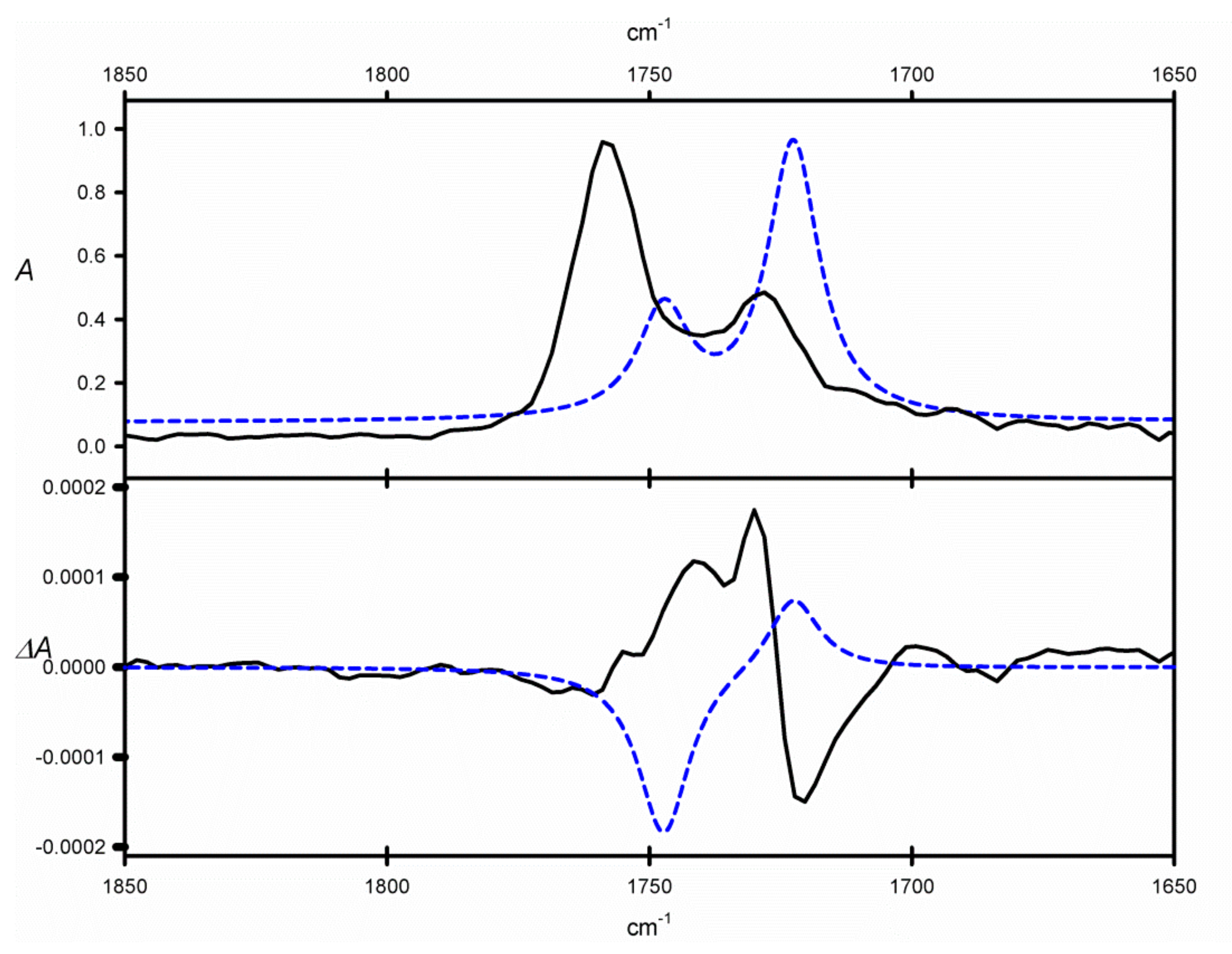
ϕ angle gives a chance for observation of non-zero CE in the region of C=O stretches in the VCD spectrum of
9. In fact, the VCD spectrum of
9 measured in CCl
4, shows a bisignate couplet in the region of C=O absorption (1700–1800 cm
−1, see
Figure 3) that may be ascribed to through-space interactions of two C=O chromophores. However, the negative sign of this couplet remains inconsistent with the twist of the neighboring vicinal C=O bonds. Similarly, the DFT calculations gave results that are opposite to the experimental ones, regardless of the calculation method used. A possible explanation for this discrepancy could follow from the underestimation of structures characterized by (+)-
sp or (+)-
synclinal ((+)-
sc) conformations of the O=C-C=O angle. Despite these disappointing results, this particular compound is an interesting example of shortcomings in the experimental/theoretical approach to structural studies with the use of VCD spectroscopy. Further studies on this and related compounds will be undertaken in the near future.
The oxirane 11 represents a somewhat different system, in which the trityl group is connected indirectly to the stereogenic center. In this particular case, the reporter part of the molecule should be sensitive enough to distinguish distant substituents of the stereogenic center. Among the thermally accessible conformers, two of them, number 1 and number 73, dominate in conformational equilibrium and are characterized by PP0 and PPP helicities of the trityl fragment, respectively. The ECD spectra, calculated for these conformers, exhibit the same −/+/− sequence of CE, in perfect agreement with the experimental ECD spectrum. The structure of the trityl moiety in low-energy conformers of 11, is stabilized mostly by Cortho-H∙∙∙π interactions and to a lesser extent by electrostatic repulsion between the oxygen atom and the π-electron cloud of the phenyl ring facing the oxirane moiety.
2.3. X-ray Diffraction Study
Taking advantage of the ability of some compounds to form crystal phases, we have extended our work to the solid phases of the trityl-containing molecules. In particular, we were interested in the possibilities to form inclusion compounds, which in turn may lead to enantiodiscrimination. The crystal structure of compounds
rac-
3,
rac-
4, (
R)-
4,
rac-
5,
rac-
7,
8,
9,
11, and
12 has been determined and studied by means of the X-ray diffraction method (detailed information is included in
Supplementary Information). The derivatives obtained, with the exception of compound
rac-
7, crystallize without solvent. Some structural gaps are observed in the crystals of the
rac-
4, (
R)-
4, and
9, however the accessible volume is too small to include the solvent molecules. Nevertheless, in crystals of
rac-
3,
rac-
5,
8,
11, and
12, the molecules strictly fill the space, and despite the tests carried out, inclusion crystals containing at least solvent molecules have not been obtained.
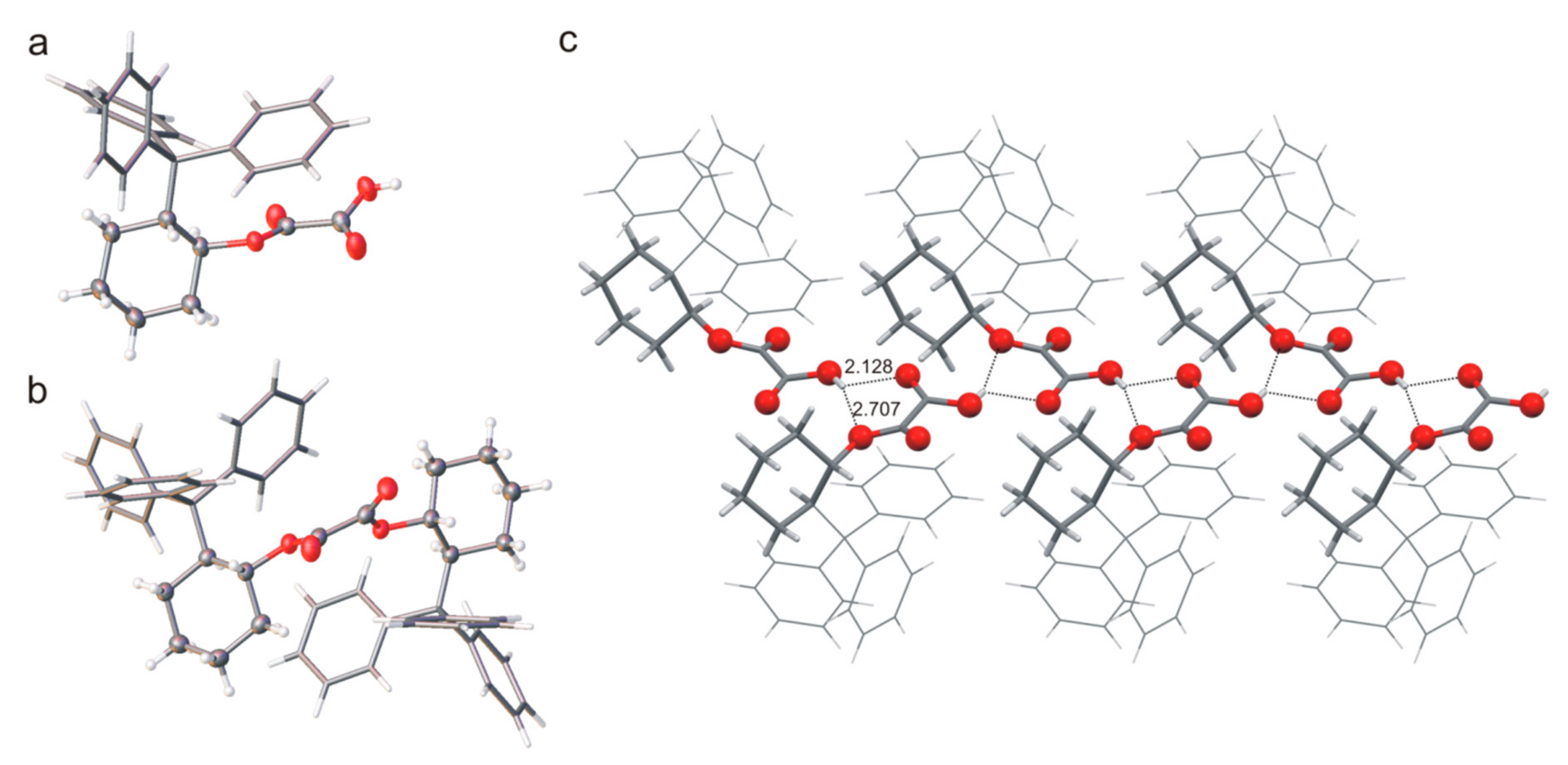
Interestingly, the two types of crystals of compound
rac-
3 (orthorhombic (
rac-
3-
α) and monoclinic (
rac-
3-
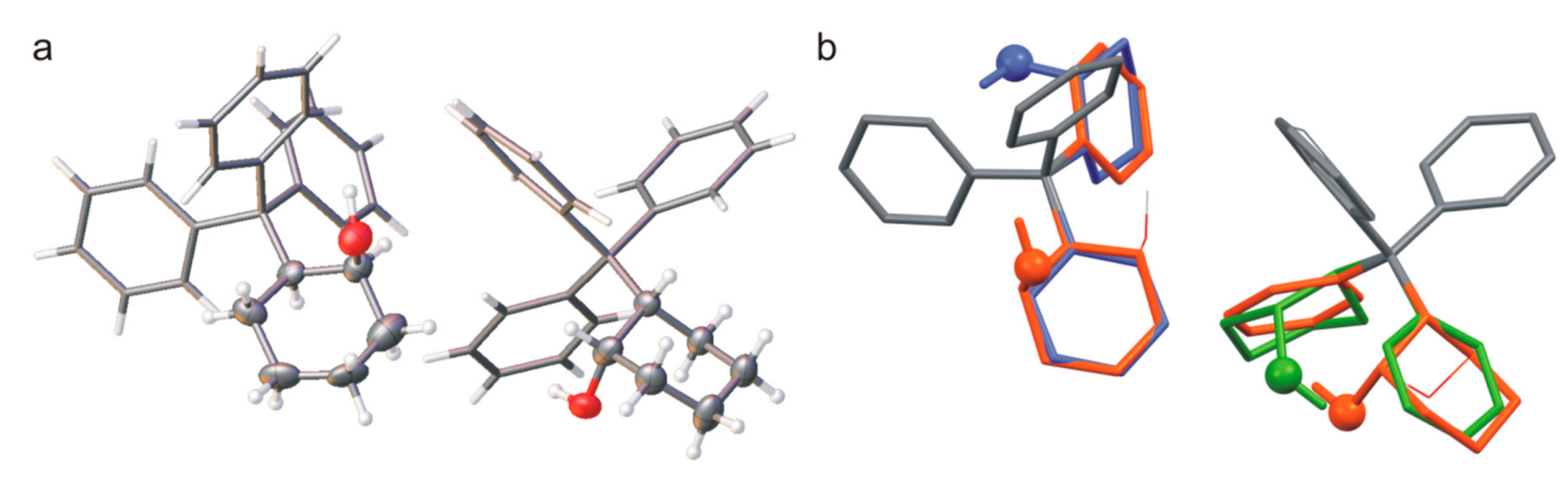
β) were obtained in one crystallization, even though the structure of the molecule is the same. Furthermore, the structural analysis showed that the polymorph
rac-3-
β is a solid solution of enantiomers (
Figure 4b), and the ratio of refined occupancy factors for oxygen atoms is 0.71: 0.29. In both orthorhombic and monoclinic crystals, the basic structural unit is a pair of (1
S,2
R) or (1
R,2
S) molecules interacting through the O-H∙∙∙O hydrogen bond. The helicity of the trityl group for all molecules observed in the crystal phase, both in
α and
β forms, is
MPM, which corresponds to the conformation of the calculated low-energy structure. A hydrogen atom of the OH group that is not involved in this hydrogen bond, is further involved in the O-H∙∙∙
π interaction, which stabilizes the structure of the molecule. In the
rac-3-
α crystals, pairs of hydrogen bonded molecules form columns stabilized by C-H∙∙∙
π interactions (
Figure 4a). However, similar columns are observed in the beta form structure where the molecules from adjacent columns interact with each other through O-H∙∙∙O hydrogen bonds (
Figure 4b). The three-dimensional structure is stabilized by weak interactions involving
π-electron systems.
Crystals of (R)-4, where the compound crystallizes with two molecules in the asymmetric unit cell (helicity MPM and PMP) where one of them is disordered (the ratio of refined occupancy factors for oxygen atoms is 0.88: 0.12) turned out to be an unusual case. For the disordered molecule, the position of the cyclohexane ring is undefined, and it mimics the phenyl ring.
Molecular disorder is also observed in the crystals of
rac-
5 (
Figure 5). The compound crystallizes in the triclinic system (symmetry
P ), with two molecules in the asymmetric unit cell where both of them are disordered—the cyclohexyl ring mimics the phenyl ring in the crystal structure. In addition, the enantiomer molecules can occupy the same positions in the crystal, and the
cis-2-tritylcyclohexanol crystallizes as a solid solution of enantiomers, as in the case of
3-
β. The helicity of the trityl group for all molecules observed in crystal is
MPM (or
PMP), which corresponds to the conformation calculated for the low-energy structure. The hydrogen atoms of the OH groups do not form strong intermolecular hydrogen bonds, as they participate in intramolecular O-H∙∙∙
π interactions stabilizing the structure of the molecule. The formation of solid solutions by compounds containing a bulky trityl group has previously been observed once and may be due to supramolecular protective properties of the substituent [
33].
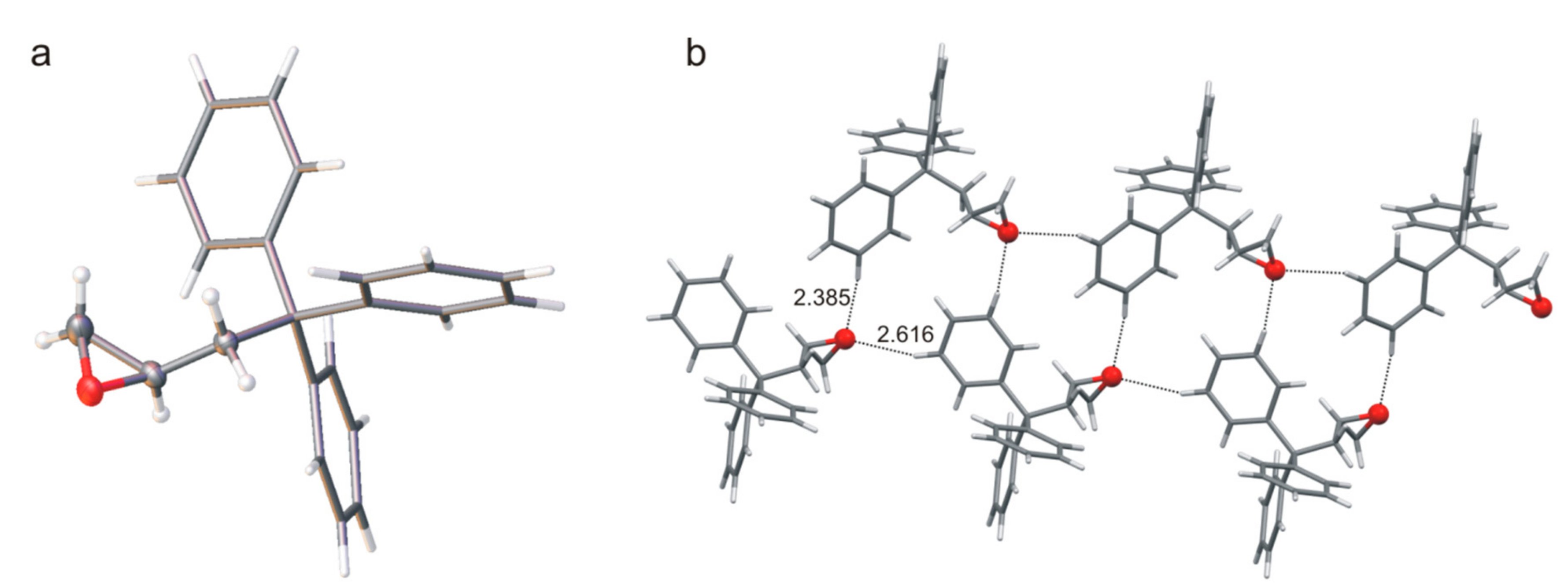
The common feature of compounds
7,
8, and
9 is the presence of an oxalyl fragment in the molecule. The conformation of this fragment is described by angle
ϕ (defined as O=C-C=O), which is −152.6(2)° for the monoester
7, 127.8(2)° for
8, and −32.1 (4)° for the diester
9. The molecular structure of monoester
7 is stabilized by intramolecular interaction of the phenyl ring and the almost parallel oxalyl part (
Figure 6). The free carboxyl group forms the bifurcate O-H∙∙∙O hydrogen bonds and participates in the formation of the supramolecular tape observed in the crystal structure. As mentioned, the ability to form inclusions was confirmed only for the compound
7 and the crystals obtained contain molecules of the solvent (chloroform). The stacking-like arrangement of phenyl ring and oxalyl part is not preserved in the structure of the diester
9, where the presence of a second
trans-2-tritylcyclohexanol unit forces a change in molecules geometry.
In the crystal structure of compound
11, the asymmetric unit cell consists of one molecule with
MMM helicity of trityl group (
PP0 for calculated the low-energy structure). The oxygen atom is involved in C-H∙∙∙O hydrogen bond and molecules form a ladder-like structural motif (
Figure 7). These C-H∙∙∙O interactions stabilize the structure of the molecule as well. The whole three-dimensional crystal structure is stabilized by weak interactions involving aromatic systems.
Compound 12 crystallizes with two molecules in the asymmetric unit cell. Interestingly, OH groups do not form strong hydrogen bonds. Cortho-H∙∙∙O interactions are observed between the molecules, while the OH groups are involved in O-H∙∙∙π interactions. The OH group has a great potential for hydrogen bonding and constitute a reliable feature in planning the crystal structure. As shown, the presence of a trityl group in close proximity to the hydroxyl group limits its availability for forming hydrogen bonds. This observation confirms that the trityl group can act as a protective group in crystal engineering.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}