Abstract
Biodegradation of contaminants is extremely complicated due to unpredictable microbial behaviors. Monitoring of microbial biodegradation drives us to determine (1) the amounts of specific degrading microbes, (2) the abundance, and (3) expression level of relevant functional genes. To this endeavor, the cultivation independent polymerase chain reaction (PCR)-based monitoring technique develops from endpoint PCR, real-time quantitative PCR, and then into novel digital PCR. In this review, we introduce these three categories of PCR techniques and summarize the timely applications of digital PCR and its superiorities than qPCR for biodegradation monitoring. Digital PCR technique, emerging as the most accurately absolute quantification method, can serve as the most promising and robust tool for monitoring of microbial biodegradation.
1. Introduction
Our living habitats are detrimentally affected by the accidentally and deliberately released multitudinous contaminants, including both affirmative conventional pollutants and emerging contaminants [1,2,3,4,5]. Microbial-based degradation, occurring with series of chemical and microbial transformations, is the ultimate fate of contaminants with economic and environmental benefits [6,7]. Until now, plenty of contaminants have shown the biodegradability, even the widely used forms of plastics once proven recalcitrant to biodegradation [8,9]. Biodegradation of contaminants is complicated due to the sophisticated interspecies interactions containing cooperation and competition, which will happen in all bioremediation processes like natural attenuation, bio-stimulation, and bioaugmentation [10,11,12,13]. Monitoring biodegradation is then of great importance for understanding the complicated processes and employing appropriate biotechniques for contaminants removal.
Biodegradation initiates when diverse contaminant catalytic enzymes in microbes participate in the redox reactions [14,15]. Parameters including the number of specific degrading microbes (i.e., microbial enumeration), the contaminants genotoxicity (i.e., mutagenicity), and the physiological activity, especially the abundance and expression level of corresponding degrading genes, reflect the biodegradation potential and efficiency [16,17]. For microbial enumeration, the number of survived cultivated bacteria could be calculated based on plate count method [18]. However, this method is with several drawbacks like long time for consuming, high variability, and inability for discerning between strains [19]. Most severely, majority of microbes (more than 99%) could hardly be cultivated in the environment [20], causing results generated from plate count method to be somehow unfaithful. Moreover, the plate-based methods are also widely applied for mutagenicity tests with limitations [21]. For physiological activity analysis, the abundance and expression level of corresponding genes determine the catalytic enzymatic activity [22], which was previously measured through diverse catalytic reactions in vitro [17,23], commanding specific reaction substrates and conditions [24]. Moreover, enzyme assays that can hardly reflect the real circumstance of environmental specimens as ecologically important microbial activities in situ may be low in magnitude, resulting in the boundedness of this method. The flow cytometer method was developed to directly differentiate and determine single cells with regards to size, shape, fluorescence, enzyme activity, etc. [25]. However, flow cytometer requires cells in heterogeneous suspension, making information of aggregated cells unavailable [26].
To resolve these challenges, polymerase chain reaction (PCR)-based techniques work by determining amplified target gene fragments and do not require microbial cultivation, thus emerging for monitoring of biodegradation [27,28]. Behaviors of microbes can be monitored by detecting the occurrence and abundance of specific gene markers. To realize quantification, the PCR-based techniques have been developing from conventional endpoint PCR to real-time quantitative PCR (qPCR) [29]. Recently, the novel digital PCR (dPCR) was reported to be more accurate for absolute quantification of target molecules [30].
The qPCR technique has been widely used for gene quantification. However, limitations still occur in accuracy, sensitivity, precision, and reproducibility [31]. It is proven that dPCR can overcome these limitations with advantages over qPCR in biochemical applications including microbial enumeration [32], low copy target detection [33], environmental DNA detection [34], rare-allele detection [35], minor mutations [36], and analysis of methylated DNA [37]. For biodegradation monitoring, it is essential to accurately quantify microbial performance along with contaminant degradation. Due to the novelty of dPCR, its applications are rarely reported. Besides, few studies clarify the superiorities of dPCR over endpoint PCR and qPCR based on their mathematical theories and technical applications in monitoring of biodegradation. Therefore, in this review, we introduce the three categories of PCR techniques and illustrate the superiorities of dPCR over qPCR for biodegradation monitoring. The dPCR technique is of promising potential on biodegradation monitoring and is expected to be widely adopted in the future.
2. Endpoint and qPCR Techniques with Their Applications in Biodegradation Monitoring
2.1. Endpoint PCR
PCR works by amplifying target fragment of DNA by forward and reverse oligonucleotide primers in multiple cycles of DNA duplexes denaturation, primers hybridization for target sequence, and elongation by DNA polymerase [38]. PCR typically repeats a series of temperature cycles with a doubling of the number of a target fragment generated after each cycle, and theoretically follows exponential amplification (i.e., 2n copies for n cycles). The logarithm of the amplified products is linear to the cycle number (Figure 1A). In practice, due to the consumption of reaction reagents, a basic PCR run mainly consists of three phases (Figure 1A): (1) Exponential phase, specific and precise amplification with 100% reaction efficiency at every cycle; (2) linear phase, slowed amplification with consumed reaction components; and (3) plateau phase, suspended amplification as PCR reagents depleted with no more products generated.
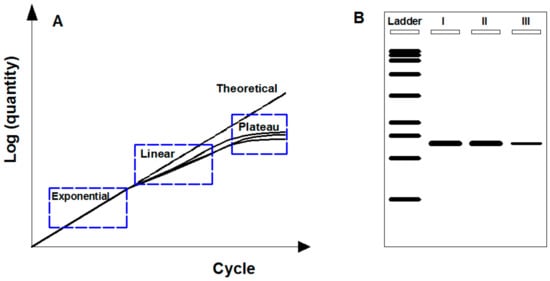
Figure 1.
Endpoint PCR-based gene monitoring. (A) Description of PCR amplification phases containing both theoretical and practical circumstances. Theoretical amplification: Logarithm of amplified products linear to cycle number. Practical amplification: Consisting of exponential, linear, and plateau phase due to the consumption of reaction reagents. (B) Scheme of agarose gel electrophoresis for endpoint amplified products. Target gene can be detected based on size discrimination but not rigorous for quantification.
Endpoint PCR means the amplified products are analyzed at the end of the reaction (i.e., plateau phase) by agarose gel electrophoresis after fluorescence staining. Through this way, comparing with DNA ladder, target genes are detected based on size discrimination. However, endpoint PCR by agarose gel electrophoresis is not rigorous for quantification due to the low gel resolution and variable reaction kinetics [39]. Gel resolution is poor mainly because of the nonquantitative staining dyes (e.g., ethidium bromide), which perform fuzzy identification for low-fold (e.g., less than 10-fold) changes [40]. Variable reaction kinetics occur when the depletion of reaction reagents at different rates after exponential phase for three replicates of a sample, resulting in plateau phase at a different point with different quantities (Figure 1A). In addition, endpoint PCR is semiquantitative by detecting the luminance levels on gel [41]. As shown in Figure 2B, it can roughly detect that the target PCR products of the type III samples were less than those of the types I and II samples but hardly for distinguishing the difference between type I and type II samples.
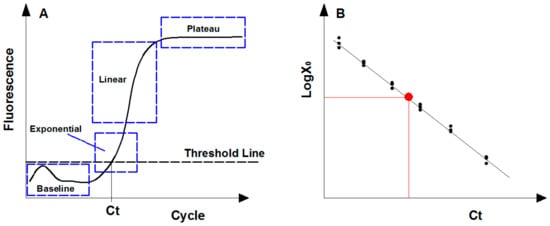
Figure 2.
Description of real-time quantitative PCR assay. (A) Fluorescence signal levels in four qPCR amplification phases due to the consumption of reaction reagents. (B) Standard curve generated by plotting the cycle threshold (Ct) value of diluted standards. The red point represents the target sample that can be calculated following the standard curve.
Endpoint PCR is usually applied for gene detection, molecular cloning, and genotyping through a quick yes or no answer. Besides, endpoint PCR is relatively simple for operation, thus can serve as the pre-step before gene quantification using other accurate techniques by giving the rough results. For biodegradation monitoring, endpoint PCR is applied for verifying the existence of corresponding genes to determine the occurrence of specific microbes’ and contaminants’ biodegradation. For instance, Mauffrey et al. [42] applied endpoint PCR to detect the occurrence of corresponding genes involved in herbicide degradation (i.e., trz, atz, phn, and puh genes), suggesting microbial degradation contributing to pesticide dissipation. For anaerobic hydrocarbon degradation, the analogues of genes alkylsuccinate synthases, assA [43], and benzylsuccinate synthase, bssA [44], were measured by endpoint PCR with degenerate primers, to determine the presence of hydrocarbon degrading bacteria.
Based on endpoint PCR, the denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) techniques were developed to explore the microbial community change [45]. They can separate the PCR amplicons of the same length with differences in GC contents and distributions. For biodegradation monitoring, Luo et al. [46] did the PCR-DGGE for the V3 region of bacterial 16S rRNA to analyze the structure of microbial consortium capable of degrading benzo(a)pyrene. However, because DGGE and TGGE techniques are gel electrophoresis-based, results generated are quantitative and microbes with low abundance can hardly be detected.
2.2. Real-Time Quantitative PCR
To satisfy the demands of gene quantification, real-time quantitative PCR (qPCR) was then developed to accurately determine the concentration of target molecular fragments. As its name implies, qPCR means amplification of target DNA fragment is detected in real time during the whole PCR process using fluorescence reporters [47].
For gene quantification, the exponential phase is the optimal point for collecting and analyzing data due to its most efficient amplification, and theoretically the amount of PCR products follows Equation (1) [48]. However, the fluorescence signal of PCR products in early cycles (i.e., baseline phase) is always disordered since the amplification remains at the background level (Figure 2A). To eliminate the background fluorescence signal, the threshold line was applied and set above the background within the exponential phase [49]. The cycle threshold (Ct) is the cycle number at which the amplification plot intersects the threshold line, and the amplified products at Ct cycles follows Equation (2). By logarithmic conservation, the logarithm of initial templates is negatively linear to Ct value, as shown in Equation (3).
The abundance of genes of interest in an environmental specimen are calculated following the calibration curve generated from artificial standards with known quantity (Figure 2B) [50]. Standards used are usually desired nominal gradient diluted genomic DNA (gDNA) with known sequences, plasmids containing target gene constructed commonly through digestion-ligation methods, or artificially synthetic DNA fragments [51]. The 95% confidence intervals (CI) of the target gene concentration by absolute quantification is calculated as Equation (4) [52].
where n is amplification cycle number, is initial templates amount, is amplification efficiency, is amplified products after n cycles, is cycle threshold, is amplified products after cycles, is the average of replicated value, and is the standard deviation of . Constant 4.30 was calculated from the student’s t distribution when the of replicated qPCR follows normal distribution [53], and constant 3 represents the number of replicate values.
The qPCR technique is widely applied in diverse contaminants’ biodegradation monitoring. For example, Laurie and Lloyd-Jones [54] quantified phnAc and nahAc genes to monitor the polycyclic aromatic hydrocarbons (PAHs) contaminations in soil. Li et al. [55] found that decreased microcystin degradation correlated to inhibited abundance of mlrA gene coding the enzyme responsible for the initial cleavage of cyclic microcystin through qPCR. For the degradation of vinyl chloride, Wilson et al. [56] determined that the abundance of two functional genes (etnC and etnE) was associated with aerobic degradation in groundwater. However, results generated from qPCR are relative to standard curve and thus could hardly accurately reflect the abundance of target genes. It is promising if we could achieve directly absolute quantification.
3. Digital PCR and Its Advantages over Previous PCR Techniques
3.1. Digital PCR Systems
The term “digital PCR”, a novel method for the absolute quantification of target nucleic acids without the requirement for standard curves, was first described by Vogelstein and Kinzler [57]. Unlike qPCR, digital PCR hinges on the distribution of template analytes into many replicate microreactors at limiting dilution with most reactions containing one or zero molecules [58]. No reference standards or endogenous controls are needed for dPCR due to its calculation methods, with a sufficiently large number of partitions following Poisson distribution [59].
Briefly, dPCR starts from DNA/RNA extraction in a similar fashion as qPCR. Then, the assembled reaction is partitioned into enormous independent PCR subreactions. PCR amplification is performed to endpoint and the absolute quantification of target molecules is calculated following the Poisson distribution statistical analysis (Figure 3) [60,61].
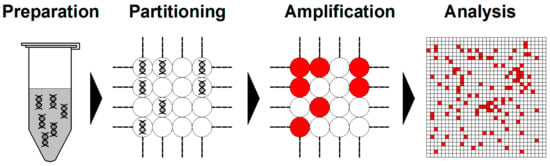
Figure 3.
Schemes of typical digital PCR (dPCR) workflow. Generally, dPCR is conducted following the steps of preparation, partitioning, amplification, and analysis.
Poisson distribution enables dPCR for accurate quantification of target molecules [62]. The precision of dPCR increases with an increasing number of partitions. The basic process of dPCR is the random distribution of m molecules into n partitions. The average number of targets per partition (λ) follows Equation (5), which can be simulated by Poisson distribution as Equation (6). The m and of an unknown sample can be easily calculated from the percentage of empty partitions P following Equations (7) and (8). The confidence interval of Poisson distribution is strongly affected by the probability of an empty partition P and the average number of targets per partition λ, and the 95% CI for the expected concentration is calculated according to Equation (9) [52,63].
where is average number of targets per partition, is the number of targets in the sample, is the number of partitions, is the sample concentration, is the partition volume, is the probability of an empty partition, is confidence interval, and 1.96 is a constant for a 95% confidence interval.
In qPCR, some unavoidable inhibitors and amplification competition of templates could seriously affect the detection accuracy since instruments could hardly resolve small differences in emitted fluorescence. Digital assays can overcome these drawbacks as the detection instrument only needs to determine whether amplification occurred in each partition by identifying the individual endpoint fluorescence, making data analysis less dependent on the detector or assay chemistry [59]. However, several sources of variations during dPCR workflow may affect the reliability, among which subsampling and partitioning errors are the most dominant [64]. Subsampling errors arise with the assays when pipetting part volume of the full sample for analysis, resulting in statistical variation between replicates. As modeled by Bizouarn [65], errors dominate when conducting subsampling for the original sample with few targets to detect (i.e., high P or low λ value) (Figure 4). Partitioning errors occur since the distribution of targets among partitions may differ between each experiment. Dube et al. [66] models the partitioning as a binominal process, with the standard deviation of the probability of an empty partition P propagating to the errors in λ and P. Partition errors dominate when very few partitions are empty (i.e., high λ or low P value) and nearly all are empty (i.e., high P or low λ value) (Figure 4). Therefore, when there are few targets contained in the original samples (i.e., high P or low λ value) subsampling deviations incorporate with partitioning errors, while few partitions are empty (i.e., high λ or low P value) partitioning errors dominate (Figure 4). Considering errors from subsampling and partitioning, preparing samples to the number of copies within the optimal range is the pre-step to improve measurement accuracy.
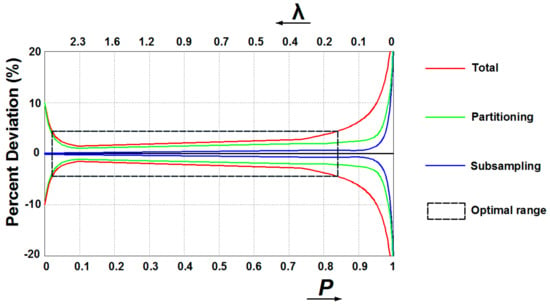
Figure 4.
Schemes of assumed total, partitioning, and subsampling errors in digital PCR assays.
Two general approaches are commercially adopted for partitioning the initial nucleic acids samples into plenty of individual microreactions, i.e., the chip-based dPCR (cdPCR) and droplet-based dPCR (ddPCR) [67]. For cdPCR, the chip is composed of physically isolated chambers or wells. Applied Biosystems’ QuantStudioTM 3D digital PCR is a commercial representative, with a microchip containing 20,000 microwells and capable of partitioning and allowing separate PCR amplification reactions in these individual microreactors. Detection of the fluorescence of an amplified microchip can thus provide us with the amount of positive and negative reactions for statistical analysis. The second approach, ddPCR, partitions PCR test into sufficiently individual droplets in a water–oil emulsion, with the use of flow cytometry to count positive PCR reactions. Two requirements are needed for the droplet-based reaction. Firstly, the oil should be nonreactive and can form stable microreactors to prevent the diffusion of the reaction reagents. Secondly, appropriate surfactants should be added to stabilize the water–oil interface and prevent oil coalescence. Commercial representative of ddPCR is Bio-Rad’s QX200TM Droplet Digital PCR system.
3.2. Fluorescence Reporters in dPCR Systems
Commercially used fluorescence reporters for gene quantification in dPCR system are SYBR Green I and TaqMan assays [68]. SYBR Green I dye is nonspecific fluorescence dye that intercalates with double stranded DNA (dsDNA), while TaqMan-based detection uses a fluorogenic probe specific to target genes.
SYBR Green I dye fluoresces almost 1000-fold greater than its free in solution when it binds to the minor grooves of dsDNA [69]. During denaturation, SYBR Green I dye is released with drastically reduced fluorescence (Figure 5). Annealing and extension generated double stranded PCR products with SYBR Green I binding to it, resulting in a net increase in fluorescence for detection. This method is relatively cost beneficial and easy for operation. The primary disadvantage of SYBR Green I is that it may generate false positive signals since SYBR dye can also bind to nonspecific double stranded DNA sequences. Therefore, it is extremely important to have well-designed primers that do not amplify nontarget sequences, and that melt curve analysis be performed.
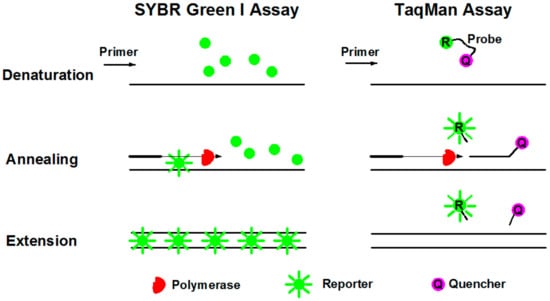
Figure 5.
Schematic diagrams of SYBR Green I and TaqMan assays during PCR procedures of denaturation, annealing, and extension.
TaqMan assay is more specific with an oligonucleotide probe designed for target sequence hybridization [70]. An oligonucleotide probe is constructed of a fluorescent reporter (R) on the 5’ end and a quencher (Q) on the 3’ end (Figure 5). While the probe is intact, the proximity of the quencher dye greatly reduces the fluorescence emitted by the reporter dye by fluorescence resonance energy transfer (FRET). During each annealing and extension, the DNA polymerase cleaves the reporter dye from the probe, leading to the emission of its fluorescence. Due to specific hybridization of the probe, TaqMan assay is more accurate for quantification than SYBR Green I. In addition, probes can be labeled with distinguishable reporter dyes, allowing detection of two distinct sequences in one reaction. The primary disadvantage is the synthesis of specific probes, which require more cost.
4. Applications of dPCR for Monitoring of Biodegradation
Biodegradation monitoring via endpoint PCR probably provides us with the occurrence of biodegradation, while qPCR gives us the quantification of these genes but always with inaccurate results. The dPCR can generate accurate results through absolute quantification. In this section, we introduce dPCR application and its advantages over qPCR for monitoring of microbial biodegradation in enumeration of specific degrading microbes, as well as the abundance and expression level regarding functional genes as listed in Table 1.

Table 1.
The dPCR-based monitoring applications and its superiorities over qPCR.
4.1. Microbial Enumeration
Specific degrading microbes directly affect the degradation efficiency. Monitoring the number of these microbes (i.e., microbial enumeration) can then help us understand the biodegradation potential.
Microbial enumeration requires the quantification of specific regions of DNA, with typically used segmental ribosomal markers targeting 16S rRNA or 23S rRNA for bacteria, and 18s rRNA for fungi, internal transcribed spacers (ITS), and other target markers [71,72]. For example, Pornwongthong et al. [73] targeted the 16S rRNA sequence to quantify the abundance of Pseudonocardia dioxanivorans CB1190 during the biodegradation of 1,4-dioxane under the effect of transition metals and organic ligands in the field. Bücker et al. [74] quantified the V4 and V5 regions of 18S rRNA to determine the abundance of fungi during diesel storage. Besides, during the PAHs biodegradation in the soil, quantification of different ITS regions was used for determining the abundance of each degrading microbe [75]. In addition, Richardson et al. [76] used the B subunit of ribosomal polymerase (rpoB) gene as a marker gene to determine the total microbial population in the fuel-contaminated soil as it is present in almost all cells.
To some contents, qPCR cannot meet the demands in microbial enumeration. The first is the detection limit of key microbial indicators during biodegradation. Krolicka et al. [77] quantified 16S rRNA and the GyrB markers to assess temporal microbial variability of oil contaminants in seawater. However, GryB gene is a single-copy gene, which is hardly used for quantification by qPCR at low abundance, and they mentioned to employ ddPCR to overcome the challenge. The second limitation is the enumeration calculated using qPCR as calculated from the standard curve, causing the relative but not absolute quantification. Hence, dPCR should be more suitable for microbial enumeration as it can achieve accurate and absolute quantification without calibration.
Recently, some researches have applied dPCR for microbial enumeration during contaminants biodegradation. The dPCR has been successfully used to determine the survival of bioaugmented phthalic acid esters degrading strain Mycobacterium sp. YC-RL4 in soil through quantifying the V3-V4 region of 16S rRNA gene [78]. For dichloromethane dichlorination, Chang et al. [79] used dPCR to quantify the 16S rRNA gene from 15 key degraders’ genomes to uncover the microbial population dynamics in a given culture. Besides, studies also reported that dPCR was applied in microbial abundance and population dynamics analysis in soil and marine sediment environment [80,81].
Microbial enumeration through dPCR is also suitable to other research, especially for water quality monitoring, i.e., the microbial pathogens detection. United States Environmental Protection Agency (USEPA) recommends using qPCR to quantify enterococci 23S rRNA as the fecal indicator for water-quality monitoring [82]. However, underestimation always occurs and dPCR could eliminate the weakness caused by qPCR [52]. Besides, dPCR also enables the simultaneous detections of diverse pathogens [83,84] and harmful bloom-forming cyanobacteria [85] through the TaqMan assay.
4.2. Functional Gene Abundance Quantification
Microbial enumeration can reflect the abundance of specific degrading microbes. However, it is hard only depending on this to investigate the biodegradation potential since different microbes may harbor different degrading genes and many interactions between species, like horizontal gene transfer always occur during biodegradation. Functional genes are responsible for the synthesis of specific catalytic enzymes involved in biodegradation of contaminants. Therefore, identification and quantification of these functional genes would provide direct information about biodegradation potential in the environment.
Currently, the dPCR is emerging for absolute quantification of these functional genes without standard curve normalization. Kim et al. [86] used 3D chip-based cdPCR technique to evaluate the copy number change of alkB1 gene responsible for alkanes’ degradation to assess microbial response under seasonal freeze-thaw condition. Dong et al. [87] employed ddPCR to quantify the functional genes involved in the nitrification and denitrification in the natural environment by determining amoA gene in ammonia oxidizing bacteria (AOB) and archaea (AOA) and nirS and nosZ genes in the denitrifiers. In addition, antibiotic-resistant genes (ARGs) are also the emerging contaminants in diverse environmental matrices [88], and researchers applied dPCR for these genes’ quantification in both soil [89] and atmosphere [90] environment to realize accurate measurement more than qPCR.
Horizontal gene transfer always occurs when microbes harboring conjugative or mobilizable plasmids containing genes coding catalytic enzymes are introduced into donor bacteria without contaminants degrading capacity [91]. It was proven that horizontal gene transfer assessment is effective during biostimulation [92] and bioaugmentation [93] monitoring. Through biostimulation, diversity of pollutant-degrading bacteria and the effective transfer of petroleum hydrocarbon degrading genes (i.e., alkB and phnAc) among resident microorganisms was enhanced for petroleum hydrocarbons degradation [92]. For bioaugmentation, augmented Rhodococcus sp. strain p52 harboring catalyzing dioxin degradation genes (i.e., dbfA and dfdA) located in broad-host conjugative plasmids would transfer the catalytic capacity to other bacteria without degradation ability to enhance biodegradation, while the strain itself would disappear for its unfitness for the environment [93].
However, we have not found any research that utilized the dPCR technique to monitor horizontal gene transfer during contaminants’ degradation. Typically, the ratio of functional gene and target gene (i.e., specific gene sequence for microbial enumeration [93]) abundance is a constant. We usually detect the horizontal gene transfer through measuring the change of this ratio. Traditional qPCR technique measures the two genes separately, resulting in large errors. The accuracy of measurement can improve if we could measure the two genes simultaneously. It is proven that dPCR technique shows the superiority that enables multigene analysis of individual environmental bacteria [94] and many researchers use dPCR for multigene quantifications. For example, Cao, Raith, and Griffith [84] used dPCR for simultaneous quantification of fecal indicators to assess water quality with improved precision and repeatability over qPCR. Moreover, accurate transgene quantification between crop plants was determined by dPCR with high reliability [95]. Therefore, dPCR is more appropriate for horizontal gene transfer monitoring, and applications of dPCR in this area will boost soon.
4.3. Gene Expressing Determination
DNA-based quantification always gives a distorted view during biodegradation monitoring, probably overestimating the pollutant degrading ability as it not only presents in active bacterial populations but also in dead microbes. The transcription level of these functional groups of genes coding multicomponent catalytic enzymes would constitute more reliable and accurate biomarkers for the biodegradation monitoring since expression occurs only in metabolically active microbes [96].
Effective biomarkers perform to a relatively high degree of correlation between expression of the functional gene and the rates of contaminants mineralization [97]. Until now, many biomarkers were identified for biodegradation monitoring. For the degradation of 1,2,3- and 1,2,4-trichlorobenzene (TCB), Wagner et al. [98] determined the transcriptions of 32 reductive dehalogenase homologous genes in Dehalococcoides stain, suggesting using cbrA gene to characterize natural dehalogenation potential. For the degradation of phenoxy acid, the transcripts of tfdA gene coding α-ketoglutarate-dependent dioxygenase functions as a molecular marker [97]. The gene bssA encoding benzylsuccinate synthase that catalyzes the first step in toluene biodegradation can be employed as a biomarker for biodegradation of toluene [99]. For the emerging contaminant, 1–4 dioxane biodegradation, expression of specific bacterial monooxygenase and dehydrogenase together in Pseudonocardia dioxanivorans CB1190 can serve as effective biomarkers to monitoring biodegradation in the environment [96].
Functional gene expression quantification lies in the effectively and specifically messenger RNA (mRNA) detection. Reverse transcription (RT) is the first step that transcribing RNA into complementary DNA (cDNA) for downstream measurement. The conjugated methods, i.e., RT-dPCR or RT-qPCR, are then developed to study gene expression variations. However, RT-qPCR can hardly reflect the actual cDNA amount in the sample and RT operation always introduces the contaminants and inhibitors that impact qPCR analysis. The most acceptable method is employing reference genes, which are assumed constitutively and evenly transcribed across diverse environmental conditions, to normalize and reduce variabilities across samples [100]. Because of the ability of dPCR to absolutely quantify the number of molecules present within a sample without the impact of contaminant inhibition, the use of reference genes and calibration curve seems not obligatory in dPCR. When no effect of measured amount of DNA/RNA is applied to each sample, RT-dPCR can realize directly absolute quantification without normalization [101,102]. For example, during nitrate degradation, Zhang et al. [103] measured the expression level of denitrification-associated genes (i.e., narG, nirK, and nirS) per gram DNA using RT-dPCR and found that reactor with bioaugmented denitrifer strain Diaphorobacter could enhance denitrification performance. For nitrogen cycle monitoring, Segawa et al. [104] analyzed the abundance and expression of biomarker genes for nitrogen fixation, nitrification, and denitrification using RT-dPCR, and only gene markers for nitrification and denitrification were highly expressed, indicating this process is the predominant occurrence. In addition, RT-dPCR has lower variability and better reproducibility than RT-qPCR counterpart [105] and the accuracy of RT-dPCR does not rely on amplification efficiency [106]. It thus out-performs RT-qPCR by consistent and precise quantification.
However, in most biodegradation conditions, quality of environmental specimen is highly variable, and all the technical issues associated with the RT step could cause significantly diverse cDNA input for dPCR quantification [107]. The strategy of employing reference genes conducted in RT-qPCR is also beneficial to RT-dPCR application especially in time course experiment [108].
Reference genes applied in RT-dPCR for monitoring of biodegradation is rarely reported. Comparing the superiorities and weaknesses between RT-dPCR and RT-qPCR for diverse applications is the most recent topic, and RT-dPCR is not widely accepted yet. Besides, we found RT-dPCR was applied to monitor lignin degradation by large fungus. Vasina et al. [109] absolutely quantified the expression of corresponding 18 lignin-degrading peroxidases using tubulin as reference gene. The expression level of degrading genes was calculated by plotting the absolute concentration of target degrading gene to reference gene. Quantification of biomarkers’ expression relative to references through RT-dPCR will also be adopted for accurate biodegradation monitoring soon.
5. Limitations of Existing Applications and Future Perspectives
For biodegradation monitoring, many factors should be taken into account for gene quantification, e.g., the purity and concentration of nucleic acids, the theoretical and practical accuracy, the time and commercial consumption. The dPCR is less affected than qPCR by these factors, with the great potential to be applied in monitoring of microbial biodegradation.
The first factor is the chemical inhibition due to the complexity of environmental samples. Nucleic acids’ extraction is the primary step, which shall unavoidably bring in contaminants to downstream PCR reactions. The qPCR assays are especially vulnerable to contamination since the detection is conducted through the real-time process. It was reported that complex biomolecules, such as humic acid, can significantly inhibit PCR reactions [110]. The dPCR can overcome the shortage due to its endpoint quantification, and the contaminant calcium was reported with less inhabitation for dPCR than qPCR [52]. Besides, dPCR can relieve the effects through increasing number of thermal cycles. Nowadays, widespread emerging contaminants is the global issue. Hence, monitoring the microbial performance for these molecules’ degradation is indispensable. Nevertheless, few researchers discussed whether emerging contaminants may affect the results of gene quantification. From this review, it showed signs that dPCR can be the replaceable toolkit of qPCR for diverse emerging contaminants’ biodegradation monitoring.
The second one is the theoretical and practical accuracy. The qPCR quantification technology highly relies on the reference curve, resulting in relative quantification, while dPCR achieved the absolute quantification through Poisson distribution. For theoretical accuracy, at environmental relevant concentrations, it was proven that dPCR is more precise, with narrow 95% confidence interval, than qPCR quantification [52]. For practical accuracy, results generated from qPCR were relative to calibration curve and were not the actual number of copies in a sample itself. However, different structure types of standard DNA may affect the quantification accuracy. It was proven that qPCR assay is seriously overestimated by using circular plasmid standard as standard when quantifying microalgal pcna gene [111]. Besides, the amplification efficiency was instrument dependent among commercially used Eppendorf RealPlex, BioRad CFX96, AB StepOne, AB 7500Fast, Corbett Rotorgene I, and Roche LC480 systems [112], hence qPCR could hardly yield acceptable precision or reproducibility. Errors generated from dPCR were mainly from subsampling and partitioning errors considered into Poisson model, while some other errors, like the partitioning volume [113], should also be taken into the model to improve the accuracy of dPCR application. To standardize experimental protocols and improve the reproducibility of data, researchers should carefully follow the Digital Minimum Information for Publication of Quantitative Digital PCR Experiments (dMIQE) Guidelines [100].
The third one is the low abundant gene quantification. The 16S rRNA gene is usually used for quantifying the amounts of bacteria. However, multiple copies of this gene are often present in a given bacterium with intragenomic copies differing in sequence, leading to identification of multiple ribosome types [114]. Quantification of single-copy gene (e.g., rpoB, GyrB markers) is thus promising for microbial enumeration. As a matter of fact, qPCR could hardly quantify low abundant genes, limiting its detection in microbial variability [115,116]. For functional gene monitoring, this circumstance should also be taken in account. The dPCR could absolutely quantify the low-copy number genes, thus the promising future for accurate biodegradation process monitoring.
The fourth one is the commercial cost. Though the qPCR instrument is relatively cheaper than dPCR, it is time consuming and requires standard curve calibration. Standard curve preparation wastes a lot of time and some reagents are extremely expensive. The instrument cost for cdPCR is relative to qPCR instrument (almost $20,000–$40,000), and they are both cheaper than ddPCR (almost $100,000). For cdPCR, the reaction chip can only hold one sample, with the cost of $12 each. If measuring multiple samples, the consumption of chips will raise the monitoring cost. Selecting the appropriate dPCR system strongly depends on the samples numbers. We also believe, with the technology development, the cost will significantly decrease to satisfy the need of the researchers.
With the rising trend of globalized emerging contaminants, microbial biodegradation monitoring technique should be further standardized. The dPCR exhibited the potential for standardizing the data due to its lab independence and absolute quantification. The next generation sequencing (NGS) is the other important technique for microbial community analysis during biodegradation. Results generated from dPCR and NGS can help us better understand the global pollutions and, therewith, the appropriate actions to face these contaminants.
Author Contributions
Y.C., M.Y., and G.D. made the literature search. Y.C. and M.Y. wrote the manuscripts. Y.C. and G.D. prepared the figures and formulas. B.Z. and B.C. helped review the manuscript and provide constructive suggestions. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), Fisheries and Oceans Canada (DFO), Canadian Foundation of Innovation (CFI), and Canada Research Chair (CRC) program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, B.; Matchinski, E.J.; Chen, B.; Ye, X.; Jing, L.; Lee, K. Marine oil spills—Oil pollution, sources and effects. In World Seas: An Environmental Evaluation, 2nd ed.; Sheppard, C., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2019; Volume 3, Chapter 21; pp. 391–406. [Google Scholar]
- Petrie, B.; Barden, R.; Kasprzyk-Hordern, B. A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring. Water Res. 2015, 72, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Boufadel, M.; Chen, B.; Foght, J.; Hodson, P.; Swanson, S.; Venosa, A. Expert Panel Report on the Behaviour and Environmental Impacts of Crude Oil Released into Aqueous Environments; Royal Society of Canada: Ottawa, ON, Canada, 2015; ISBN 978-1-928140-02-3. [Google Scholar]
- Xin, X.; Huang, G.; An, C.; Raina-Fulton, R.; Weger, H. Insights into Long-Term Toxicity of Triclosan to Freshwater Green Algae in Lake Erie. Environ. Sci. Technol. 2019, 53, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Huang, G.; An, C.; Feng, R. Interactive Toxicity of Triclosan and Nano-TiO2 to Green Alga Eremosphaera viridis in Lake Erie: A New Perspective Based on Fourier Transform Infrared Spectromicroscopy and Synchrotron-Based X-ray Fluorescence Imaging. Environ. Sci. Technol. 2019, 53, 9884–9894. [Google Scholar] [CrossRef] [PubMed]
- Lui, M.Y.; Wong, C.Y.Y.; Choi, A.W.-T.; Mui, Y.F.; Qi, L.; Horváth, I.T. Valorization of Carbohydrates of Agricultural Residues and Food Wastes: A Key Strategy for Carbon Conservation. ACS Sustain. Chem. Eng. 2019, 7, 17799–17807. [Google Scholar] [CrossRef]
- Cao, Y.-Q.; Li, Q.; Xia, P.-F.; Wei, L.-J.; Guo, N.; Li, J.-W.; Wang, S.-G. AraBAD based toolkit for gene expression and metabolic robustness improvement in Synechococcus elongatus. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Chauhan, D.; Agrawal, G.; Deshmukh, S.; Roy, S.S.; Priyadarshini, R. Biofilm formation by Exiguobacterium sp. DR11 and DR14 alter polystyrene surface properties and initiate biodegradation. RSC Adv. 2018, 8, 37590–37599. [Google Scholar] [CrossRef]
- Raddadi, N.; Fava, F. Biodegradation of oil-based plastics in the environment: Existing knowledge and needs of research and innovation. Sci. Total Environ. 2019, 679, 148–158. [Google Scholar] [CrossRef]
- McKew, B.A.; Coulon, F.; Yakimov, M.M.; Denaro, R.; Genovese, M.; Smith, C.J.; Osborn, A.M.; Timmis, K.N.; McGenity, T.J. Efficacy of intervention strategies for bioremediation of crude oil in marine systems and effects on indigenous hydrocarbonoclastic bacteria. Environ. Microbiol. 2007, 9, 1562–1571. [Google Scholar] [CrossRef]
- Chen, Z.; An, C.; Boufadel, M.; Owens, E.; Chen, Z.; Lee, K.; Cao, Y.; Cai, M. Use of Surface-Washing Agents for the Treatment of Oiled Shorelines: Research Advancements, Technical Applications and Future Challenges. Chem. Eng. J. 2019. [Google Scholar] [CrossRef]
- McGenity, T.J.; Folwell, B.D.; McKew, B.A.; Sanni, G.O. Marine crude-oil biodegradation: A central role for interspecies interactions. Aquat. Biosyst. 2012, 8, 10. [Google Scholar] [CrossRef]
- Trueba-Santiso, A.; Fernández-Verdejo, D.; Marco-Rius, I.; Soder-Walz, J.M.; Casabella, O.; Vicent, T.; Marco-Urrea, E. Interspecies interaction and effect of co-contaminants in an anaerobic dichloromethane-degrading culture. Chemosphere 2020, 240, 124877. [Google Scholar] [CrossRef] [PubMed]
- Nikel, P.I.; Pérez-Pantoja, D.; de Lorenzo, V. Pyridine nucleotide transhydrogenases enable redox balance of Pseudomonas putida during biodegradation of aromatic compounds. Environ. Microbiol. 2016, 18, 3565–3582. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, Y.; Feng, H.; Wang, J.; Yang, X.; Wang, Z. Genome-guided identification and characterization of bacteria for simultaneous degradation of polycyclic aromatic hydrocarbons and resistance to hexavalent chromium. Int. Biodeterior. Biodegrad. 2019, 138, 78–86. [Google Scholar] [CrossRef]
- Varjani, S.J. Microbial degradation of petroleum hydrocarbons. Bioresour. Technol. 2017, 223, 277–286. [Google Scholar] [CrossRef]
- Emadian, S.M.; Onay, T.T.; Demirel, B. Biodegradation of bioplastics in natural environments. Waste Manag. 2017, 59, 526–536. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Liu, P.-W.G.; Whang, L.-M.; Wu, Y.-J.; Cheng, S.-S. Biodegradability and microbial community investigation for soil contaminated with diesel blending with biodiesel. Process Saf. Environ. 2019, 130, 115–125. [Google Scholar] [CrossRef]
- Hansen, S.J.; Morovic, W.; DeMeules, M.; Stahl, B.; Sindelar, C.W. Absolute enumeration of probiotic strains Lactobacillus acidophilus NCFM® and Bifidobacterium animalis subsp. lactis Bl-04® via chip-based digital PCR. Front. Microbiol. 2018, 9, 704. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Nesslany, F. The current limitations of in vitro genotoxicity testing and their relevance to the in vivo situation. Food Chem. Toxicol. 2017, 106, 609–615. [Google Scholar] [CrossRef]
- Rocca, J.D.; Hall, E.K.; Lennon, J.T.; Evans, S.E.; Waldrop, M.P.; Cotner, J.B.; Nemergut, D.R.; Graham, E.B.; Wallenstein, M.D. Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J. 2015, 9, 1693. [Google Scholar] [CrossRef]
- Meng, L.; Li, W.; Bao, M.; Sun, P. Promoting the treatment of crude oil alkane pollution through the study of enzyme activity. Int. J. Biol. Macromol. 2018, 119, 708–716. [Google Scholar] [CrossRef]
- Scopes, R.K. Enzyme activity and assays. Encycl. Life Sci. 2002, 1–6. [Google Scholar] [CrossRef]
- Szczepaniak, Z.; Cyplik, P.; Juzwa, W.; Czarny, J.; Staninska, J.; Piotrowska-Cyplik, A. Antibacterial effect of the Trichoderma viride fungi on soil microbiome during PAH’s biodegradation. Int. Biodeterior. Biodegrad. 2015, 104, 170–177. [Google Scholar] [CrossRef]
- Jahan-Tigh, R.R.; Ryan, C.; Obermoser, G.; Schwarzenberger, K. Flow cytometry. J. Investig. Dermatol. 2012, 132, e1. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Duan, G.-L.; Ding, K.; Huang, F.-Y.; Zhu, Y.-G. DNA stable-isotope probing identifies uncultivated members of Pseudonocardia associated with biodegradation of pyrene in agricultural soil. FEMS Microbiol. Ecol. 2018, 94, fiy026. [Google Scholar] [CrossRef]
- Steffan, R.; Atlas, R. Polymerase chain reaction: Applications in environmental microbiology. Annu. Rev. Microbiol. 1991, 45, 137–161. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Vandesompele, J.; Kubista, M. Real-time PCR: Current technology and applications. In Data Analysis Softw.; Logan, J., Edwards, K., Saunders, N., Eds.; Caister Academic Press: Norfold, UK, 2009; pp. 65–83. [Google Scholar]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003. [Google Scholar] [CrossRef]
- Hall Sedlak, R.; Jerome, K.R. The potential advantages of digital PCR for clinical virology diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 501–507. [Google Scholar] [CrossRef]
- Yang, R.; Paparini, A.; Monis, P.; Ryan, U. Comparison of next-generation droplet digital PCR (ddPCR) with quantitative PCR (qPCR) for enumeration of Cryptosporidium oocysts in faecal samples. Int. J. Parasitol. 2014, 44, 1105–1113. [Google Scholar] [CrossRef]
- Jones, M.; Williams, J.; Gärtner, K.; Phillips, R.; Hurst, J.; Frater, J. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, definetherain. J. Virol. Methods 2014, 202, 46–53. [Google Scholar] [CrossRef]
- Doi, H.; Takahara, T.; Minamoto, T.; Matsuhashi, S.; Uchii, K.; Yamanaka, H. Droplet digital polymerase chain reaction (PCR) outperforms real-time PCR in the detection of environmental DNA from an invasive fish species. Environ. Sci. Technol. 2015, 49, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rizaldos, E.; Paweletz, C.; Song, C.; Oxnard, G.R.; Mamon, H.; Jänne, P.A.; Makrigiorgos, G.M. Enhanced ratio of signals enables digital mutation scanning for rare allele detection. J. Mol. Diagn. 2015, 17, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, P.; Zeng, G.; Yang, C.; Huang, D.; Zhang, J. Bioremediation of soils contaminated with polycyclic aromatic hydrocarbons, petroleum, pesticides, chlorophenols and heavy metals by composting: Applications, microbes and future research needs. Biotechnol. Adv. 2015, 33, 745–755. [Google Scholar] [CrossRef]
- Wiencke, J.K.; Bracci, P.M.; Hsuang, G.; Zheng, S.; Hansen, H.; Wrensch, M.R.; Rice, T.; Eliot, M.; Kelsey, K.T. A comparison of DNA methylation specific droplet digital PCR (ddPCR) and real time qPCR with flow cytometry in characterizing human T cells in peripheral blood. Epigenetics 2014, 9, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Erlich, H. Polymerase chain reaction strategy. Annu. Rev. Biochem. 1992, 61, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Kainz, P. The PCR plateau phase–towards an understanding of its limitations. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1494, 23–27. [Google Scholar] [CrossRef]
- Deschaght, P.; De Baere, T.; Van Simaey, L.; De Baets, F.; De Vos, D.; Pirnay, J.-P.; Vaneechoutte, M. Comparison of the sensitivity of culture, PCR and quantitative real-time PCR for the detection of Pseudomonas aeruginosa in sputum of cystic fibrosis patients. BMC Microbiol. 2009, 9, 244. [Google Scholar] [CrossRef]
- Antiabong, J.F.; Ngoepe, M.G.; Abechi, A.S. Semi-quantitative digital analysis of polymerase chain reaction-electrophoresis gel: Potential applications in low-income veterinary laboratories. Vet. World 2016, 9, 935. [Google Scholar] [CrossRef]
- Mauffrey, F.; Baccara, P.-Y.; Gruffaz, C.; Vuilleumier, S.; Imfeld, G. Bacterial community composition and genes for herbicide degradation in a stormwater wetland collecting herbicide runoff. Water Air Soil Pollut. 2017, 228, 452. [Google Scholar] [CrossRef]
- Andrade, L.L.; Leite, D.C.; Ferreira, E.M.; Ferreira, L.Q.; Paula, G.R.; Maguire, M.J.; Hubert, C.R.; Peixoto, R.S.; Domingues, R.M.; Rosado, A.S. Microbial diversity and anaerobic hydrocarbon degradation potential in an oil-contaminated mangrove sediment. BMC Microbiol. 2012, 12, 186. [Google Scholar] [CrossRef]
- Oka, A.R.; Phelps, C.D.; Zhu, X.; Saber, D.L.; Young, L. Dual biomarkers of anaerobic hydrocarbon degradation in historically contaminated groundwater. Environ. Sci. Technol. 2011, 45, 3407–3414. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 1999, 2, 317–322. [Google Scholar] [CrossRef]
- Luo, Y.; Tian, Y.; Huang, X.; Yan, C.; Hong, H.; Lin, G.; Zheng, T. Analysis of community structure of a microbial consortium capable of degrading benzo (a) pyrene by DGGE. Mar. Pollut. Bull. 2009, 58, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; van Pelt-Verkuil, E.; Gunst, Q.D.; Ruijter, J.M.; van den Hoff, M.J. Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol. Detect. Quantif. 2017, 14, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Feretzaki, M.; Lingner, J. A practical qPCR approach to detect TERRA, the elusive telomeric repeat-containing RNA. Methods 2017, 114, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Larionov, A.; Krause, A.; Miller, W. A standard curve based method for relative real time PCR data processing. BMC Bioinform. 2005, 6, 62. [Google Scholar] [CrossRef]
- Dhanasekaran, S.; Doherty, T.M.; Kenneth, J.; Group, T.T.S. Comparison of different standards for real-time PCR-based absolute quantification. J. Immunol. Methods 2010, 354, 34–39. [Google Scholar] [CrossRef]
- Wang, D.; Yamahara, K.M.; Cao, Y.; Boehm, A.B. Absolute quantification of Enterococcal 23S rRNA gene using digital PCR. Environ. Sci. Technol. 2016, 50, 3399–3408. [Google Scholar] [CrossRef]
- Wang, D.; Green, H.C.; Shanks, O.C.; Boehm, A.B. New performance metrics for quantitative polymerase chain reaction-based microbial source tracking methods. Environ. Sci. Technol. Lett. 2013, 1, 20–25. [Google Scholar] [CrossRef]
- Laurie, A.D.; Lloyd-Jones, G. Quantification of phnAc andnahAc in contaminated New Zealand soils by competitive PCR. Appl. Environ. Microbiol. 2000, 66, 1814–1817. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shimizu, K.; Sakharkar, M.K.; Utsumi, M.; Zhang, Z.; Sugiura, N. Comparative study for the effects of variable nutrient conditions on the biodegradation of microcystin-LR and concurrent dynamics in microcystin-degrading gene abundance. Bioresour. Technol. 2011, 102, 9509–9517. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.P.; Liu, X.; Mattes, T.E.; Cupples, A.M. Nocardioides, Sediminibacterium, Aquabacterium, Variovorax, and Pseudomonas linked to carbon uptake during aerobic vinyl chloride biodegradation. Environ. Sci. Pollut. Res. 2016, 23, 19062–19070. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Majumdar, N.; Banerjee, S.; Pallas, M.; Wessel, T.; Hegerich, P. Poisson plus quantification for digital PCR systems. Sci. Rep. 2017, 7, 9617. [Google Scholar] [CrossRef]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. dPCR: A technology review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef]
- Whale, A.S.; Cowen, S.; Foy, C.A.; Huggett, J.F. Methods for applying accurate digital PCR analysis on low copy DNA samples. PLoS ONE 2013, 8, e58177. [Google Scholar] [CrossRef] [PubMed]
- Morley, A.A. Digital PCR: A brief history. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef]
- Groth, S.F.D.S. The evaluation of limiting dilution assays. J. Immunol. Methods 1982, 49, R11–R23. [Google Scholar] [CrossRef]
- Jacobs, B.K.; Goetghebeur, E.; Clement, L. Impact of variance components on reliability of absolute quantification using digital PCR. BMC Bioinform. 2014, 15, 283. [Google Scholar] [CrossRef] [PubMed]
- Bizouarn, F. Introduction to digital PCR. Methods Mol. Biol. 2014, 1160, 27–41. [Google Scholar] [PubMed]
- Dube, S.; Qin, J.; Ramakrishnan, R. Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS ONE 2008, 3, e2876. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Cui, X.; Hu, J.; Li, Z.; Choi, J.R.; Yang, Q.; Lin, M.; Hui, L.Y.; Xu, F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2017, 90, 459–474. [Google Scholar] [CrossRef]
- Broeders, S.; Huber, I.; Grohmann, L.; Berben, G.; Taverniers, I.; Mazzara, M.; Roosens, N.; Morisset, D. Guidelines for validation of qualitative real-time PCR methods. Trends Food Sci. Technol. 2014, 37, 115–126. [Google Scholar] [CrossRef]
- Dragan, A.; Pavlovic, R.; McGivney, J.; Casas-Finet, J.; Bishop, E.; Strouse, R.; Schenerman, M.; Geddes, C. SYBR Green I: Fluorescence properties and interaction with DNA. J. Fluoresc. 2012, 22, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, X.; Su, F.; Wang, L.; Liu, C.; Duan, X.; Li, Z. Real-time fluorescence ligase chain reaction for sensitive detection of single nucleotide polymorphism based on fluorescence resonance energy transfer. Biosens. Bioelectron. 2015, 74, 705–710. [Google Scholar] [CrossRef]
- Martin-Sanchez, P.M.; Gorbushina, A.A.; Toepel, J. Quantification of microbial load in diesel storage tanks using culture-and qPCR-based approaches. Int. Biodeterior. Biodegrad. 2018, 126, 216–223. [Google Scholar] [CrossRef]
- Brown, M.V.; Fuhrman, J.A. Marine bacterial microdiversity as revealed by internal transcribed spacer analysis. Aquat. Microb. Ecol. 2005, 41, 15–23. [Google Scholar] [CrossRef]
- Pornwongthong, P.; Mulchandani, A.; Gedalanga, P.B.; Mahendra, S. Transition metals and organic ligands influence biodegradation of 1, 4-dioxane. Appl. Biochem. Biotechnol. 2014, 173, 291–306. [Google Scholar] [CrossRef]
- Bücker, F.; de Moura, T.M.; da Cunha, M.E.; de Quadros, P.D.; Beker, S.A.; Cazarolli, J.C.; Caramão, E.B.; Frazzon, A.P.G.; Bento, F.M. Evaluation of the deteriogenic microbial community using qPCR, n-alkanes and FAMEs biodegradation in diesel, biodiesel and blends (B5, B10, and B50) during storage. Fuel 2018, 233, 911–917. [Google Scholar] [CrossRef]
- Fayeulle, A.; Veignie, E.; Schroll, R.; Munch, J.C.; Rafin, C. PAH biodegradation by telluric saprotrophic fungi isolated from aged PAH-contaminated soils in mineral medium and historically contaminated soil microcosms. J. Soils Sediments 2019, 19, 3056–3067. [Google Scholar] [CrossRef]
- Richardson, E.L.; King, C.K.; Powell, S.M. The use of microbial gene abundance in the development of fuel remediation guidelines in polar soils. Integr. Environ. Assess. 2015, 11, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Krolicka, A.; Boccadoro, C.; Nilsen, M.M.; Demir-Hilton, E.; Birch, J.; Preston, C.; Scholin, C.; Baussant, T. Identification of microbial key-indicators of oil contamination at sea through tracking of oil biotransformation: An Arctic field and laboratory study. Sci. Total Environ. 2019, 696, 133715. [Google Scholar] [CrossRef]
- Ren, L.; Jia, Y.; Ruth, N.; Qiao, C.; Wang, J.; Zhao, B.; Yan, Y. Biodegradation of phthalic acid esters by a newly isolated Mycobacterium sp. YC-RL4 and the bioprocess with environmental samples. Environ. Sci. Pollut. Res. 2016, 23, 16609–16619. [Google Scholar] [CrossRef]
- Chang, H.-W.; Sung, Y.; Kim, K.-H.; Nam, Y.-D.; Roh, S.W.; Kim, M.-S.; Jeon, C.O.; Bae, J.-W. Development of microbial genome-probing microarrays using digital multiple displacement amplification of uncultivated microbial single cells. Environ. Sci. Technol. 2008, 42, 6058–6064. [Google Scholar] [CrossRef]
- Hoshino, T.; Inagaki, F. Molecular quantification of environmental DNA using microfluidics and digital PCR. Syst. Appl. Microbiol. 2012, 35, 390–395. [Google Scholar] [CrossRef]
- Kim, T.G.; Jeong, S.-Y.; Cho, K.-S. Comparison of droplet digital PCR and quantitative real-time PCR for examining population dynamics of bacteria in soil. Appl. Biochem. Biotechnol. 2014, 98, 6105–6113. [Google Scholar] [CrossRef]
- US EPA. Method 1611: Enterococci in Water by TaqMan® Quantitative Polymerase Chain Reaction (qPCR) Assay; US EPA: Washington, DC, USA, 2012.
- Bian, X.; Jing, F.; Li, G.; Fan, X.; Jia, C.; Zhou, H.; Jin, Q.; Zhao, J. A microfluidic droplet digital PCR for simultaneous detection of pathogenic Escherichia coli O157 and Listeria monocytogenes. Biosens. Bioelectron. 2015, 74, 770–777. [Google Scholar] [CrossRef]
- Cao, Y.; Raith, M.R.; Griffith, J.F. Droplet digital PCR for simultaneous quantification of general and human-associated fecal indicators for water quality assessment. Water Res. 2015, 70, 337–349. [Google Scholar] [CrossRef]
- Te, S.H.; Chen, E.Y.; Gin, K.Y.-H. Comparison of quantitative PCR and droplet digital PCR multiplex assays for two genera of bloom-forming cyanobacteria, Cylindrospermopsis and Microcystis. Appl. Environ. Microbiol. 2015, 81, 5203–5211. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, A.H.; Chang, W. Enhanced bioremediation of nutrient-amended, petroleum hydrocarbon-contaminated soils over a cold-climate winter: The rate and extent of hydrocarbon biodegradation and microbial response in a pilot-scale biopile subjected to natural seasonal freeze-thaw temperatures. Sci. Total Environ. 2018, 612, 903–913. [Google Scholar] [PubMed]
- Dong, L.; Meng, Y.; Wang, J.; Liu, Y. Evaluation of droplet digital PCR for characterizing plasmid reference material used for quantifying ammonia oxidizers and denitrifiers. Anal. Bioanal. Chem. 2014, 406, 1701–1712. [Google Scholar] [CrossRef]
- Ren, S.; Boo, C.; Guo, N.; Wang, S.; Elimelech, M.; Wang, Y. Photocatalytic reactive ultrafiltration membrane for removal of antibiotic resistant bacteria and antibiotic resistance genes from wastewater effluent. Environ. Sci. Technol. 2018, 52, 8666–8673. [Google Scholar] [CrossRef]
- Cavé, L.; Brothier, E.; Abrouk, D.; Bouda, P.S.; Hien, E.; Nazaret, S. Efficiency and sensitivity of the digital droplet PCR for the quantification of antibiotic resistance genes in soils and organic residues. Appl. Microbiol. Biotechnol. 2016, 100, 10597–10608. [Google Scholar] [CrossRef]
- Gao, M.; Qiu, T.; Sun, Y.; Wang, X. The abundance and diversity of antibiotic resistance genes in the atmospheric environment of composting plants. Environ. Int. 2018, 116, 229–238. [Google Scholar] [CrossRef]
- Liu, Z.; He, Z.; Huang, H.; Ran, X.; Oluwafunmilayo, A.O.; Lu, Z. pH stress-induced cooperation between Rhodococcus ruber YYL and Bacillus cereus MLY1 in biodegradation of tetrahydrofuran. Front. Microbiol. 2017, 8, 2297. [Google Scholar] [CrossRef]
- Shahi, A.; Aydin, S.; Ince, B.; Ince, O. Evaluation of microbial population and functional genes during the bioremediation of petroleum-contaminated soil as an effective monitoring approach. Ecotoxicol. Environ. Saf. 2016, 125, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Wang, Y.; Tian, L.; Chen, M.; Sun, J.; Li, L. Genetic bioaugmentation of activated sludge with dioxin-catabolic plasmids harbored by Rhodococcus sp. strain p52. Environ. Sci. Technol. 2018, 52, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.A.; Hong, J.W.; Quake, S.R.; Leadbetter, J.R. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science 2006, 314, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.; Dasgupta, K.; Xing, Y.P.; Hernandez, B.T.; Shao, M.; Rohozinski, D.; Kovak, E.; Lin, J.; de Oliveira, M.L.P.; Stover, E. Accurate measurement of transgene copy number in crop plants using droplet digital PCR. Plant J. 2017, 90, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Gedalanga, P.B.; Pornwongthong, P.; Mora, R.; Chiang, S.-Y.D.; Baldwin, B.; Ogles, D.; Mahendra, S. Identification of biomarker genes to predict biodegradation of 1, 4-dioxane. Appl. Environ. Microbiol. 2014, 80, 3209–3218. [Google Scholar] [CrossRef] [PubMed]
- Bælum, J.; Nicolaisen, M.H.; Holben, W.E.; Strobel, B.W.; Sørensen, J.; Jacobsen, C.S. Direct analysis of tfdA gene expression by indigenous bacteria in phenoxy acid amended agricultural soil. ISME J. 2008, 2, 677. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Adrian, L.; Kleinsteuber, S.; Andreesen, J.R.; Lechner, U. Transcription analysis of genes encoding homologues of reductive dehalogenases in Dehalococcoides sp. strain CBDB1 by using terminal restriction fragment length polymorphism and quantitative PCR. Appl. Environ. Microbiol. 2009, 75, 1876–1884. [Google Scholar] [CrossRef]
- Von Netzer, F.; Kuntze, K.; Vogt, C.; Richnow, H.H.; Boll, M.; Lueders, T. Functional gene markers for fumarate-adding and dearomatizing key enzymes in anaerobic aromatic hydrocarbon degradation in terrestrial environments. J. Mol. Microbiol. Biotechnol. 2016, 26, 180–194. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef]
- Kaitu’u-Lino, T.; Hastie, R.; Cannon, P.; Lee, S.; Stock, O.; Hannan, N.J.; Hiscock, R.; Tong, S. Stability of absolute copy number of housekeeping genes in preeclamptic and normal placentas, as measured by digital PCR. Placenta 2014, 35, 1106–1109. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, X.; Wang, X.; Qiu, T.; Gao, M.; Sun, Y.; Cheng, S.; Zhang, Q. Bioaugmentation with Diaphorobacter polyhydroxybutyrativorans to enhance nitrate removal in a poly (3-hydroxybutyrate-co-3-hydroxyvalerate)-supported denitrification reactor. Bioresour. Technol. 2018, 263, 499–507. [Google Scholar] [CrossRef]
- Segawa, T.; Ishii, S.; Ohte, N.; Akiyoshi, A.; Yamada, A.; Maruyama, F.; Li, Z.; Hongoh, Y.; Takeuchi, N. The nitrogen cycle in cryoconites: Naturally occurring nitrification-denitrification granules on a glacier. Environ. Microbiol. 2014, 16, 3250–3262. [Google Scholar] [CrossRef]
- Basu, A.S. Digital assays part I: Partitioning statistics and digital PCR. SLAS Technol. 2017, 22, 369–386. [Google Scholar] [CrossRef]
- Sedlak, R.H.; Nguyen, T.; Palileo, I.; Jerome, K.R.; Kuypers, J. Superiority of digital reverse transcription-PCR (RT-PCR) over real-time RT-PCR for quantitation of highly divergent human rhinoviruses. J. Clin. Microbiol. 2017, 55, 442–449. [Google Scholar] [CrossRef]
- Suslov, O.; Steindler, D.A. PCR inhibition by reverse transcriptase leads to an overestimation of amplification efficiency. Nucleic Acids Res. 2005, 33, e181. [Google Scholar] [CrossRef]
- Zmienko, A.; Samelak-Czajka, A.; Goralski, M.; Sobieszczuk-Nowicka, E.; Kozlowski, P.; Figlerowicz, M. Selection of reference genes for qPCR-and ddPCR-based analyses of gene expression in senescing barley leaves. PLoS ONE 2015, 10, e0118226. [Google Scholar] [CrossRef]
- Vasina, D.V.; Moiseenko, K.V.; Fedorova, T.V.; Tyazhelova, T.V. Lignin-degrading peroxidases in white-rot fungus Trametes hirsuta 072. Absolute expression quantification of full multigene family. PLoS ONE 2017, 12, e0173813. [Google Scholar] [CrossRef] [PubMed]
- Green, H.C.; Field, K.G. Sensitive detection of sample interference in environmental qPCR. Water Res. 2012, 46, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhang, H.; Miranda, L.; Lin, S. Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: Microalgal pcna as the model gene. PLoS ONE 2010, 5, e9545. [Google Scholar] [CrossRef] [PubMed]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2011, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Case, R.J.; Boucher, Y.; Dahllöf, I.; Holmström, C.; Doolittle, W.F.; Kjelleberg, S. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl. Environ. Microbiol. 2007, 73, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Dube, S.; Mir, A.; Qin, J.; Sun, G.; Ramakrishnan, R.; Jones, R.C.; Livak, K.J. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods 2010, 50, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. Quantitative analysis of copy number variants based on real-time LightCycler PCR. Curr. Protoc. Hum. Genet. 2014, 80, 7–21. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).