Formal [3+2] Cycloaddition Reactions of Electron-Rich Aryl Epoxides with Alkenes under Lewis Acid Catalysis Affording Tetrasubstituted Tetrahydrofurans


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
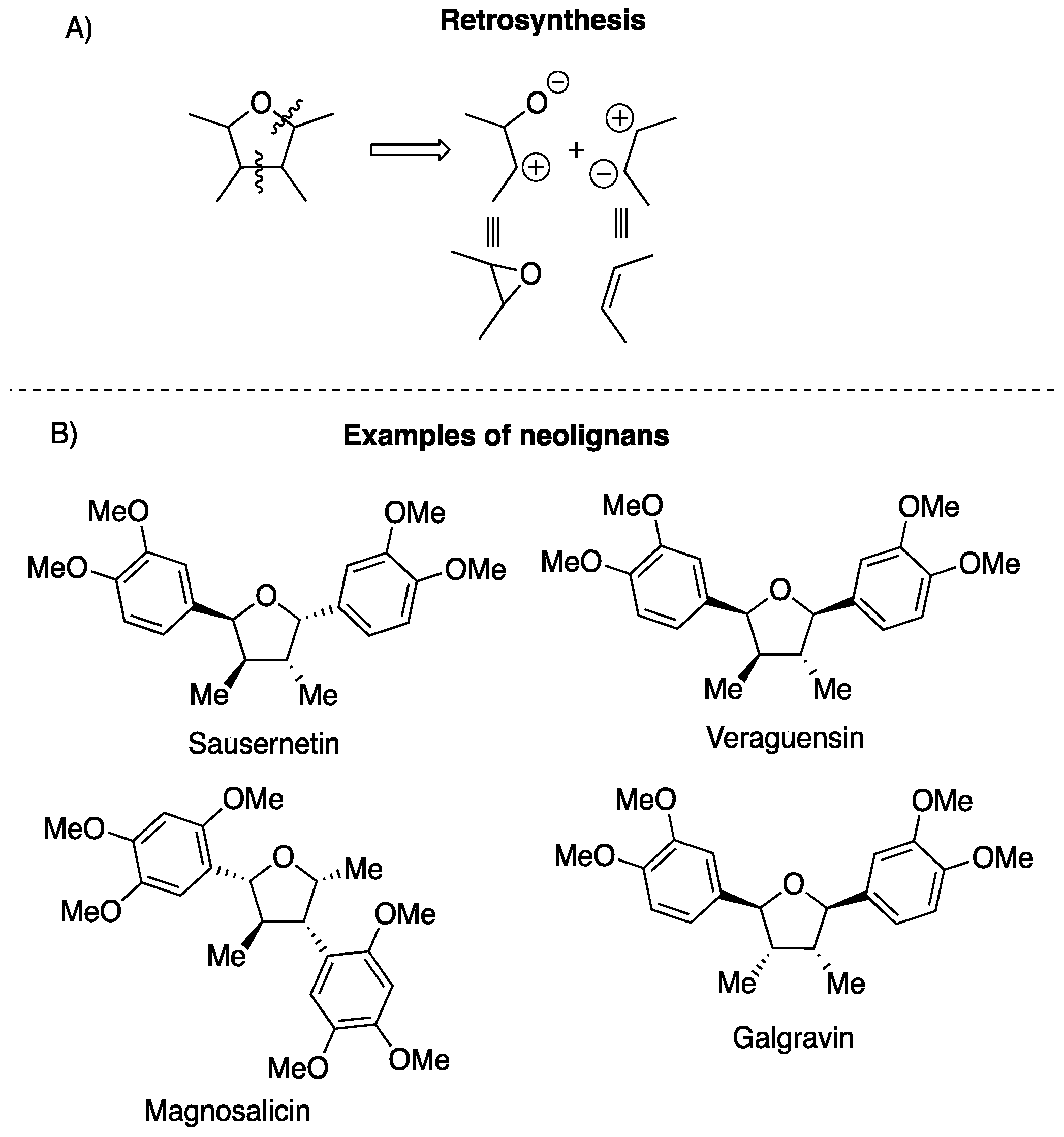
1. Introduction
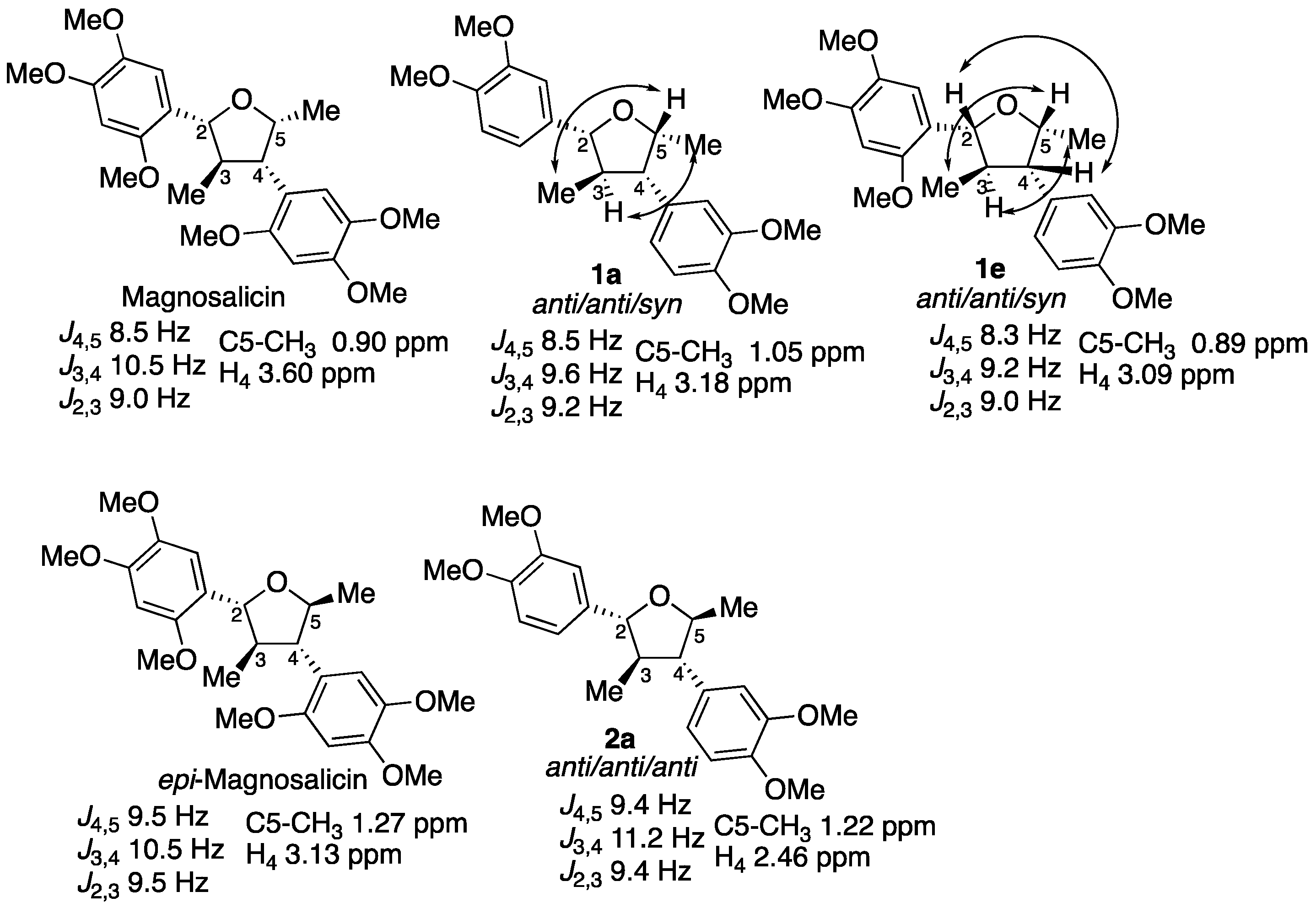
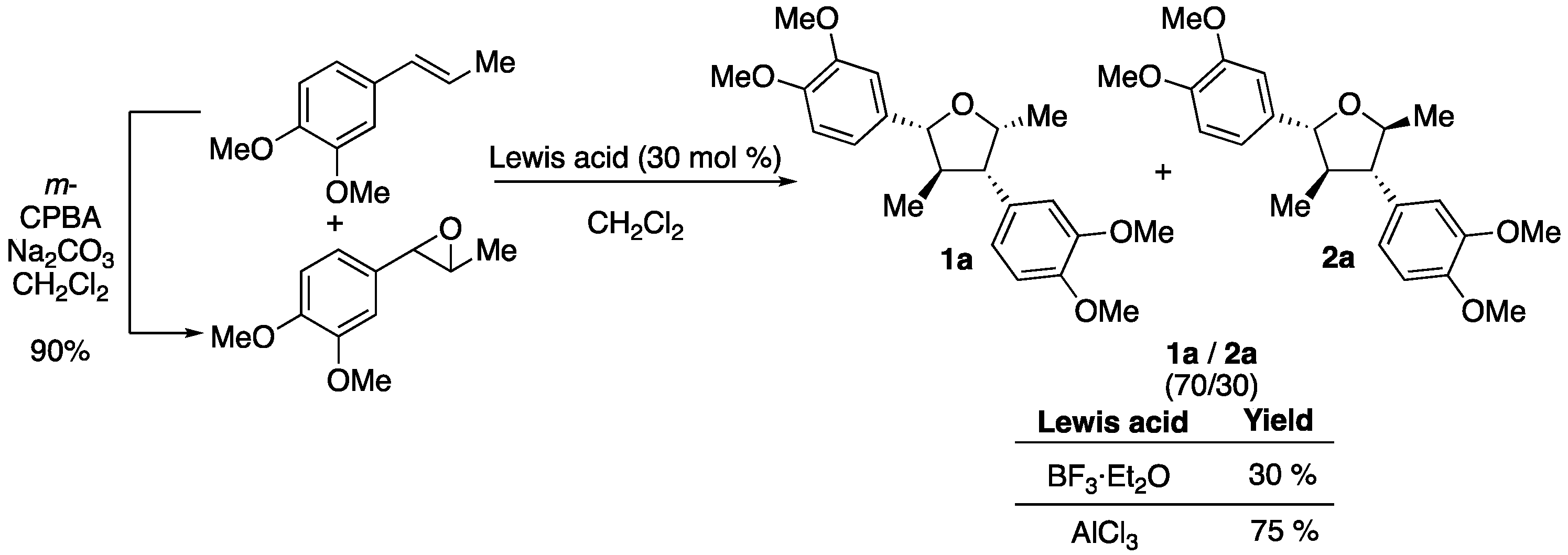
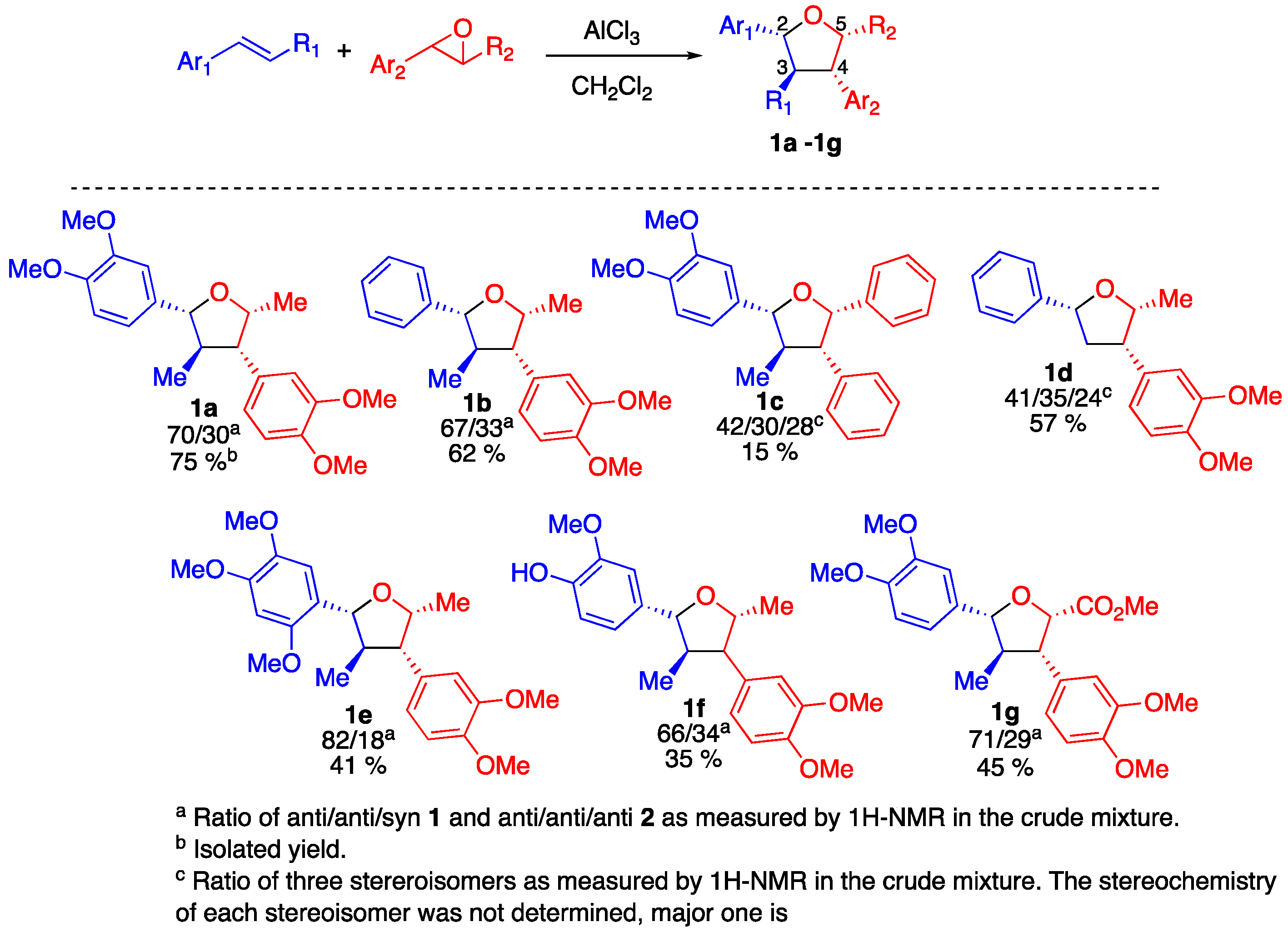
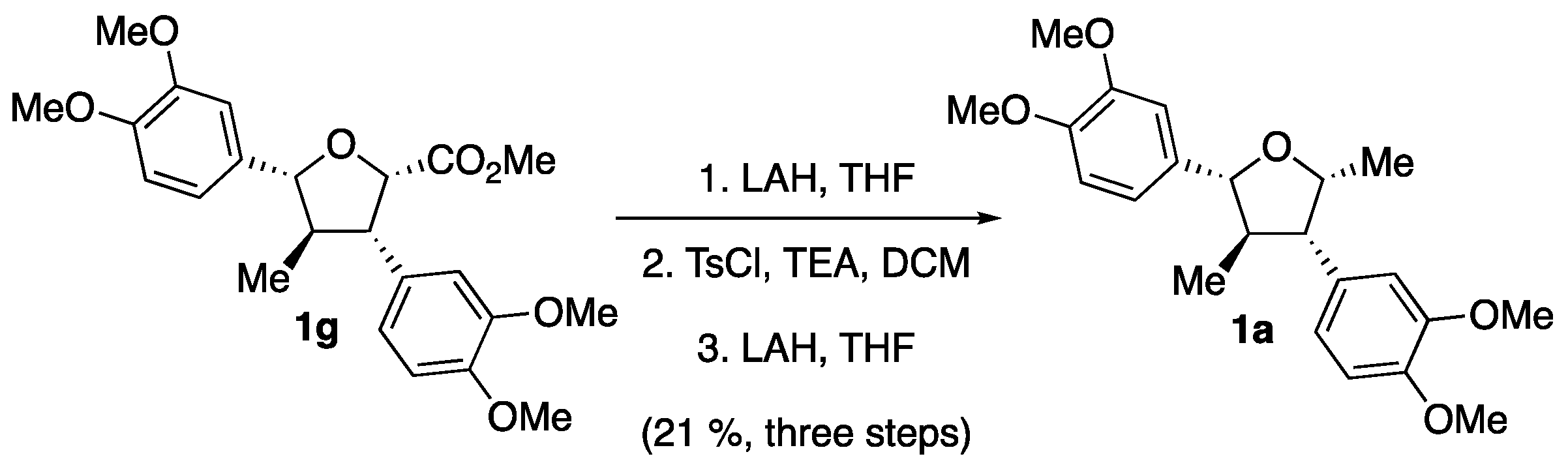
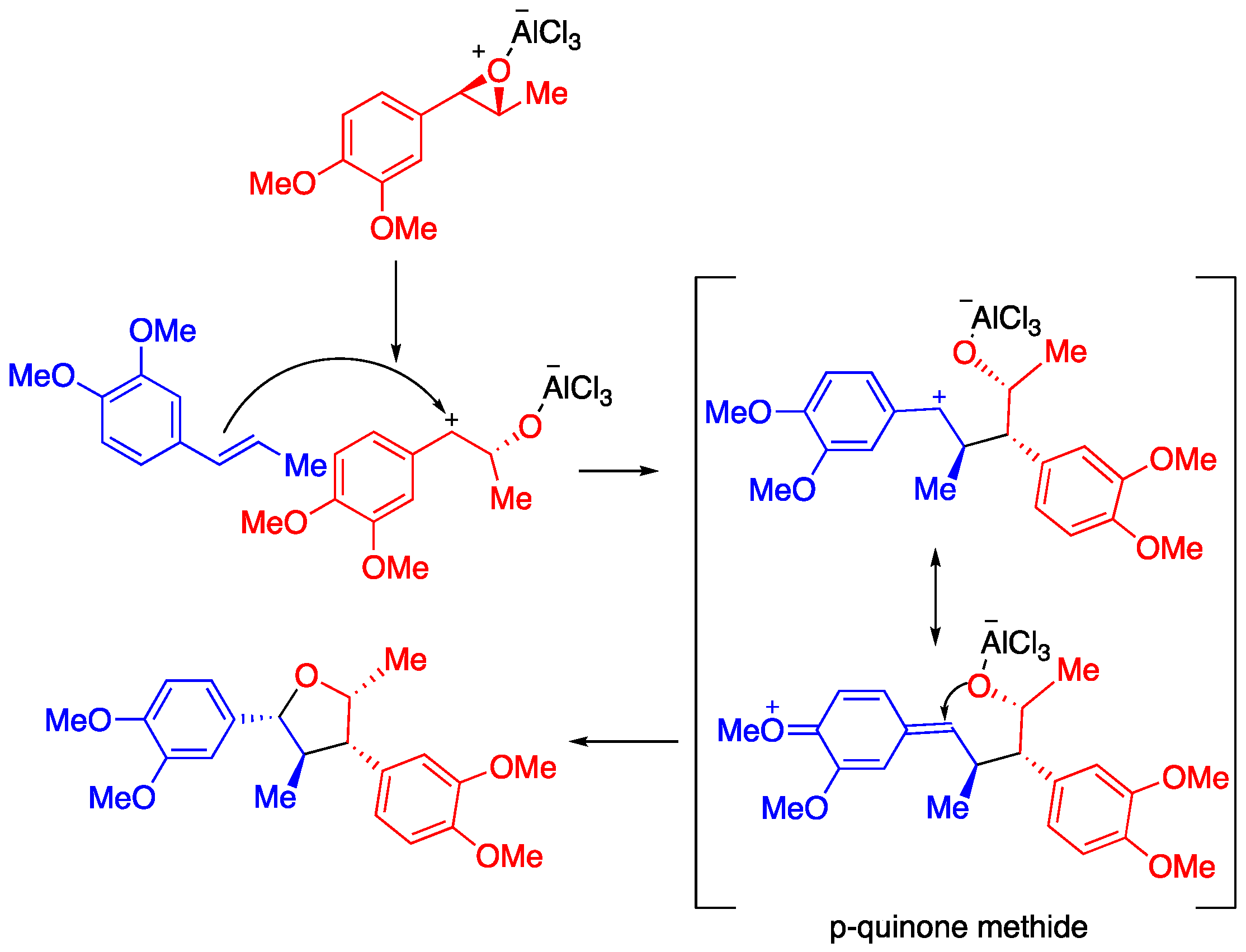
2. Results
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Preparation of Epoxides 2-(3,4-dimethoxyphenyl)-3-methyloxirane and 2,3-diphenyloxirane
4.3. Preparation of Methyl 3-(3,4-dimethoxyphenyl)oxirane-2-carboxylate
4.4. General Experimental Procedure for the Preparation of Tetrahydrofurans
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saleem, M.; Kim, H.J.; Ali, M.S.; Lee, Y.S. An update on bioactive plant lignans. Nat. Prod. Rep. 2005, 22, 696–716. [Google Scholar] [CrossRef] [PubMed]
- Harmatha, J.; Dinan, L. Biological activities of lignans and stilbenoids associated with plant-insect chemical interactions. Phytochem. Rev. 2003, 2, 321–330. [Google Scholar] [CrossRef]
- Ward, R.S. Lignans neolignans, and related compounds. Nat. Prod. Rep. 1995, 12, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Macías-Villamizar, V.; Cuca-Suárez, L.; González, F.V.; Rodríguez, S. Lignoids Isolated from Nectandra turbacensis (Kunth) Nees (Lauraceae). Rec. Nat. Prod. 2016, 10, 654–658. [Google Scholar]
- Fang, X.; Hu, X. Advances in the Synthesis of Lignan Natural Products. Molecules 2018, 23, 3385. [Google Scholar] [CrossRef] [PubMed]
- Shizuri, Y. Total Synthesis of Lignans and Neolignans. J. Synth. Org. Chem. 1984, 42, 889–899. [Google Scholar] [CrossRef]
- Angle, S.R.; Choi, I. Regioselective and stereoselective synthesis of tetrahydrofurans from a functionalized allylic silane and an aldehyde via formal [3+2]-cycloaddition reaction. Tetrahedron Lett. 2008, 49, 6245–6249. [Google Scholar] [CrossRef]
- Liu, J.; Li, M.; Qu, B.; Lu, L.; Xiao, W. A photoinduced Wolff rearrangement/Pd-catalyzed [3+2] cycloaddition sequence: An unexpected route to tetrhydrofurans. Chem. Comm. 2019, 55, 2031–2034. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Huang, Z.; Murhade, G.M. Catalytic palladium-oxyallyl cycloaddition. Science 2018, 362, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.; Rodríguez, S.; González, F.V. Regioselective Ring Opening and Isomerization Reactions of 3,4-Epoxyesters Catalyzed by Boron Trifluoride. Org. Lett. 2011, 13, 3856–3859. [Google Scholar] [CrossRef] [PubMed]
- Shuler, W.G.; Combee, L.A.; Falk, I.D.; Hilinski, M.K. Intermolecular Electrophilic Addition of Epoxides to Alkenes: [3+2] Cycloadditions Catalyzed by Lewis Acids. Eur. J. Org. Chem. 2016, 20, 3335–3338. [Google Scholar] [CrossRef]
- Vilotijevic, I.; Jamison, T.F. Epoxide-Opening Cascades in the Synthesis of Polycyclic Polyether Natural Products. Angew. Chem. Int. Ed. Engl. 2009, 48, 5250–5281. [Google Scholar] [CrossRef] [PubMed]
- Tsuruga, T.; Ebizuka, Y.; Nakajima, J.; Chun, Y.; Noguchi, H.; Iitaka, Y.; Sankawa, U.; Seto, H. Isolation of a new neolignan, magnosalicin, from magnolia salicifolia. Tetrahedron Lett. 1984, 25, 4129–4132. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–1g are available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macías-Villamizar, V.E.; Cuca-Suárez, L.; Rodríguez, S.; González, F.V. Formal [3+2] Cycloaddition Reactions of Electron-Rich Aryl Epoxides with Alkenes under Lewis Acid Catalysis Affording Tetrasubstituted Tetrahydrofurans. Molecules 2020, 25, 692. https://doi.org/10.3390/molecules25030692
Macías-Villamizar VE, Cuca-Suárez L, Rodríguez S, González FV. Formal [3+2] Cycloaddition Reactions of Electron-Rich Aryl Epoxides with Alkenes under Lewis Acid Catalysis Affording Tetrasubstituted Tetrahydrofurans. Molecules. 2020; 25(3):692. https://doi.org/10.3390/molecules25030692
Chicago/Turabian StyleMacías-Villamizar, Víctor E., Luís Cuca-Suárez, Santiago Rodríguez, and Florenci V. González. 2020. "Formal [3+2] Cycloaddition Reactions of Electron-Rich Aryl Epoxides with Alkenes under Lewis Acid Catalysis Affording Tetrasubstituted Tetrahydrofurans" Molecules 25, no. 3: 692. https://doi.org/10.3390/molecules25030692
APA StyleMacías-Villamizar, V. E., Cuca-Suárez, L., Rodríguez, S., & González, F. V. (2020). Formal [3+2] Cycloaddition Reactions of Electron-Rich Aryl Epoxides with Alkenes under Lewis Acid Catalysis Affording Tetrasubstituted Tetrahydrofurans. Molecules, 25(3), 692. https://doi.org/10.3390/molecules25030692