
Organocatalytic Asymmetric Aldol Reaction of Arylglyoxals and Hydroxyacetone: Enantioselective Synthesis of 2,3-Dihydroxy-1,4-diones


Abstract
1. Introduction
2. Results and Discussion
2.1. Optimization of Reaction Conditions
2.2. Substrate Scope Study
2.3. Scale-Up Experiment and Crystal Structure of Compound 4j
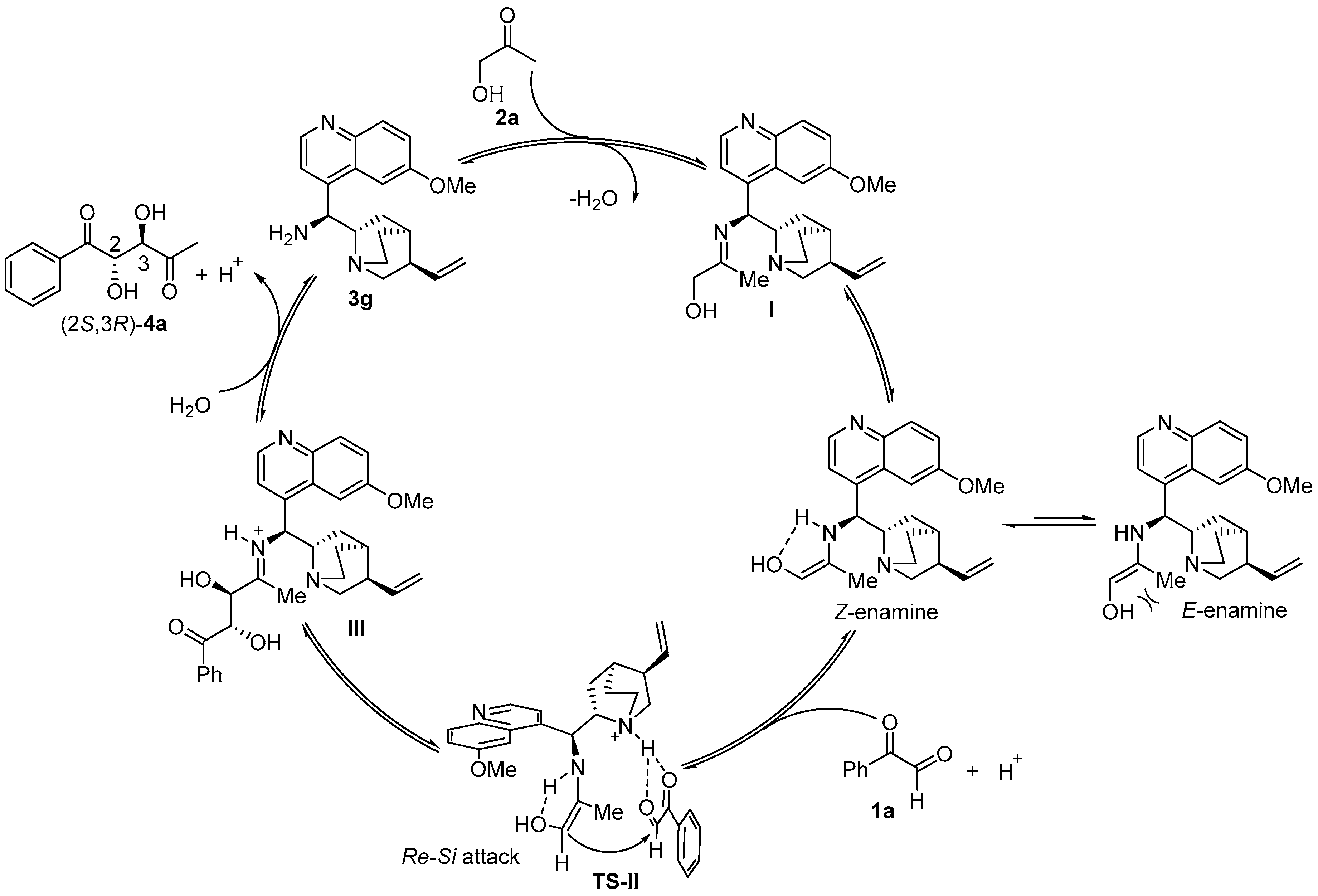
2.4. Plausible Reaction Mechanism and Transition States
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of 2,3-Dihydroxy-1,4-dione Diketone Products 4a–4p
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mukaiyama, T. Directed aldol reaction. Org. React. 1982, 28, 203–331. [Google Scholar]
- Mahrwald, R. (Ed.) Modern Aldol Reactions; Wiley-VCH: Weinheim, Germany, 2004; Volume 1–2. [Google Scholar]
- Palomo, C.; Oiarbide, M.; García, J.M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Liu, X.; Zheng, K.; Li, W.; Hu, X.; Lin, L.; Feng, X. Catalytic asymmetric synthesis of 3-(α-Hydroxy-β-carbonyl) oxindoles by a ScIII-catalyzed direct aldol-type reaction. Chem. Eur. J. 2010, 16, 3736–3742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zheng, K.; Yang, Y.; Shi, J.; Lin, L.; Liu, X.; Feng, X. Asymmetric mukaiyama aldol reaction catalyzed by C2-symmetric N,N′-dioxide-Ni(II) complex. Synlett 2011, 7, 903–906. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yasui, Y.; Kojima, M.; Kawamura, T.; Ishikawa, H. Diarylprolinol in an asymmetric aldol reaction of an α-alkyl-α-oxo aldehyde as an electrophile. Chem. Commun. 2012, 48, 4570. [Google Scholar] [CrossRef]
- Alberg, D.G.; Poulsen, T.B.; Bertelsen, S.; Christensen, K.L.; Birkler, R.D.; Johannsen, M.; Jørgensen, K.A. Organocatalysis with endogenous compounds: Towards novel non-enzymatic reactions. Bioorg. Med. Chem. Lett. 2009, 19, 3888. [Google Scholar] [CrossRef]
- Hayashi, Y.; Kojima, M. Asymmetric aldol reaction of glyoxal catalyzed by diarylprolinol. ChemCatChem 2013, 5, 2883. [Google Scholar] [CrossRef]
- Moles, F.J.N.; Guillena, G.; Nájera, C. Aqueous enantioselective aldol reaction of methyl- and phenylglyoxal organocatalyzed by N-Tosyl-(Sa)-binam-l-prolinamide. Synlett 2015, 26, 656–660. [Google Scholar] [CrossRef]
- Konda, S.; Guo, Q.-S.; Abe, M.; Huang, H.; Arman, H.; Zhao, J.C.-G. Organocatalyzed Asymmetric Aldol Reactions of Ketones and β,γ-Unsaturated α-Ketoesters and Phenylglyoxal Hydrates. J. Org. Chem. 2015, 80, 806–815. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Yang, X.-C.; Lu, H.-J.; Wang, M.-C. Enantioselective direct synthesis of syn- and anti-α,β-dihydroxy γ-keto esters using a dinuclear zinc-AzePhenol complex. Eur. J. Org. Chem. 2018, 785–793. [Google Scholar] [CrossRef]
- Ren, L.; Lian, X.-L.; Gong, L.-Z. Brønsted acid/Rhodium(II) cooperative catalytic asymmetric three-component aldol-type reaction for the synthesis of 3-amino oxindoles. Chem. Eur. J. 2013, 19, 3315–3318. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Yamaguchi, Y.; Tanaka, Y.; Maruoka, K. syn-Selective and enantioselective direct cross-aldol reactions between aldehydes catalyzed by an axially chiral amino sulfonamide. Angew. Chem. Int. Ed. 2007, 46, 1738–1740. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Yamaguchi, Y.; Maruoka, K. A designer axially chiral amino sulfonamide as an efficient organocatalyst for direct asymmetric anti-selective mannich reactions and syn-selective cross-aldol reactions. Chem. Eur. J. 2009, 15, 6678–6687. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, F.; Dong, S.; Liu, X.; Feng, X. Asymmetric bisprolinamide-catalyzed cross-aldol reaction of aldehydes. Synlett. 2008, 1, 73. [Google Scholar]
- Guo, Q.; Bhanushali, M.; Zhao, C.-G. Quinidine thiourea-catalyzed aldol reaction of unactivated ketones: Highly enantioselective synthesis of 3-alkyl-3-hydroxyindolin-2-ones. Angew. Chem. Int. Ed. 2010, 49, 9460–9464. [Google Scholar] [CrossRef]
- Guang, J.; Guo, Q.; Zhao, J.C.-G. Acetylphosphonate as a surrogate of acetate or acetamide in organocatalyzed enantioselective aldol reactions. Org. Lett. 2012, 14, 3174–3177. [Google Scholar] [CrossRef]
- Gioia, C.; Ricci, A.; Bernardi, L.; Bourahla, K.; Tanchoux, N.; Robitzer, M.; Quignard, F. Chitosan aerogel beads as a heterogeneous organocatalyst for the asymmetric aldol reaction in the presence of water: An assessment of the effect of additives. Eur. J. Org. Chem. 2013, 588–594. [Google Scholar] [CrossRef]
- Quintard, A.; Rodriguez, J. Didecarboxylative Iron-catalyzed bidirectional aldolization towards diversity-oriented ketodiol synthesis. Chem. Eur. J. 2015, 21, 14717–14722. [Google Scholar] [CrossRef]
- Gijsen, H.J.M.; Qiao, L.; Fitz, W.; Wong, C.-H. Recent advances in the chemoenzymatic synthesis of carbohydrates and carbohydrate mimetics. Chem. Rev. 1996, 96, 443–473. [Google Scholar] [CrossRef]
- Enders, D.; Voith, M.; Lenzen, A. The dihydroxyacetone unit—A versatile C3 building block in organic synthesis. Angew. Chem. Int. Ed. 2005, 44, 1304–1325. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ramasastry, S.S.V.; Zhang, H.; Tanaka, F.; Barbas, C.F., III. Direct catalytic asymmetric synthesis of anti-1,2-Amino alcohols and syn-1,2-diols through organocatalytic anti-mannich and syn-aldol reactions. J. Am. Chem. Soc. 2007, 129, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Xu, H.; Li, J.; Zhang, L.; Cheng, J.-P. A simple primary-tertiary diamine-Brønsted acid catalyst for asymmetric direct aldol reactions of linear aliphatic ketones. J. Am. Chem. Soc. 2007, 129, 3074–3075. [Google Scholar] [CrossRef]
- Crotti, S.; Iorio, N.D.; Artusi, C.; Mazzanti, A.; Righi, P.; Bencivenni, G. Direct access to alkylideneoxindoles via axially enantioselective knoevenagel condensation. Org. Lett. 2019, 21, 3013–3017. [Google Scholar] [CrossRef]
- CCDC 1974588 (4j) Contains the Supplementary Crystallographic Data for This Paper. Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 29 December 2019).
- Czarnecki, P.; Plutecka, A.; Gawroński, J.; Kacprzak, K. Simple and practical direct asymmetric aldol reaction of hydroxyacetone catalyzed by 9-amino Cinchona alkaloid tartrates. Green Chem. 2011, 13, 1280–1287. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Solvent | Time (h) | Yield b (%) | dr c(anti:syn) | ee c (%) |
|---|---|---|---|---|---|---|
| 1 d,e | L-Pro | DMF | 24 | n.r. | n.d. | n.d. |
| 2 d,e | L-Ser | DMF | 60 | n.r. | n.d. | n.d. |
| 3 d,e | L-Thr | DMF | 60 | n.r. | n.d. | n.d. |
| 4 | 3a | CHCl3 | 143 | trace | n.d. | n.d. |
| 5 | 3b | CHCl3 | 234 | trace | n.d. | n.d. |
| 6 | 3c | CHCl3 | 16 | 49 | 80:20 | 94 |
| 7 | 3d | CHCl3 | 13 | 85 | 80:20 | 94 |
| 8 | 3e | CHCl3 | 12 | 82 | 86:14 | 92 |
| 9 | 3f | CHCl3 | 80 | 62 | 57:43 | −57 |
| 10 e | 3g | CHCl3 | 13 | 84 | 89:11 | 94 |
| 11 e,f | 3g | CHCl3 | 84 | 74 | 75:25 | 92 |
| 12 e,g | 3g | CHCl3 | 15 | 75 | 86:14 | 90 |
| 13 e,h | 3g | CHCl3 | 46 | 89 | 93:7 | 96 |
| 14 e,h | 3h | CHCl3 | 60 | 85 | 87:13 | 89 |
| 15 h | 3g | CHCl3 | 60 | 83 | 93:7 | 96 |
| 16 h,i | 3g | CHCl3 | 108 | 59 | 93:7 | 96 |
| 17 h | 3g | CH2Cl2 | 60 | 17 | 87:13 | 95 |
| 18 h | 3g | DCE | 60 | 30 | 87:13 | 94 |
| 19 h | 3g | PhCH3 | 60 | 48 | 87:13 | 88 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.-H.; Zhang, Y.-Z.; Wu, Z.-L.; Cai, T.; Wen, W.; Guo, Q.-X. Organocatalytic Asymmetric Aldol Reaction of Arylglyoxals and Hydroxyacetone: Enantioselective Synthesis of 2,3-Dihydroxy-1,4-diones. Molecules 2020, 25, 648. https://doi.org/10.3390/molecules25030648
Zhou Y-H, Zhang Y-Z, Wu Z-L, Cai T, Wen W, Guo Q-X. Organocatalytic Asymmetric Aldol Reaction of Arylglyoxals and Hydroxyacetone: Enantioselective Synthesis of 2,3-Dihydroxy-1,4-diones. Molecules. 2020; 25(3):648. https://doi.org/10.3390/molecules25030648
Chicago/Turabian StyleZhou, Yu-Hao, Yu-Zu Zhang, Zhu-Lian Wu, Tian Cai, Wei Wen, and Qi-Xiang Guo. 2020. "Organocatalytic Asymmetric Aldol Reaction of Arylglyoxals and Hydroxyacetone: Enantioselective Synthesis of 2,3-Dihydroxy-1,4-diones" Molecules 25, no. 3: 648. https://doi.org/10.3390/molecules25030648
APA StyleZhou, Y.-H., Zhang, Y.-Z., Wu, Z.-L., Cai, T., Wen, W., & Guo, Q.-X. (2020). Organocatalytic Asymmetric Aldol Reaction of Arylglyoxals and Hydroxyacetone: Enantioselective Synthesis of 2,3-Dihydroxy-1,4-diones. Molecules, 25(3), 648. https://doi.org/10.3390/molecules25030648