White Matter and Neuroprotection in Alzheimer’s Dementia


 , , ,
, , ,
Abstract
1. Introduction
2. White Matter Plasticity in the Adult Brain, Ageing, Cognitive Decline, and Alzheimer’s Dementia: Focus on Oligodendrocyte Precursor Cells (OPC)
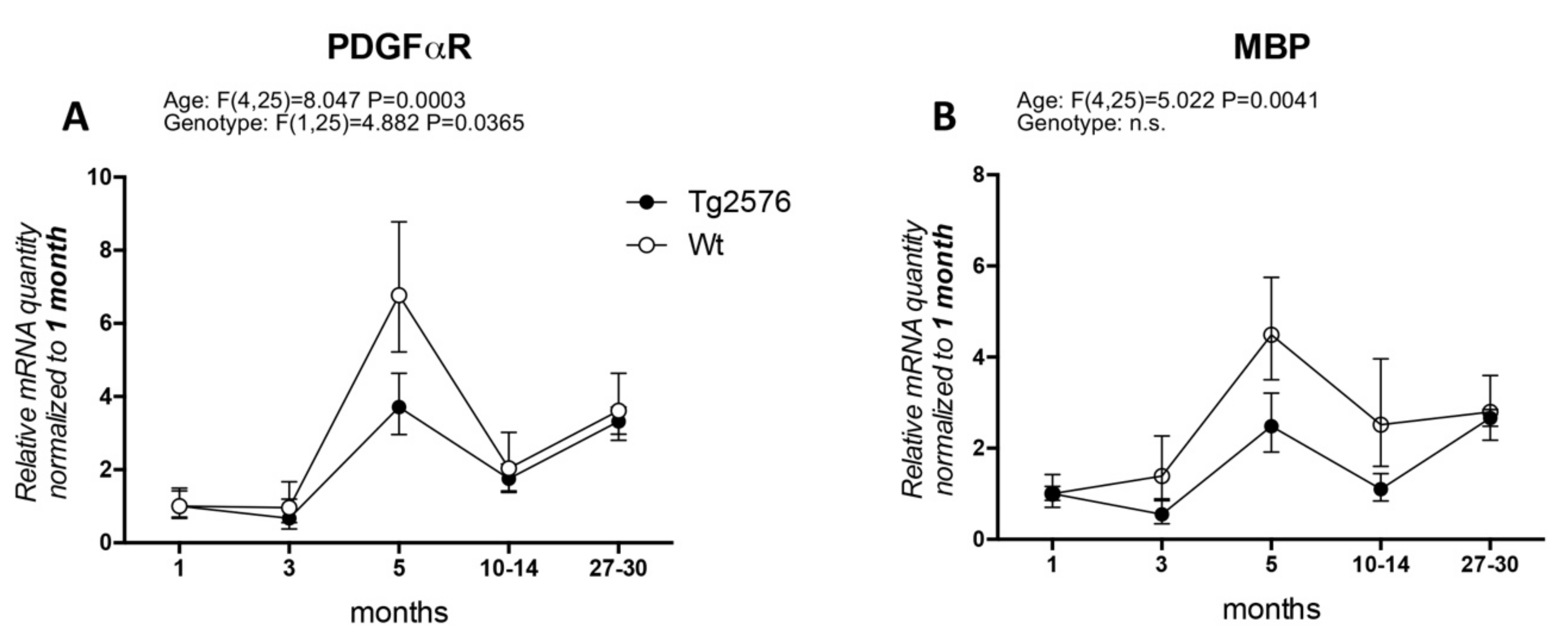
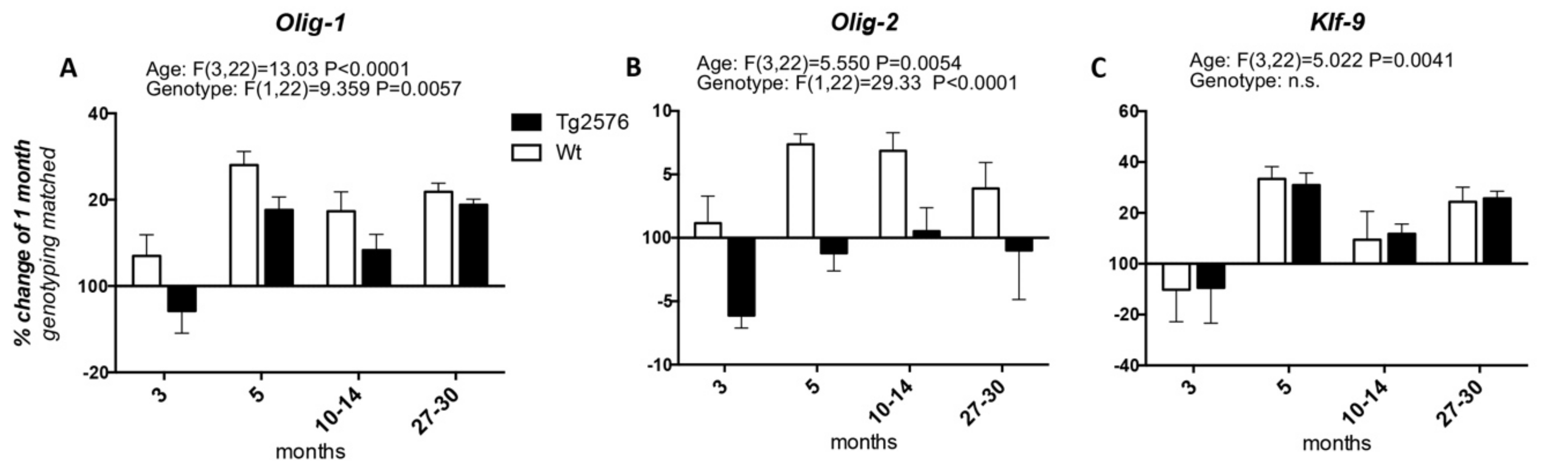
3. OPCs and Mature Oligodendrocytes in Alzheimer’s Disease and Animal Models
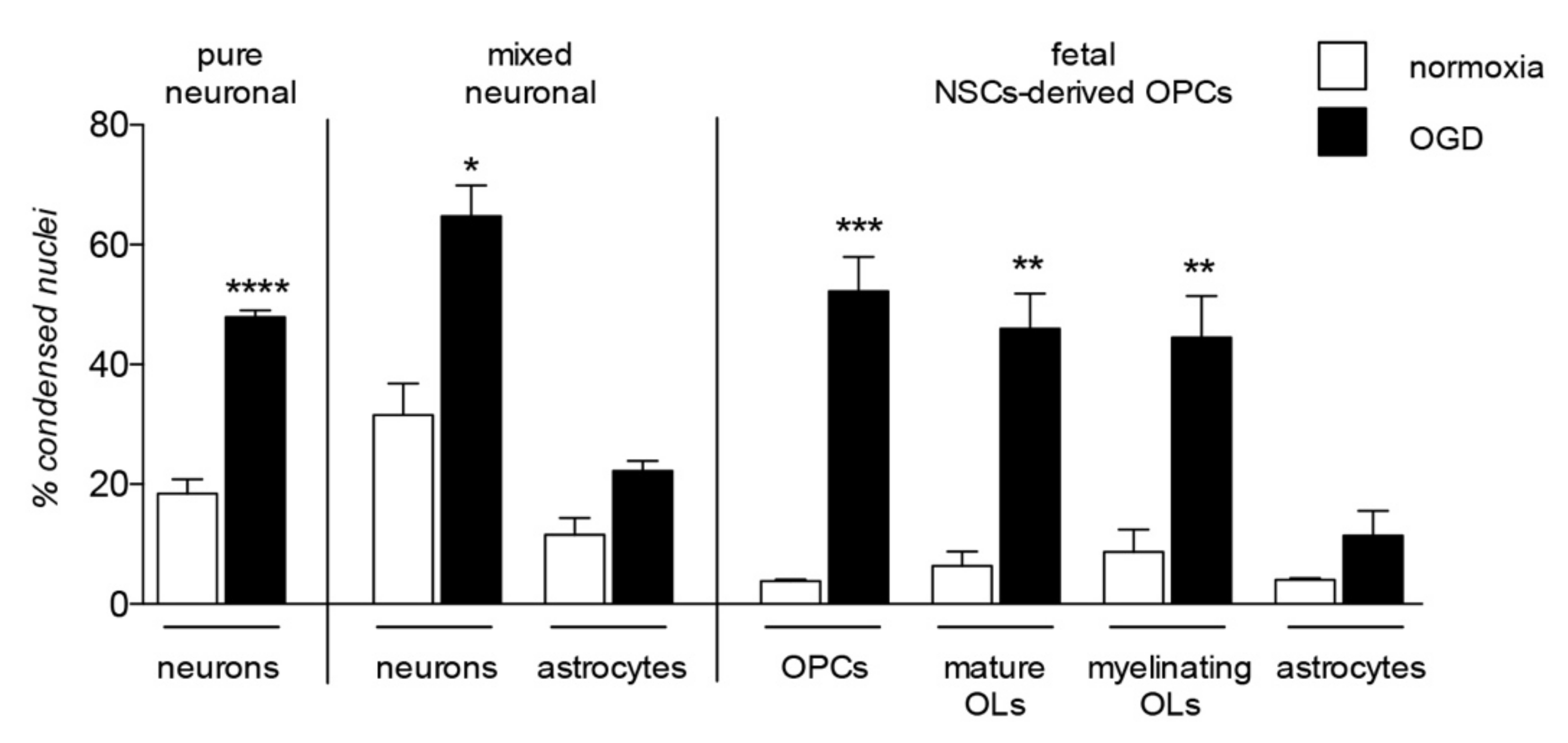
4. OPC Vulnerability in Neurodegenerative Diseases
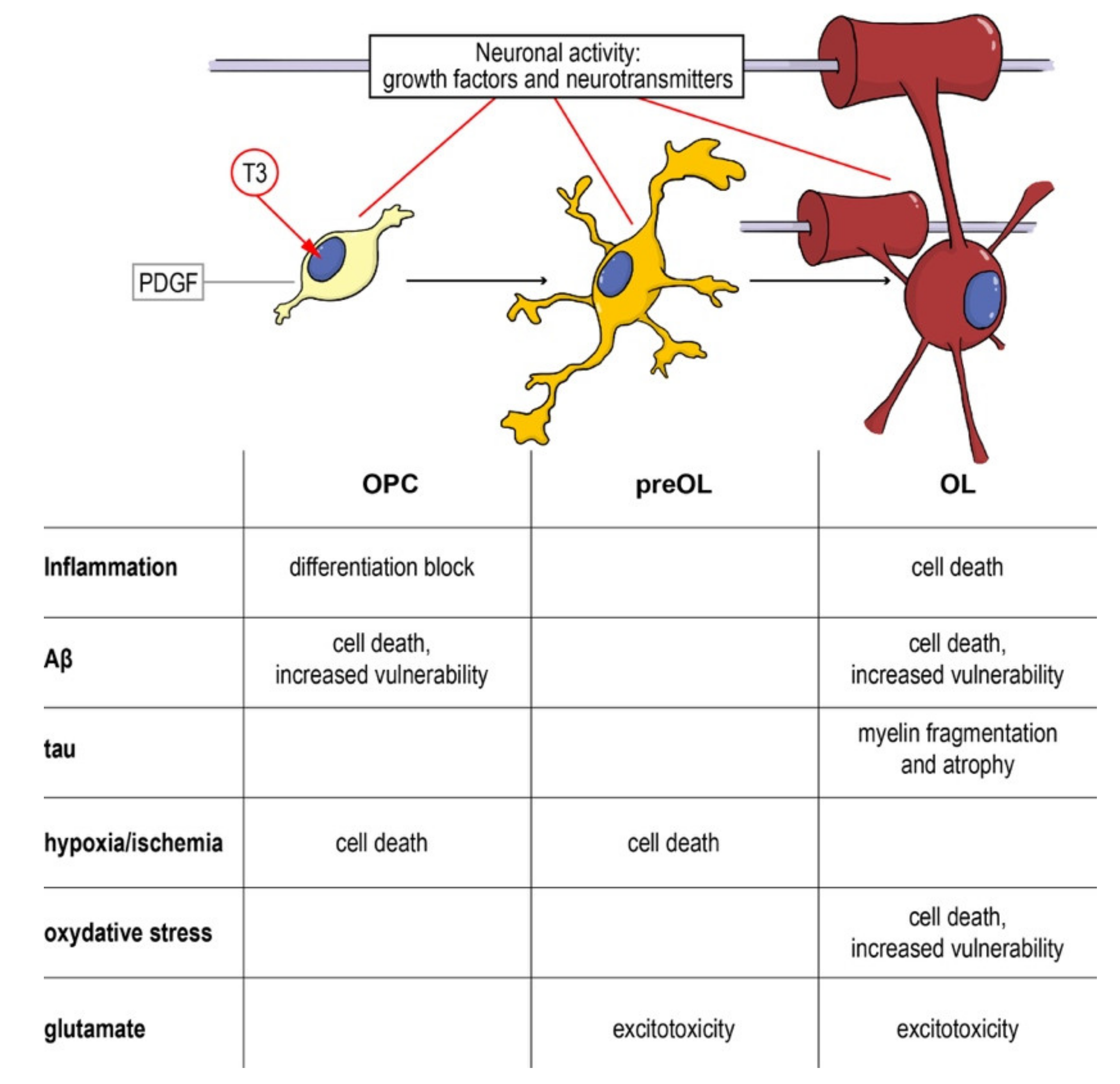
5. OPCs as Target for Neuroprotection
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix A1. Materials and Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NCBI Acc. N. | Primer Sequences |
|---|---|---|
| GAPDH | NM_17701 | F: 5′-ggcaagttcaatggcacagtcaag-3′ R: 5′-acatactcagcaccagcatcacc-3′ |
| Klf-9 | NM_010638.4 | F: 5′-agtggcttcgaaggggaaac-3′ R: 5′-tccgagcgcgagaacttttt-3′ |
| MBP | NM_001025251 | F: 5′-gcctgtccctcagcagattt-3′ R: 5′-gtcgtaggcccccttgaatc-3′ |
| Olig-1 | NM_ 016968 | F: 5′-ccgccccagatgtactatgc-3′ R: 5′-aacccaccagctcatacagc-3′ |
| Olig-2 | NM_016967.2 | F: 5′-gcttagatcatccctggggc-3′ R: 5′-agatcatcgggttctgggga-3′ |
| PDGFαR | NM_001083316.2 | F: 5′-cggaacctcagagagaatcgg-3′ R: 5′-tccccatagctcctgagacc-3′ |
References
- Lüders, E.; Steinmetz, H.; Jäncke, L. Brain size and grey matter volume in the healthy human brain. Neuroreport 2002, 13, 2371–2374. [Google Scholar] [CrossRef]
- Ozgen, H.; Baron, W.; Hoekstra, D.; Kahya, N. Oligodendroglial membrane dynamics in relation to myelin biogenesis. Cell. Mol. Life Sci. 2016, 73, 3291–3310. [Google Scholar] [CrossRef]
- Wang, S.; Young, K.M. White matter plasticity in adulthood. Neuroscience 2014, 276, 148–160. [Google Scholar] [CrossRef]
- Swire, M.; Ffrench-Constant, C. Seeing Is Believing: Myelin Dynamics in the Adult CNS. Neuron 2018, 98, 684–686. [Google Scholar] [CrossRef]
- Kato, D.; Wake, H. Activity-Dependent Myelination. Adv. Exp. Med. Biol. 2019, 1190, 43–51. [Google Scholar] [PubMed]
- Zhang, K.; Sejnowski, T.J. A universal scaling law between gray matter and white matter of cerebral cortex. Proc. Natl. Acad. Sci. USA 2000, 97, 5621–5626. [Google Scholar] [CrossRef] [PubMed]
- Filley, C.M.; Fields, R.D. White matter and cognition: Making the connection. J. Neurophysiol. 2016, 116, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Nave, K.-A. Myelin dynamics: protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef]
- Stassart, R.M.; Möbius, W.; Nave, K.-A.; Edgar, J.M. The Axon-Myelin Unit in Development and Degenerative Disease. Front. Neurosci. 2018, 12, 467. [Google Scholar] [CrossRef]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and axonal support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the central nervous system: Structure, function, and pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.H.; Chambers, C.; Franklin, R.J.M. Remyelination: The true regeneration of the central nervous system. J. Comp. Pathol. 2013, 149, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Trotter, J.; Karram, K.; Nishiyama, A. NG2 cells: Properties, progeny and origin. Brain Res. Rev. 2010, 63, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Papanikolaou, M.; Rivera, A. Physiology of Oligodendroglia. In Neuroglia in Neurodegenerative Diseases; Verkhratsky, A., Ho, M., Zorec, R., Parpura, V., Eds.; Springer: Singapore, Singapore, 2019; pp. 117–128. [Google Scholar]
- Barres, B.A.; Raff, M.C. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 1993, 361, 258–260. [Google Scholar] [CrossRef]
- Bergles, D.E.; Roberts, J.D.B.; Somogyl, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef]
- Gallo, V.; Armstrong, R.C. Myelin repair strategies: A cellular view. Curr. Opin. Neurol. 2008, 21, 278–283. [Google Scholar] [CrossRef]
- Hill, R.A.; Nishiyama, A. NG2 cells (polydendrocytes): Listeners to the neural network with diverse properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef]
- Fields, R.D. Imaging learning: The search for a memory trace. Neuroscientist 2011, 17, 185–196. [Google Scholar] [CrossRef]
- Liu, H.; Yang, Y.; Xia, Y.; Zhu, W.; Leak, R.K.; Wei, Z.; Wang, J.; Hu, X. Aging of cerebral white matter. Ageing Res. Rev. 2017, 34, 64–76. [Google Scholar] [CrossRef]
- Lemaître, H.; Crivello, F.; Grassiot, B.; Alpérovitch, A.; Tzourio, C.; Mazoyer, B. Age- and sex-related effects on the neuroanatomy of healthy elderly. Neuroimage 2005, 26, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Shenkin, S.D.; Bastin, M.E.; MacGillivray, T.J.; Deary, I.J.; Starr, J.M.; Rivers, C.S.; Wardlaw, J.M. Cognitive Correlates of Cerebral White Matter Lesions and Water Diffusion Tensor Parameters in Community-Dwelling Older People. Cerebrovasc. Dis. 2005, 20, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Marner, L.; Nyengaard, J.R.; Tang, Y.; Pakkenberg, B. Marked loss of myelinated nerve fibers in the human brain with age. J. Comp. Neurol. 2003, 462, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Casaccia, P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 2010, 33, 193–201. [Google Scholar] [CrossRef]
- Mito, R.; Raffelt, D.; Dhollander, T.; Vaughan, D.N.; Tournier, J.D.; Salvado, O.; Brodtmann, A.; Rowe, C.C.; Villemagne, V.L.; Connelly, A. Fibre-specific white matter reductions in Alzheimer’s disease and mild cognitive impairment. Brain 2018, 141, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, E.; Geerlings, M.I.; Biessels, G.J.; Nederkoorn, P.J.; Kloppenborg, R.P. White Matter Hyperintensities and Cognition in Mild Cognitive Impairment and Alzheimer’s Disease: A Domain-Specific Meta-Analysis. J. Alzheimers Dis. 2018, 63, 515–527. [Google Scholar] [CrossRef]
- Dean, D.C.; Hurley, S.A.; Kecskemeti, S.R.; O’Grady, J.P.; Canda, C.; Davenport-Sis, N.J.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Asthana, S.; et al. Association of Amyloid Pathology With Myelin Alteration in Preclinical Alzheimer Disease. JAMA Neurol. 2017, 74, 41–49. [Google Scholar] [CrossRef]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef]
- Baldassarro, V.A.; Bighinati, A.; Sannia, M.; Giardino, L.; Calzà, L.. Brain susceptibility to hypoxia/hypoxiemia and metabolic dysfunction in Alzheimer’s disease: Insights from animal and in vitro models. In The Neuroscience of Dementia: Genetics, Neurology, Behavior and Diet; Preedy, V.R., Martin, C.R., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Balducci, C.; Mehdawy, B.; Mare, L.; Giuliani, A.; Lorenzini, L.; Sivilia, S.; Giardino, L.; Calzà, L.; Lanzillotta, A.; Sarnico, I.; et al. The γ-secretase modulator CHF5074 restores memory and hippocampal synaptic plasticity in plaque-free Tg2576 mice. J. Alzheimer’s Dis. 2011, 24, 799–816. [Google Scholar] [CrossRef]
- Giuliani, A.; Sivilia, S.; Baldassarro, V.A.; Gusciglio, M.; Lorenzini, L.; Sannia, M.; Calza, L.; Giardino, L. Age-related changes of the neurovascular unit in the cerebral cortex of Alzheimer disease mouse models: A neuroanatomical and molecular study. J. Neuropathol. Exp. Neurol. 2019, 78, 101–112. [Google Scholar] [CrossRef]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calza, L. Vulnerability of primary neurons derived from Tg2576 Alzheimer mice to oxygen and glucose deprivation: Role of intraneuronal amyloid-β accumulation and astrocytes. DMM Dis. Model. Mech. 2017, 10, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Beggiato, S.; Baldassarro, V.A.; Mangano, C.; Giardino, L.; Imbimbo, B.P.; Antonelli, T.; Calzà, L.; Ferraro, L. CHF5074 restores visual memory ability and pre-synaptic cortical acetylcholine release in pre-plaque Tg2576 mice. J. Neurochem. 2013, 124, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Giuliani, A.; Sivilia, S.; Lorenzini, L.; Antonelli, T.; Imbimbo, B.P.; Giardino, L.; Calzà, L.; Ferraro, L. CHF5074 and LY450139 sub-acute treatments differently affect cortical extracellular glutamate levels in pre-plaque Tg2576 mice. Neuroscience 2014, 266, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, G.; Baer, K.; Buffo, A.; Curtis, M.A.; Faull, R.L.; Rees, M.I.; Götz, M.; Dimou, L. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 2013, 61, 273–286. [Google Scholar] [CrossRef]
- Simpson, J.E.; Fernando, M.S.; Clark, L.; Ince, P.G.; Matthews, F.; Forster, G.; O’Brien, J.T.; Barber, R.; Kalaria, R.N.; Brayne, C.; et al. White matter lesions in an unselected cohort of the elderly: Astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol. Appl. Neurobiol. 2007, 33, 410–419. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef]
- Zhan, X.; Jickling, G.C.; Ander, B.P.; Stamova, B.; Liu, D.; Kao, P.F.; Zelin, M.A.; Jin, L.W.; Decarli, C.; Sharp, F.R. Myelin basic protein associates with AβPP, Aβ1-42, and Amyloid plaques in cortex of Alzheimer’s disease brain. J. Alzheimers Dis. 2015, 44, 1213–1229. [Google Scholar] [CrossRef]
- Papuć, E.; Kurys-Denis, E.; Krupski, W.; Tatara, M.; Rejdak, K. Can antibodies against glial derived antigens be early biomarkers of hippocampal demyelination and memory loss in Alzheimer’s disease? J. Alzheimers Dis. 2015, 48, 115–121. [Google Scholar] [CrossRef]
- Tognatta, R.; Miller, R.H. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology 2016, 110, 539–547. [Google Scholar] [CrossRef]
- Barateiro, A.; Brites, D.; Fernandes, A. Oligodendrocyte Development and Myelination in Neurodevelopment: Molecular Mechanisms in Health and Disease. Curr. Pharm. Des. 2016, 22, 656–679. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calzà, L. Differential effects of glucose deprivation on the survival of fetal versus adult neural stem cells-derived oligodendrocyte precursor cells. Glia 2019. [Google Scholar] [CrossRef] [PubMed]
- Baldassarro, V.A.; Marchesini, A.; Facchinetti, F.; Villetti, G.; Calzà, L.; Giardino, L. Cell death in pure-neuronal and neuron-astrocyte mixed primary culture subjected to oxygen-glucose deprivation: The contribution of poly(ADP-ribose) polymerases and caspases. Microchem. J. 2018, 136, 215–222. [Google Scholar] [CrossRef]
- Dombrowski, Y.; O’Hagan, T.; DIttmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; Van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Setzu, A.; Lathia, J.D.; Zhao, C.; Wells, K.; Rao, M.S.; Ffrench-Constant, C.; Franklin, R.J.M. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia 2006, 54, 297–303. [Google Scholar] [CrossRef]
- Schaeffer, J.; Cossetti, C.; Mallucci, G.; Pluchino, S. Multiple Sclerosis. In Neurobiology of Brain Disorders: Biological Basis of Neurological and Psychiatric Disorders; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 497–520. [Google Scholar]
- El Behi, M.; Sanson, C.; Bachelin, C.; Guillot-Noël, L.; Fransson, J.; Stankoff, B.; Maillart, E.; Sarrazin, N.; Guillemot, V.; Abdi, H.; et al. Adaptive human immunity drives remyelination in a mouse model of demyelination. Brain 2017, 140, 967–980. [Google Scholar] [CrossRef]
- Matthews, P.M. Chronic inflammation in multiple sclerosis—Seeing what was always there. Nat. Rev. Neurol. 2019, 15, 582–593. [Google Scholar] [CrossRef]
- Fernández, M.; Baldassarro, V.A.; Sivilia, S.; Giardino, L.; Calzà, L. Inflammation severely alters thyroid hormone signaling in the central nervous system during experimental allergic encephalomyelitis in rat: Direct impact on OPCs differentiation failure. Glia 2016, 64, 1573–1589. [Google Scholar] [CrossRef]
- Lee, J.Y.; Petratos, S. Thyroid Hormone Signaling in Oligodendrocytes: From Extracellular Transport to Intracellular Signal. Mol. Neurobiol. 2016, 53, 6568–6583. [Google Scholar] [CrossRef] [PubMed]
- Verden, D.; Macklin, W.B. Neuroprotection by central nervous system remyelination: Molecular, cellular, and functional considerations. J. Neurosci. Res. 2016, 94, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Fern, R.F.; Matute, C.; Stys, P.K. White matter injury: Ischemic and nonischemic. Glia 2014, 62, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.-J.; DeBoy, C.A. Pharmacological approaches to intervention in hypomyelinating and demyelinating white matter pathology. Neuropharmacology 2016, 110, 605–625. [Google Scholar] [CrossRef]
- Amaral, A.I.; Hadera, M.G.; Tavares, J.M.; Kotter, M.R.N.; Sonnewald, U. Characterization of glucose-related metabolic pathways in differentiated rat oligodendrocyte lineage cells. Glia 2016, 64, 21–34. [Google Scholar] [CrossRef]
- Lee, J.T.; Xu, J.; Lee, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Zhang, W.; Yao, Z. Demyelination Takes Place Prior To Neuronal Damage Following Intracerebroventricular Injection of Amyloid-Beta Oligomer. Neuropsychiatry 2018, 8, 1770–1785. [Google Scholar]
- Roth, A.D.; Ramírez, G.; Alarcón, R.; Von Bernhardi, R. Oligodendrocytes damage in Alzheimer’s disease: Beta amyloid toxicity and inflammation. Biol. Res. 2005, 38, 381–387. [Google Scholar] [CrossRef]
- Quintela-López, T.; Ortiz-Sanz, C.; Serrano-Regal, M.P.; Gaminde-Blasco, A.; Valero, J.; Baleriola, J.; Sánchez-Gómez, M.V.; Matute, C.; Alberdi, E. Aβ oligomers promote oligodendrocyte differentiation and maturation via integrin β1 and Fyn kinase signaling. Cell Death Dis. 2019, 10, 445. [Google Scholar] [CrossRef]
- Pak, K.; Chan, S.L.; Mattson, M.P. Presenilin-1 mutation sensitizes oligodendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. NeuroMolecular Med. 2003, 3, 53–64. [Google Scholar] [CrossRef]
- Kahlson, M.A.; Colodner, K.J. Glial tau pathology in tauopathies: Functional consequences. J. Exp. Neurosci. 2015, 9 (Suppl. S2), 43–50. [Google Scholar] [CrossRef]
- Ossola, B.; Zhao, C.; Compston, A.; Pluchino, S.; Franklin, R.J.M.; Spillantini, M.G. Neuronal expression of pathological tau accelerates oligodendrocyte progenitor cell differentiation. Glia 2016, 64, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Rybnikova, E.A. Editorial: Brain Hypoxia and Ischemia: New Insights Into Neurodegeneration and Neuroprotection. Front. Neurosci. 2019, 13, 770. [Google Scholar] [CrossRef]
- Rocha-Ferreira, E.; Hristova, M. Plasticity in the Neonatal Brain following Hypoxic-Ischaemic Injury. Neural Plast. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, B.; Scafidi, J.; Aguirre, A.; Vaccarino, F.; Nguyen, V.; Borok, E.; Horvath, T.L.; Rowitch, D.H.; Gallo, V. Oligodendrocyte Regeneration after Neonatal Hypoxia Requires FoxO1-Mediated p27Kip1 Expression. J. Neurosci. 2012, 32, 14775–14793. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective Vulnerability of Late Oligodendrocyte Progenitors to Hypoxia-Ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Baldassarro, V.A.; Krężel, W.; Fernández, M.; Schuhbaur, B.; Giardino, L.; Calzà, L. The role of nuclear receptors in the differentiation of oligodendrocyte precursor cells derived from fetal and adult neural stem cells. Stem Cell Res. 2019, 37, 101443. [Google Scholar] [CrossRef] [PubMed]
- Tse, K.H.; Cheng, A.; Ma, F.; Herrup, K. DNA damage-associated oligodendrocyte degeneration precedes amyloid pathology and contributes to Alzheimer’s disease and dementia. Alzheimers Dement. 2018, 14, 664–679. [Google Scholar] [CrossRef]
- Dewar, D.; Underhill, S.M.; Goldberg, M.P. Oligodendrocytes and Ischemic Brain Injury. J. Cereb. Blood Flow Metab. 2003, 23, 263–274. [Google Scholar] [CrossRef]
- Giacci, M.K.; Bartlett, C.A.; Smith, N.M.; Iyer, K.S.; Toomey, L.M.; Jiang, H.; Guagliardo, P.; Kilburn, M.R.; Fitzgerald, M. Oligodendroglia are particularly vulnerable to oxidative damage after neurotrauma in vivo. J. Neurosci. 2018, 38, 6491–6504. [Google Scholar] [CrossRef]
- Kolodziejczyk, K.; Saab, A.S.; Nave, K.A.; Attwell, D. Why do oligodendrocyte lineage cells express glutamate receptors? F1000 Biol. Rep. 2010, 2, 57. [Google Scholar] [PubMed]
- Tekkök, S.B.; Goldberg, M.P. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J. Neurosci. 2001, 21, 4237–4248. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Yue, Q.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Oligodendrocyte excitotoxicity determined by local glutamate accumulation and mitochondrial function. J. Neurochem. 2006, 98, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; Van Bruggen, D.; Falcão, A.M.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef]
- Zeisel, A.; Moz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; Manno, G.L.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C.; Edgar, J.M.; Smith, K.J. Neuroprotection and repair in multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 624–634. [Google Scholar] [CrossRef]
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.-P.; Küry, P.; Kremer, D. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019, 8, 825. [Google Scholar] [CrossRef]
- Bothwell, M. Mechanisms and Medicines for Remyelination. Annu. Rev. Med. 2017, 68, 431–443. [Google Scholar] [CrossRef]
- Cole, K.L.H.; Early, J.J.; Lyons, D.A. Drug discovery for remyelination and treatment of MS. Glia 2017, 65, 1565–1589. [Google Scholar] [CrossRef]
- Skaper, S.D. Oligodendrocyte precursor cells as a therapeutic target for demyelinating diseases. In Progress in Brain Research; Elsevier B.V.: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Huang, J.K.; Jarjour, A.A.; Oumesmar, B.N.; Kerninon, C.; Williams, A.; Krezel, W.; Kagechika, H.; Bauer, J.; Zhao, C.; Evercooren, A.B.V.; et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat. Neurosci. 2011, 14, 45–55. [Google Scholar] [CrossRef]
- Lariosa-Willingham, K.D.; Rosler, E.S.; Tung, J.S.; Dugas, J.C.; Collins, T.L.; Leonoudakis, D. A high throughput drug screening assay to identify compounds that promote oligodendrocyte differentiation using acutely dissociated and purified oligodendrocyte precursor cells. BMC Res. Notes 2016, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Bove, R.M.; Green, A.J. Remyelinating Pharmacotherapies in Multiple Sclerosis. Neurotherapeutics 2017, 14, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Veto, S.; Acs, P.; Bauer, J.; Lassmann, H.; Berente, Z.; Setalo, G.; Borgulya, G.; Sumegi, B.; Komoly, S.; Gallyas, F.; et al. Inhibiting poly(ADP-ribose) polymerase: A potential therapy against oligodendrocyte death. Brain 2010, 133, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calzà, L. PARP activity and inhibition in fetal and adult oligodendrocyte precursor cells: Effect on cell survival and differentiation. Stem Cell Res. 2017, 22, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Porrini, V.; Lanzillotta, A.; Branca, C.; Benarese, M.; Parrella, E.; Lorenzini, L.; Calzà, L.; Flaibani, R.; Spano, P.F.; Imbimbo, B.P.; et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience 2015, 302, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Sharma, S.; Winston, J.; Nunez, M.; Bottini, G.; Franceschi, M.; Scarpini, E.; Frigerio, E.; Fiorentini, F.; Fernandez, M.; et al. CHF5074 Reduces Biomarkers of Neuroinflammation in Patients with Mild Cognitive Impairment: A 12-Week, Double-Blind, Placebo- Controlled Study. Curr. Alzheimer Res. 2013, 10, 742–753. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzini, L.; Fernandez, M.; Baldassarro, V.A.; Bighinati, A.; Giuliani, A.; Calzà, L.; Giardino, L. White Matter and Neuroprotection in Alzheimer’s Dementia. Molecules 2020, 25, 503. https://doi.org/10.3390/molecules25030503
Lorenzini L, Fernandez M, Baldassarro VA, Bighinati A, Giuliani A, Calzà L, Giardino L. White Matter and Neuroprotection in Alzheimer’s Dementia. Molecules. 2020; 25(3):503. https://doi.org/10.3390/molecules25030503
Chicago/Turabian StyleLorenzini, Luca, Mercedes Fernandez, Vito Antonio Baldassarro, Andrea Bighinati, Alessandro Giuliani, Laura Calzà, and Luciana Giardino. 2020. "White Matter and Neuroprotection in Alzheimer’s Dementia" Molecules 25, no. 3: 503. https://doi.org/10.3390/molecules25030503
APA StyleLorenzini, L., Fernandez, M., Baldassarro, V. A., Bighinati, A., Giuliani, A., Calzà, L., & Giardino, L. (2020). White Matter and Neuroprotection in Alzheimer’s Dementia. Molecules, 25(3), 503. https://doi.org/10.3390/molecules25030503