Towards Deep Neural Network Models for the Prediction of the Blood–Brain Barrier Permeability for Diverse Organic Compounds


Abstract
1. Introduction
2. Results and Discussion
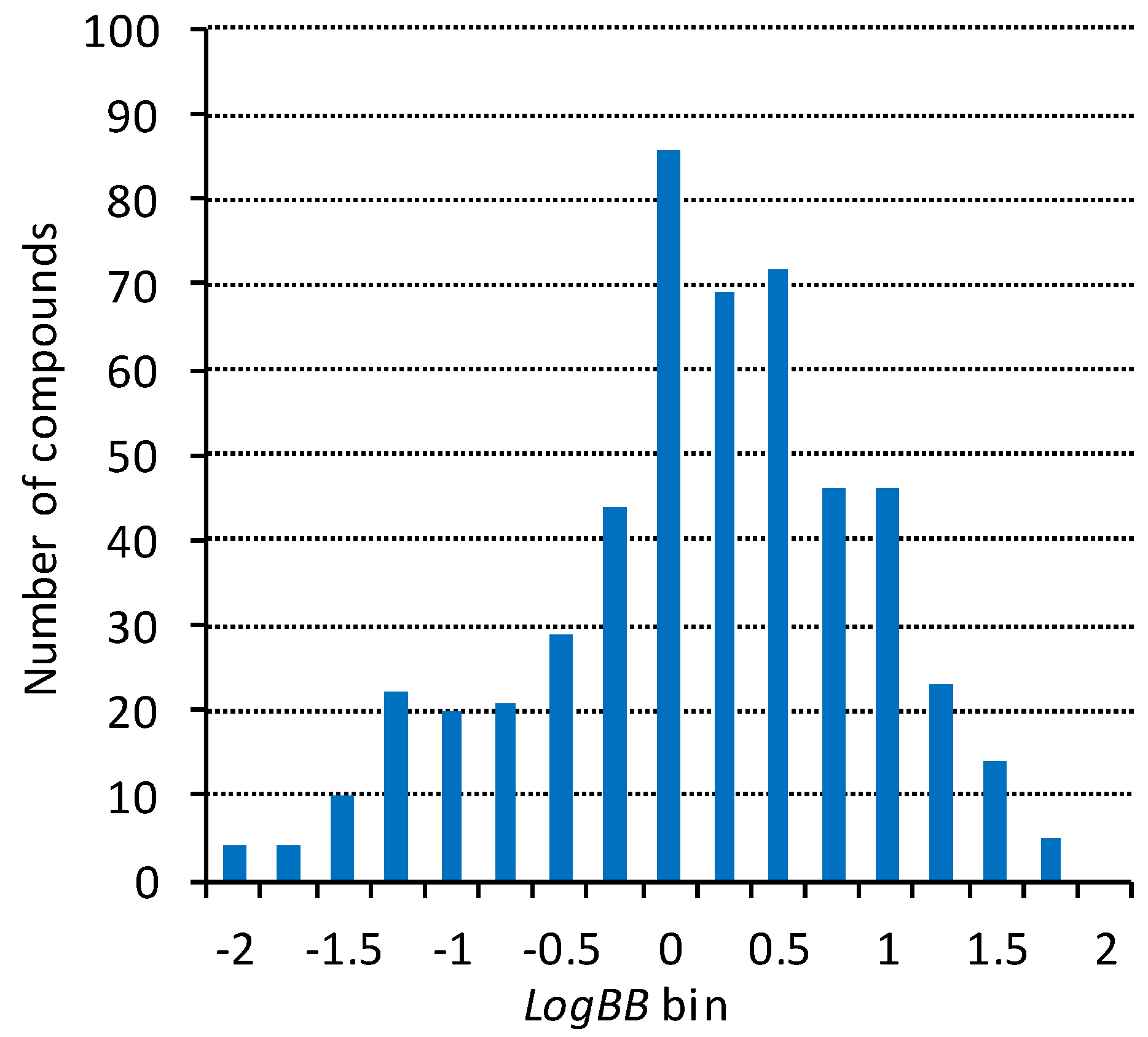
2.1. Blood–Brain Barrier Permeability Dataset
2.2. Molecular Descriptors
2.3. Neural Network Modeling Procedure
2.3.1. General Modeling Approach
2.3.2. Data Preprocessing and Descriptor Selection
- Selection of a specified number of descriptors with the highest F-values in the univariate linear regression (f_regression) or non-parametric mutual information scores [73] (mutual_info_regression) between the descriptor and the endpoint;
- Recursive feature elimination (RFE) [74] based on the descriptor importance scores from the Partial Least Squares (PLSR), Random Forest, linear Support Vector Machine, ElasticNet or Lasso regression models;
- Stepwise descriptor selection procedure, wherein a multiple linear or Partial Least Squares regression is iteratively refined by adding descriptors with the highest F-value or mutual information scores with the residual endpoint.
2.3.3. Hyperparameter Optimization
2.3.4. Prediction and Applicability Control
- The predicted values are calculated for each individual model in the ensemble and transformed back to the original endpoint scale;
- For each predicted value, a sanity check is performed to ensure that it lies within a reasonable range ( for LogBB). Values outside of this range (extended compared to the training dataset) most probably indicate that the compound is beyond the model applicability domain limits and the individual predicted value cannot be trusted;
- If such failed predictions are obtained from more than a specified fraction of the ensemble models (usually 50%), a prediction failure is reported;
- The individual predicted values are clipped to a specified acceptable range ( for LogBB);
- Mean and standard deviation of the individual predicted values are computed;
- If the standard deviation is greater than a specified fraction of the acceptable range (usually 30%), a prediction failure is reported;
- Otherwise, the mean and standard deviation values are reported.
2.4. Predictive LogBB Model
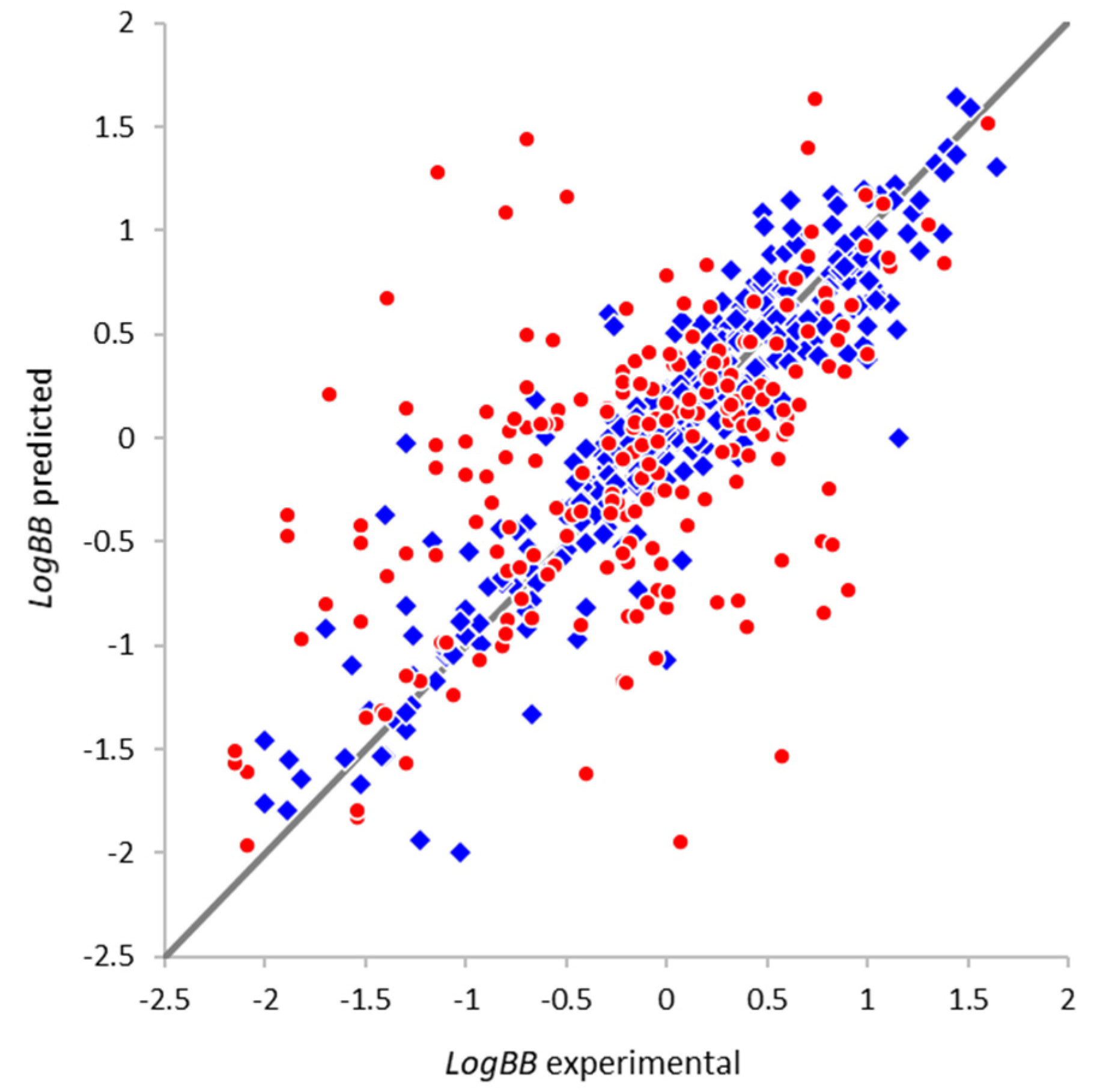
2.4.1. Optimal Architecture and Model Quality
2.4.2. Model Interpretation
2.4.3. External Validation
3. Materials and Methods
3.1. Blood–Brain Barrier Permeability Datasets
3.2. Modeling Workflow
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di, L.; Rong, H.; Feng, B. Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 2013, 56, 2–12. [Google Scholar] [CrossRef]
- Wager, T.T.; Liras, J.L.; Mente, S.; Trapa, P. Strategies to minimize CNS toxicity: In vitro high-throughput assays and computational modeling. Expert Opin. Drug Metab. Toxicol. 2012, 8, 531–542. [Google Scholar] [CrossRef]
- Summerfield, S.G.; Zhang, Y.; Liu, H. Examining the uptake of central nervous system drugs and candidates across the blood-brain barrier. J. Pharmacol. Exp. Ther. 2016, 358, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Birngruber, T.; Ghosh, A.; Perez-Yarza, V.; Kroath, T.; Ratzer, M.; Pieber, T.R.; Sinner, F. Cerebral open flow microperfusion: A new in vivo technique for continuous measurement of substance transport across the intact blood-brain barrier. Clin. Exp. Pharmacol. Physiol. 2013, 40, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Allen, D.D.; Bloomquist, J.R. Novel models for assessing blood-brain barrier drug permeation. Expert Opin. Drug Metab. Toxicol. 2012, 8, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Alavijeh, M.S. Overview of experimental models of the blood-brain barrier in CNS drug discovery. Curr. Protoc. Pharmacol. 2013, 62, 7.15.1–7.15.30. [Google Scholar] [CrossRef]
- Neuhaus, W. In vitro models of the blood-brain barrier. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Katt, M.E.; Shusta, E.V. In vitro models of the blood-brain barrier: Building in physiological complexity. Curr. Opin. Chem. Eng. 2020, 30, 42–52. [Google Scholar] [CrossRef]
- Sosnina, E.A.; Osolodkin, D.I.; Radchenko, E.V.; Sosnin, S.; Palyulin, V.A. Influence of descriptor implementation on compound ranking based on multiparameter assessment. J. Chem. Inf. Model. 2018, 58, 1083–1093. [Google Scholar] [CrossRef]
- Morales, J.F.; Montoto, S.S.; Fagiolino, P.; Ruiz, M.E. Current state and future perspectives in QSAR models to predict blood-brain barrier penetration in central nervous system drug R&D. Mini Rev. Med. Chem. 2017, 17, 247–257. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L. Blood-brain barrier. In Drug-Like Properties: Concepts, Structure Design and Methods; Kerns, E.H., Di, L., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 122–136. ISBN 978-0-12-369520-8. [Google Scholar] [CrossRef]
- Levin, V.A. Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J. Med. Chem. 1980, 23, 682–684. [Google Scholar] [CrossRef]
- Young, R.C.; Mitchell, R.C.; Brown, T.H.; Ganellin, C.R.; Griffiths, R.; Jones, M.; Rana, K.K.; Saunders, D.; Smith, I.R.; Sore, N.E.; et al. Development of a new physicochemical model for brain penetration and its application to the design of centrally acting H2 receptor histamine antagonists. J. Med. Chem. 1988, 31, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Verma, J.; Roy, N. In silico modeling for blood-brain barrier permeability predictions. In Drug Absorption Studies: In Situ, In Vitro and In Silico Models; Ehrhardt, C., Kim, K.-J., Eds.; Biotechnology: Pharmaceutical Aspects; Springer: Boston, MA, USA, 2008; pp. 510–556. ISBN 978-0-387-74901-3. [Google Scholar] [CrossRef]
- Raevsky, O.A.; Solodova, S.L.; Lagunin, A.A.; Poroikov, V.V. Computer modeling of blood brain barrier permeability for physiologically active compounds. Biochem. Mosc. Suppl. Ser. B 2013, 7, 95–107. [Google Scholar] [CrossRef]
- Lanevskij, K.; Japertas, P.; Didziapetris, R. Improving the prediction of drug disposition in the brain. Expert Opin. Drug Metab. Toxicol. 2013, 9, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dong, K.; Zhang, W.; Summerfield, S.G.; Terstappen, G.C. Prediction of brain: Blood unbound concentration ratios in CNS drug discovery employing in silico and in vitro model systems. Drug Discov. Today 2018, 23, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Lanevskij, K.; Dapkunas, J.; Juska, L.; Japertas, P.; Didziapetris, R. QSAR analysis of blood-brain distribution: The influence of plasma and brain tissue binding. J. Pharm. Sci. 2011, 100, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.-C.; Wang, G.-P.; Shan, H.-L.; Liang, W.-Q.; Gao, J.-Q. Predicting blood-brain barrier penetration from molecular weight and number of polar atoms. Eur. J. Pharm. Biopharm. 2008, 70, 462–466. [Google Scholar] [CrossRef]
- Shayanfar, A.; Soltani, S.; Jouyban, A. Prediction of blood-brain distribution: Effect of ionization. Biol. Pharm. Bull. 2011, 34, 266–271. [Google Scholar] [CrossRef][Green Version]
- Garg, P.; Verma, J. In silico prediction of blood brain barrier permeability: An Artificial Neural Network model. J. Chem. Inf. Model. 2006, 46, 289–297. [Google Scholar] [CrossRef]
- Kortagere, S.; Chekmarev, D.; Welsh, W.J.; Ekins, S. New predictive models for blood-brain barrier permeability of drug-like molecules. Pharm. Res. 2008, 25, 1836–1845. [Google Scholar] [CrossRef]
- Abraham, M.H.; Ibrahim, A.; Zhao, Y.; Acree, W.E. A data base for partition of volatile organic compounds and drugs from blood/plasma/serum to brain, and an LFER analysis of the data. J. Pharm. Sci. 2006, 95, 2091–2100. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Q.-J.; Pan, J.; Yang, Y.; Wu, X.-P. A prediction model for blood-brain barrier permeation and analysis on its parameter biologically. Comput. Methods Programs Biomed. 2009, 95, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Raevsky, O.A.; Grigorev, V.Y.; Polianczyk, D.E.; Raevskaja, O.E.; Dearden, J.C. Contribution assessment of multiparameter optimization descriptors in CNS penetration. SAR QSAR Environ. Res. 2018, 29, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Brito-Sánchez, Y.; Marrero-Ponce, Y.; Barigye, S.J.; Yaber-Goenaga, I.; Morell Pérez, C.; Le-Thi-Thu, H.; Cherkasov, A. Towards better BBB passage prediction using an extensive and curated data set. Mol. Inform. 2015, 34, 308–330. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood-Brain Barrier (BBB) score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Roy, D.; Hinge, V.K.; Kovalenko, A. To pass or not to pass: Predicting the blood-brain barrier permeability with the 3D-RISM-KH molecular solvation theory. ACS Omega 2019, 4, 16774–16780. [Google Scholar] [CrossRef]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef]
- Wang, Y.; Gallagher, E.; Jorgensen, C.; Troendle, E.P.; Hu, D.; Searson, P.C.; Ulmschneider, M.B. An experimentally validated approach to calculate the blood-brain barrier permeability of small molecules. Sci. Rep. 2019, 9, 6117. [Google Scholar] [CrossRef]
- Thai, N.Q.; Theodorakis, P.E.; Li, M.S. Fast estimation of the blood-brain barrier permeability by pulling a ligand through a lipid membrane. J. Chem. Inf. Model. 2020, 60, 3057–3067. [Google Scholar] [CrossRef]
- Kouskoura, M.G.; Piteni, A.I.; Markopoulou, C.K. A new descriptor via bio-mimetic chromatography and modeling for the blood brain barrier (Part II). J. Pharm. Biomed. Anal. 2019, 164, 808–817. [Google Scholar] [CrossRef]
- Sobańska, A.W.; Wanat, K.; Brzezińska, E. Prediction of the blood-brain barrier permeability using RP-18 thin layer chromatography. Open Chem. 2019, 17, 43–56. [Google Scholar] [CrossRef]
- Janicka, M.; Sztanke, M.; Sztanke, K. Predicting the blood-brain barrier permeability of new drug-like compounds via HPLC with various stationary phases. Molecules 2020, 25, 487. [Google Scholar] [CrossRef] [PubMed]
- Guntner, A.S.; Thalhamer, B.; Klampfl, C.; Buchberger, W. Collision cross sections obtained with ion mobility mass spectrometry as new descriptor to predict blood-brain barrier permeation by drugs. Sci. Rep. 2019, 9, 19182. [Google Scholar] [CrossRef]
- Lingineni, K.; Belekar, V.; Tangadpalliwar, S.R.; Garg, P. The role of multidrug resistance protein (MRP-1) as an active efflux transporter on blood-brain barrier (BBB) permeability. Mol. Divers. 2017, 21, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Kuanar, M.; Slavov, S.; Dobchev, D.A.; Fara, D.C.; Karelson, M.; Acree, W.E.; Solov’ev, V.P.; Varnek, A. Correlation of blood-brain penetration using structural descriptors. Bioorg. Med. Chem. 2006, 14, 4888–4917. [Google Scholar] [CrossRef] [PubMed]
- Hemmateenejad, B.; Miri, R.; Safarpour, M.A.; Mehdipour, A.R. Accurate prediction of the blood-brain partitioning of a large set of solutes using ab initio calculations and genetic neural network modeling. J. Comput. Chem. 2006, 27, 1125–1135. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, H.; Oprea, T.I.; Golbraikh, A.; Tropsha, A. QSAR modeling of the blood-brain barrier permeability for diverse organic compounds. Pharm. Res. 2008, 25, 1902–1914. [Google Scholar] [CrossRef]
- Fan, Y.; Unwalla, R.; Denny, R.A.; Di, L.; Kerns, E.H.; Diller, D.J.; Humblet, C. Insights for predicting blood-brain barrier penetration of CNS targeted molecules using QSPR approaches. J. Chem. Inf. Model. 2010, 50, 1123–1133. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Xia, Z.-N.; Qin, L.-T.; Liu, S.-S. Prediction of blood-brain partitioning: A model based on molecular electronegativity distance vector descriptors. J. Mol. Graph. Model. 2010, 29, 214–220. [Google Scholar] [CrossRef]
- Muehlbacher, M.; Spitzer, G.M.; Liedl, K.R.; Kornhuber, J. Qualitative prediction of blood-brain barrier permeability on a large and refined dataset. J. Comput.-Aided Mol. Des. 2011, 25, 1095–1106. [Google Scholar] [CrossRef]
- Chen, H.; Winiwarter, S.; Fridén, M.; Antonsson, M.; Engkvist, O. In silico prediction of unbound brain-to-plasma concentration ratio using machine learning algorithms. J. Mol. Graph. Model. 2011, 29, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, K.; Filipic, S.; Smoliński, A.; Kaliszan, R.; Agbaba, D. Partial least square and hierarchical clustering in ADMET modeling: Prediction of blood-brain barrier permeation of α-adrenergic and imidazoline receptor ligands. J. Pharm. Pharm. Sci. 2013, 16, 622–647. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zheng, F.; Zhan, C.-G. Improved prediction of blood-brain barrier permeability through machine learning with combined use of molecular property-based descriptors and fingerprints. AAPS J. 2018, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhao, J.; Zhang, Y.; Zhou, W.; Yin, L.; Wang, Y.; Fan, Y.; Chen, Y.; Liu, H. ADME properties evaluation in drug discovery: In silico prediction of blood-brain partitioning. Mol. Divers. 2018, 22, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In silico prediction of blood-brain barrier permeability of compounds by machine learning and resampling methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef]
- Majumdar, S.; Basak, S.C.; Lungu, C.N.; Diudea, M.V.; Grunwald, G.D. Finding needles in a haystack: Determining key molecular descriptors associated with the blood-brain barrier entry of chemical compounds using machine learning. Mol. Inform. 2019, 38, e1800164. [Google Scholar] [CrossRef]
- Singh, M.; Divakaran, R.; Konda, L.S.K.; Kristam, R. A classification model for blood brain barrier penetration. J. Mol. Graph. Model. 2020, 96, 107516. [Google Scholar] [CrossRef]
- Alsenan, S.; Al-Turaiki, I.; Hafez, A. A Recurrent Neural Network model to predict blood-brain barrier permeability. Comput. Biol. Chem. 2020, 89, 107377. [Google Scholar] [CrossRef]
- Shaker, B.; Yu, M.-S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.-S.; Na, D. LightBBB: Computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Zefirov, N.S.; Palyulin, V.A. Fragmental approach in QSPR. J. Chem. Inf. Comput. Sci. 2002, 42, 1112–1122. [Google Scholar] [CrossRef]
- Artemenko, N.V.; Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. Artificial neural network and fragmental approach in prediction of physicochemical properties of organic compounds. Russ. Chem. Bull. 2003, 52, 20–29. [Google Scholar] [CrossRef]
- Artemenko, N.V.; Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. Prediction of physical properties of organic compounds using artificial neural networks within the substructure approach. Dokl. Chem. 2001, 381, 317–320. [Google Scholar] [CrossRef]
- Artemenko, N.V.; Palyulin, V.A.; Zefirov, N.S. Neural-network model of the lipophilicity of organic compounds based on fragment descriptors. Dokl. Chem. 2002, 383, 114–116. [Google Scholar] [CrossRef]
- Dyabina, A.S.; Radchenko, E.V.; Palyulin, V.A.; Zefirov, N.S. Prediction of blood-brain barrier permeability of organic compounds. Dokl. Biochem. Biophys. 2016, 470, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A.; Zefirov, N.S. Prediction of human intestinal absorption of drug compounds. Russ. Chem. Bull. 2016, 65, 576–580. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Rulev, Y.A.; Safanyaev, A.Y.; Palyulin, V.A.; Zefirov, N.S. Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components. Dokl. Biochem. Biophys. 2017, 473, 128–131. [Google Scholar] [CrossRef]
- Berishvili, V.P.; Kuimov, A.N.; Voronkov, A.E.; Radchenko, E.V.; Kumar, P.; Choonara, Y.E.; Pillay, V.; Kamal, A.; Palyulin, V.A. Discovery of novel tankyrase inhibitors through molecular docking-based virtual screening and molecular dynamics simulation studies. Molecules 2020, 25, 3171. [Google Scholar] [CrossRef]
- Karlov, D.S.; Radchenko, E.V.; Palyulin, V.A.; Zefirov, N.S. Molecular design of proneurogenic and neuroprotective compounds-allosteric NMDA receptor modulators. Dokl. Biochem. Biophys. 2017, 473, 132–136. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Rudakova, E.V.; Proshin, A.N.; Serkov, I.V.; Radchenko, E.V.; Palyulin, V.A.; et al. New hybrids of 4-amino-2,3-polymethylene-quinoline and p-tolylsulfonamide as dual inhibitors of acetyl- and butyrylcholinesterase and potential multifunctional agents for Alzheimer’s disease treatment. Molecules 2020, 25, 3915. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Radchenko, E.V.; et al. Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer’s disease treatment: Synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg. Chem. 2020, 94, 103387. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Tropsha, A. A turning point for blood-brain barrier modeling. Pharm. Res. 2009, 26, 1283–1284. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify II: A practical guide to chemogenomics data curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef]
- Oprea, T.I.; Olah, M.; Ostopovici, L.; Rad, R.; Mracec, M. On the propagation of errors in the QSAR literature. In EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions; Ford, M., Livingstone, D., Dearden, J., van de Waterbeemd, H., Eds.; Blackwell Science Inc.: New York, NY, USA, 2003; pp. 314–315. ISBN 978-1-4051-2516-1. [Google Scholar]
- Baskin, I.I.; Halberstam, N.M.; Artemenko, N.V.; Palyulin, V.A.; Zefirov, N.S. NASAWIN—A universal software for QSPR/QSAR studies. In EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions; Ford, M., Livingstone, D., Dearden, J., van de Waterbeemd, H., Eds.; Blackwell Science Inc.: New York, NY, USA, 2003; pp. 260–263. ISBN 978-1-4051-2516-1. [Google Scholar]
- Filzmoser, P.; Liebmann, B.; Varmuza, K. Repeated double cross validation. J. Chemom. 2009, 23, 160–171. [Google Scholar] [CrossRef]
- Klambauer, G.; Unterthiner, T.; Mayr, A.; Hochreiter, S. Self-normalizing neural networks. In Proceedings of the 31st International Conference on Neural Information Processing Systems; NIPS’17. Curran Associates Inc.: Red Hook, NY, USA, 2017; pp. 972–981, ISBN 978-1-5108-6096-4. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ross, B.C. Mutual information between discrete and continuous data sets. PLoS ONE 2014, 9, e87357. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Bergstra, J.; Komer, B.; Eliasmith, C.; Yamins, D.; Cox, D.D. Hyperopt: A Python library for model selection and hyperparameter optimization. Comput. Sci. Discov. 2015, 8, 014008. [Google Scholar] [CrossRef]
- Baskin, I.I.; Ait, A.O.; Halberstam, N.M.; Palyulin, V.A.; Zefirov, N.S. An approach to the interpretation of backpropagation neural network models in QSAR studies. SAR QSAR Environ. Res. 2002, 13, 35–41. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood-brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, K.; Diedenhofen, M.; Klamt, A. Prediction of blood-brain partitioning and human serum albumin binding based on COSMO-RS σ-moments. J. Chem. Inf. Model. 2007, 47, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Telting-Diaz, M.; Lunte, C.E. Distribution of tacrine across the blood-brain barrier in awake, freely moving rats using in vivo microdialysis sampling. Pharm. Res. 1993, 10, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Fridén, M.; Winiwarter, S.; Jerndal, G.; Bengtsson, O.; Wan, H.; Bredberg, U.; Hammarlund-Udenaes, M.; Antonsson, M. Structure-brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J. Med. Chem. 2009, 52, 6233–6243. [Google Scholar] [CrossRef]
- Tsinman, O.; Tsinman, K.; Sun, N.; Avdeef, A. Physicochemical selectivity of the BBB microenvironment governing passive diffusion—Matching with a porcine brain lipid extract artificial membrane permeability model. Pharm. Res. 2011, 28, 337–363. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Full Set | Non-Overlapping Subset |
|---|---|---|
| Number of compounds N | 564 | 213 |
| Correlation coefficient R | 0.78 | 0.58 |
| Root mean squared error RMSE | 0.47 | 0.68 |
| Compounds with absolute error > 1.0 | 33 (6%) | 29 (14%) |
| Compounds with absolute error > 1.5 | 11 (2%) | 11 (5%) |
| Compound | LogBB Val 1 | LogBB Train 2 | LogBB Pred 3 | Notes |
|---|---|---|---|---|
| 2-(2-Aminoethyl)thiazole (YG16) | −1.40 (78) | −0.42 | −0.37 | Incorrect validation value |
| 2-(2-Dimethylaminoethyl)pyridine (YG15) | −1.30 (131) | −0.06 | −0.03 | Incorrect validation value |
| Tacrine | 1.16 (146) | −0.13 | −0.00 | Literature discrepancy |
| Warfarin | 0.00 (520) | −1.30 | −1.07 | Calculated value in source |
Sample Availability: The samples of compounds are not available from authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A. Towards Deep Neural Network Models for the Prediction of the Blood–Brain Barrier Permeability for Diverse Organic Compounds. Molecules 2020, 25, 5901. https://doi.org/10.3390/molecules25245901
Radchenko EV, Dyabina AS, Palyulin VA. Towards Deep Neural Network Models for the Prediction of the Blood–Brain Barrier Permeability for Diverse Organic Compounds. Molecules. 2020; 25(24):5901. https://doi.org/10.3390/molecules25245901
Chicago/Turabian StyleRadchenko, Eugene V., Alina S. Dyabina, and Vladimir A. Palyulin. 2020. "Towards Deep Neural Network Models for the Prediction of the Blood–Brain Barrier Permeability for Diverse Organic Compounds" Molecules 25, no. 24: 5901. https://doi.org/10.3390/molecules25245901
APA StyleRadchenko, E. V., Dyabina, A. S., & Palyulin, V. A. (2020). Towards Deep Neural Network Models for the Prediction of the Blood–Brain Barrier Permeability for Diverse Organic Compounds. Molecules, 25(24), 5901. https://doi.org/10.3390/molecules25245901