
Implication of Lactucopicrin in Autophagy, Cell Cycle Arrest and Oxidative Stress to Inhibit U87Mg Glioblastoma Cell Growth


Abstract
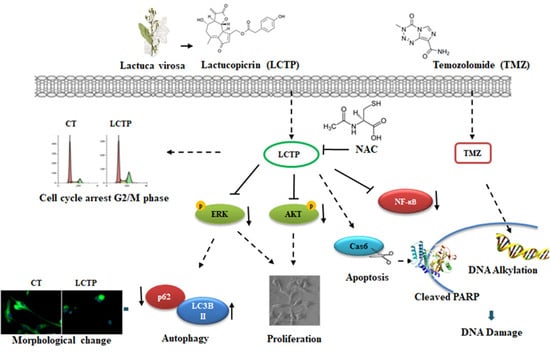
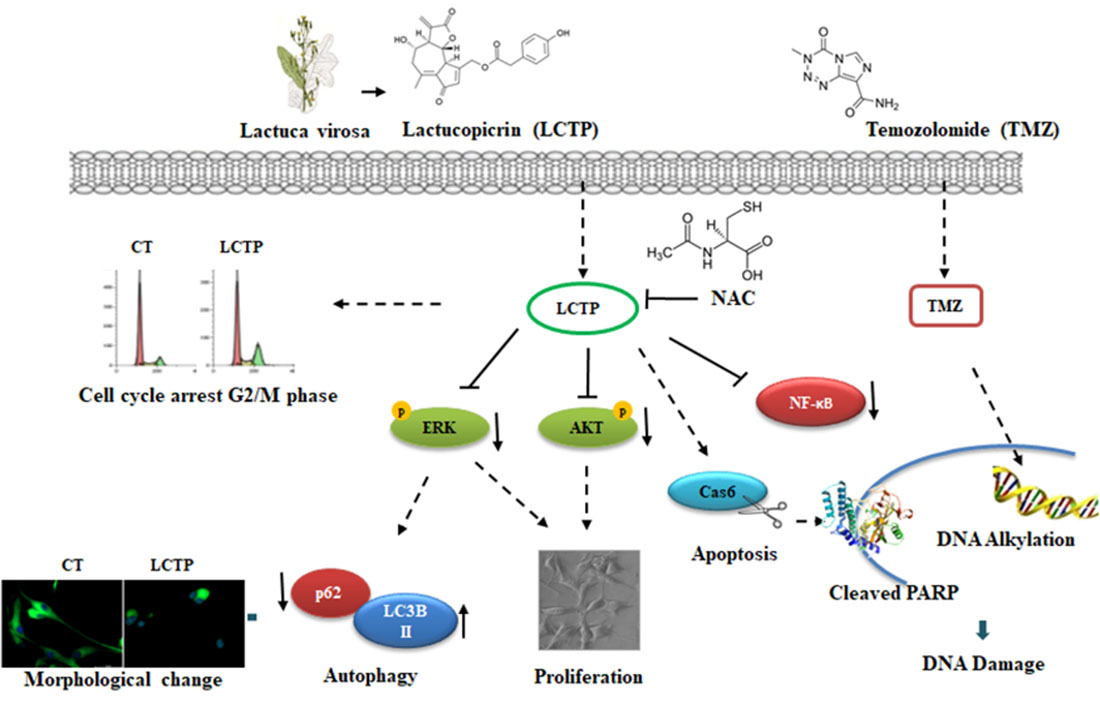{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
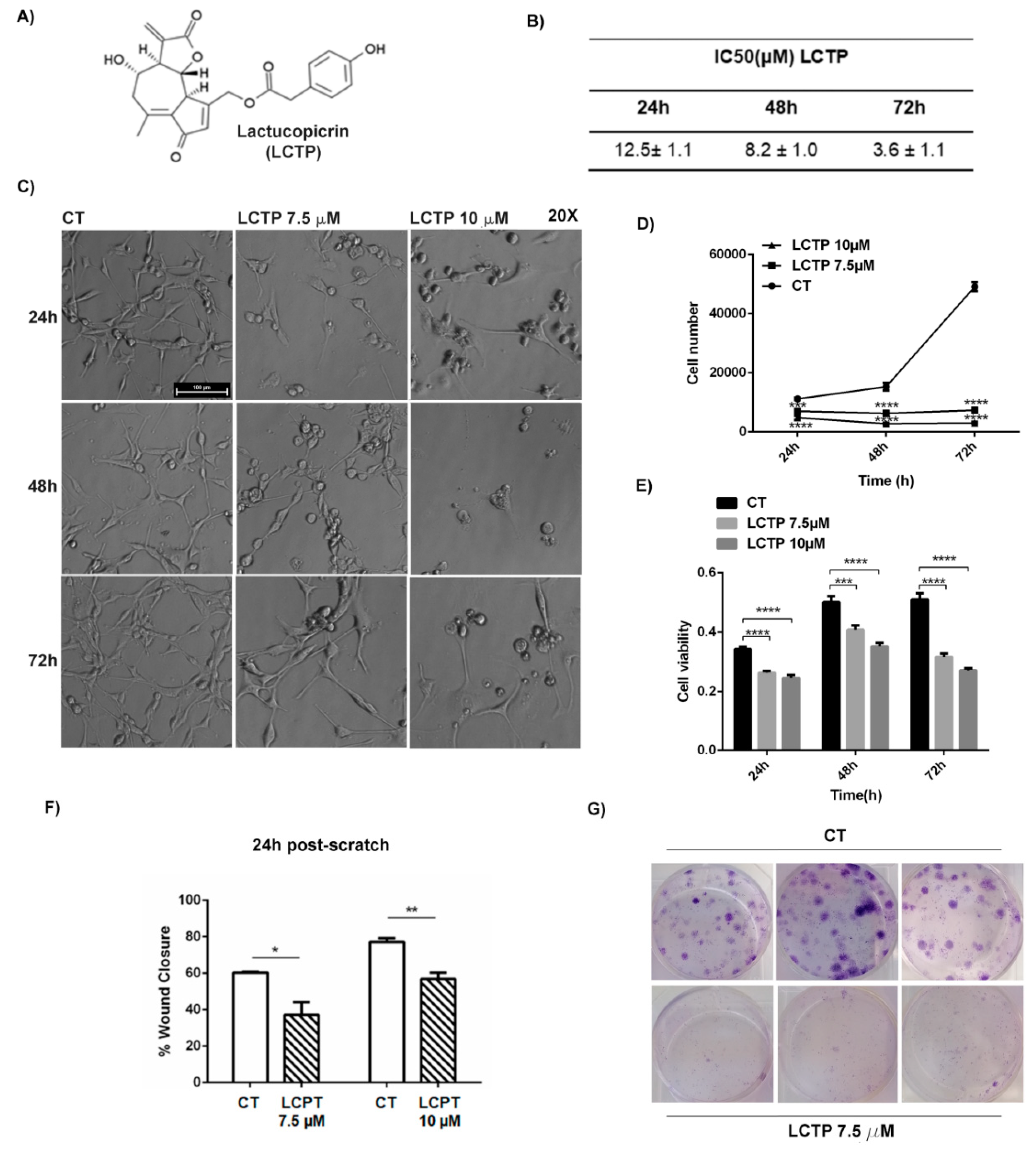
2.1. Dose-Response and Time-Course of LCTP Effects on Glioblastoma U87Mg Cells
2.2. LCTP Interferes with Clonogenic Survival and Motility Capacity of GBM Cells
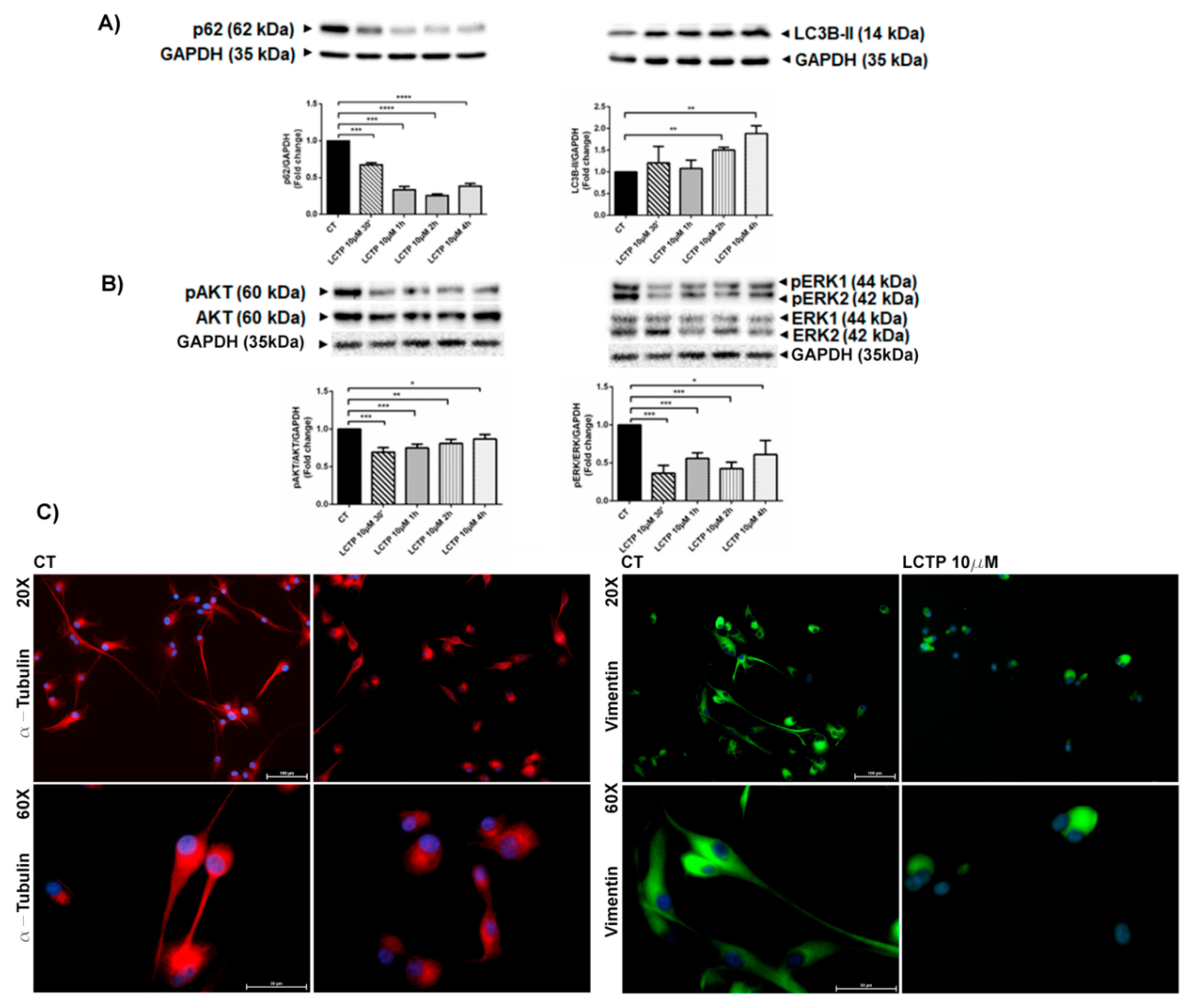
2.3. Rapid Autophagy Response of U87Mg to LCTP Treatment Potentially Remodels the Cytoskeleton Proteins
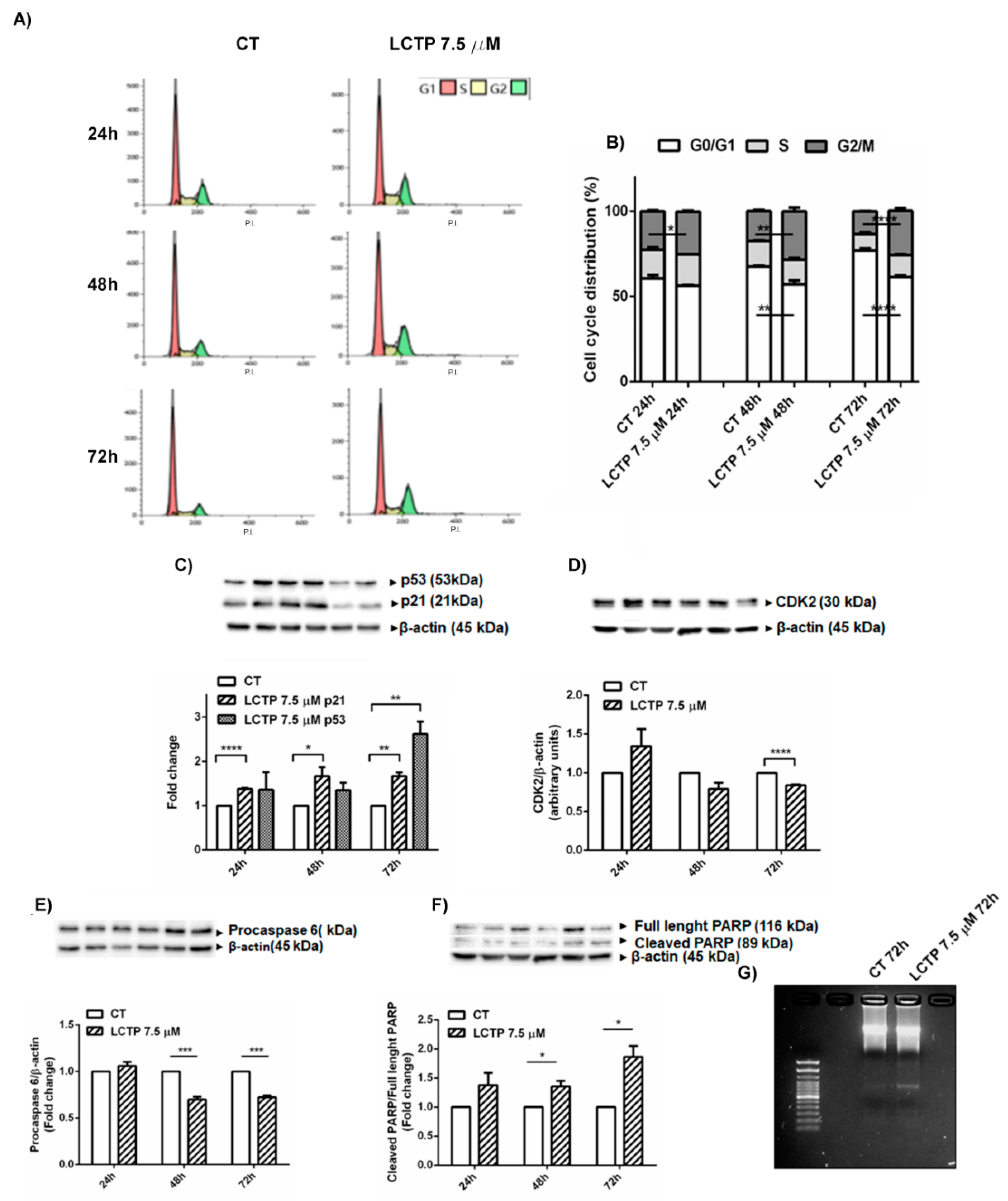
2.4. Cell Cycle Arrest in G2/M Phase and Apoptosis Induction by LCTP in U87Mg Cells
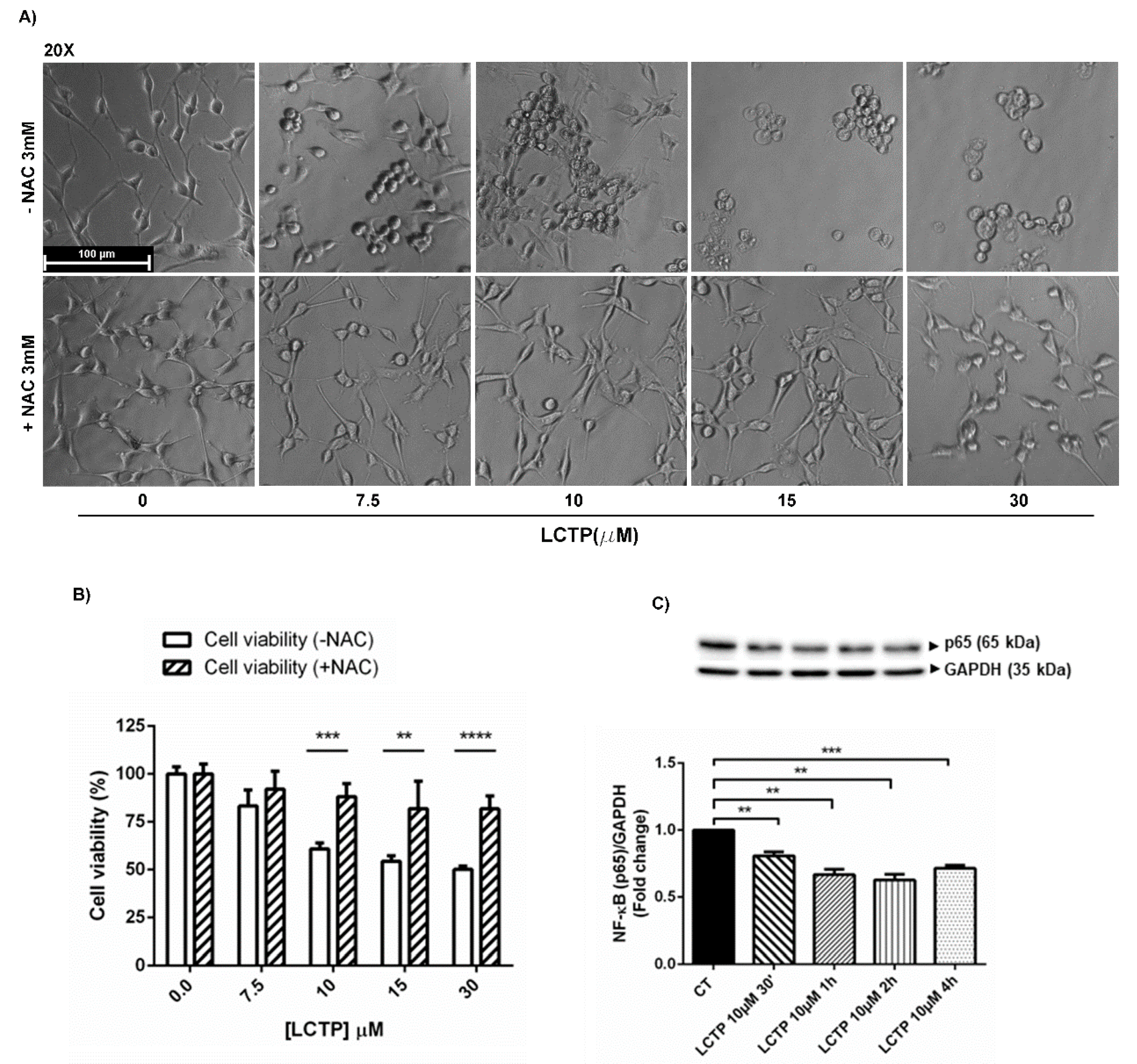
2.5. Involvement of Oxidative Stress in LCTP-Mediated Cytotoxicity in U87Mg Cells
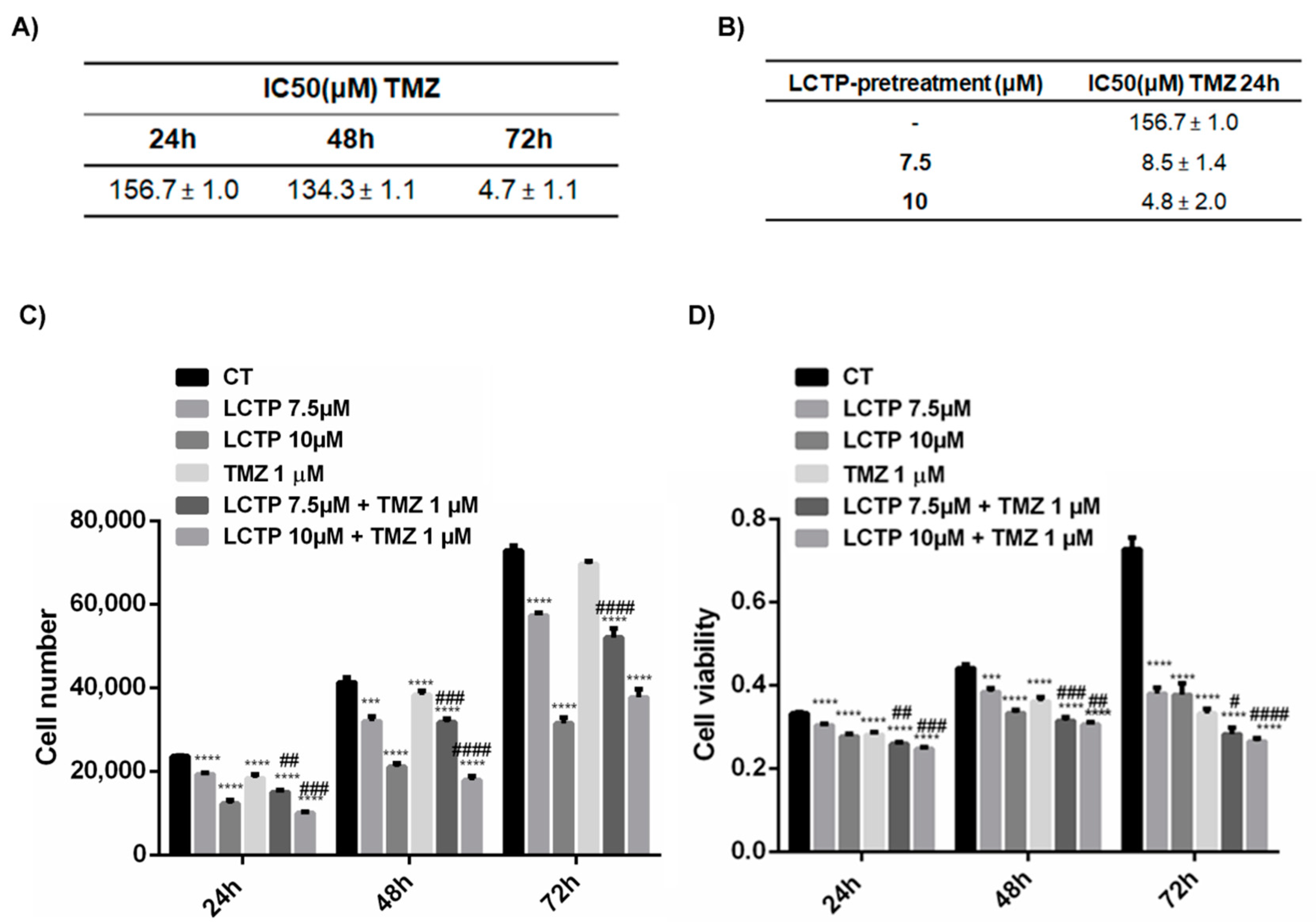
2.6. LCTP Enhances the Sensitivity of U87Mg to Canonical Therapy Temozolomide
2.7. Synergist Effect of LCTP and TMZ Affects the Cell Growth and Viability
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Estimation of Half—Maximal Inhibitory Concentration (IC50) of LCTP and TMZ in U87Mg Cells
4.3. Proliferation Assay
4.4. Cell Viability Assay
4.5. Microscopic Observation of Cell Morphology
4.6. Combined Treatment of TMZ and LCTP in Human GBM Cell Line
4.7. Estimation of IC50 of TMZ after Pre-Treatment with LCTP
4.8. Wound Healing Assay
4.9. Clonogenic Assay
4.10. Western Blot Analysis of LCTP-Treated U87Mg Cells
4.11. Western Blot of Apoptosis-Associated Proteins
4.12. Immunofluorescence
4.13. Cell Cycle Analysis by Flow Cytometry
4.14. DNA Laddering
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ciceroni, C.; Bonelli, M.; Mastrantoni, E.; Niccolini, C.; Laurenza, M.; LaRocca, L.M.; Pallini, R.; Traficante, A.; Spinsanti, P.; Ricci-Vitiani, L.; et al. Type-3 metabotropic glutamate receptors regulate chemoresistance in glioma stem cells, and their levels are inversely related to survival in patients with malignant gliomas. Cell Death Differ. 2013, 20, 396–407. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Sassi, F.; Lunardi Brunetto, A.; Schwartsmann, G.; Roesler, R.; Abujamra, A.L. Glioma Revisited: From Neurogenesis and Cancer Stem Cells to the Epigenetic Regulation of the Niche. J. Oncol. 2012, 2012, 537861. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Weber, D.C. The Role of Radio- and Chemotherapy in Glioblastoma. Oncol. Res. Treat. 2005, 28, 315–317. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Squatrito, M.; Carbajal, E.; Holland, E.C. Glioma Formation, Cancer Stem Cells, and Akt Signaling. Stem Cell Rev. Rep. 2008, 4, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Park, C.K.; Kim, J.E.; Kim, J.Y.; Song, S.W.; Kim, J.W.; Choi, S.H.; Kim, T.M.; Lee, S.H.; Kim, I.H.; Park, S. The Changes in MGMT Promoter Methylation Status in Initial and Recurrent Glioblastomas. Transl. Oncol. 2012, 5, 393–397. [Google Scholar] [CrossRef]
- Messaoudi, K.; Clavreul, A.; Lagarce, F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 2015, 20, 899–905. [Google Scholar] [CrossRef]
- Vengoji, R.; Macha, M.A.; Batra, S.K.; Shonka, N. Natural products: A hope for glioblastoma patients. Oncotarget 2018, 9, 22194–22219. [Google Scholar] [CrossRef]
- Yi, G.Z.; Xiang, W.; Feng, W.Y.; Chen, Z.Y.; Li, Y.M.; Deng, S.Z.; Guo, M.L.; Zhao, L.; Sun, X.G.; He, M.Y.; et al. Identification of Key Candidate Proteins and Pathways Associated with Temozolomide Resistance in Glioblastoma Based on Subcellular Proteomics and Bioinformatical Analysis. Biomed. Res. Int. 2018, 2018, 5238760. [Google Scholar] [CrossRef] [PubMed]
- Sestito, S.; Runfola, M.; Tonelli, M.; Chiellini, G.; Rapposelli, S. New Multitarget Approaches in the War Against Glioblastoma: A Mini-Perspective. Front. Pharmacol. 2018, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Oliva, M.A.; Sanchez, M.; Staffieri, S.; Esposito, V.; Giangaspero, F.; Cantore, G. Effects of hispolon on glioblastoma cell growth. Environ. Toxicol. 2017, 32, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Oliva, M.A.; Staffieri, S.; Sanchez, M.; Madonna, M.; Riozzi, B.; Esposito, V.; Giangaspero, F.; Frati, L. Effects of aloe emodin on U87MG glioblastoma cell growth: In vitro and in vivo study. Environ. Toxicol. 2018, 33, 1160–1167. [Google Scholar] [CrossRef]
- Kreuger, M.R.; Grootjans, S.; Biavatti, M.W.; Vandenabeele, P.; D’Herde, K. Sesquiterpene lactones as drugs with multiple targets in cancer treatment: Focus on parthenolide. Anticancer Drugs 2012, 23, 883–896. [Google Scholar]
- Amorim, M.H.; Gil da Costa, R.M.; Lopes, C.; Bastos, M.M. Sesquiterpene lactones: Adverse health effects and toxicity mechanisms. Crit. Rev. Toxicol. 2013, 43, 559–579. [Google Scholar] [CrossRef]
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids lactones: Benefits to plants and people. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Wang, J.; Yu, Z.; Wang, C.; Tian, X.; Huo, X.; Wang, Y.; Sun, C.; Feng, L.; Ma, J.; Zhang, B.; et al. Dehydrocostus lactone, a natural sesquiterpene lactone, suppresses the biological characteristics of glioma, through inhibition of the NF-kappaB/COX-2 signaling pathway by targeting IKKbeta. Am. J. Cancer Res. 2017, 7, 1270–1284. [Google Scholar]
- Wang, X.; Yu, Z.; Wang, C.; Cheng, W.; Tian, X.; Huo, X.; Wang, Y.; Sun, C.; Feng, L.; Xing, J.; et al. Alantolactone, a natural sesquiterpene lactone, has potent antitumor activity against glioblastoma by targeting IKKbeta kinase activity and interrupting NF-kappaB/COX-2-mediated signaling cascades. J. Exp. Clin. Cancer Res. 2017, 36, 93. [Google Scholar] [CrossRef] [PubMed]
- Youn, U.J.; Miklóssy, G.; Chai, X.; Wongwiwatthananukit, S.; Toyama, O.; Songsak, T.; Turkson, J.; Chang, L.C. Bioactive sesquiterpene lactones and other compounds isolated from Vernonia cinerea. Fitoterapia 2014, 93, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lan, D.; Ning, S.; Ruan, L. Anticancer action of lactucopicrin in SKMEL-5 human skin cancer cells is mediated via apoptosis induction, G2/M cell cycle arrest and downregulation of m=TOR/PI3K/AKT signalling pathway. J. BUON 2018, 23, 224–228. [Google Scholar] [PubMed]
- Meng, Q.; Tang, B.; Qiu, B. Growth inhibition of Saos-2 osteosarcoma cells by lactucopicrin is mediated via inhibition of cell migration and invasion, sub-G1 cell cycle disruption, apoptosis induction and Raf signalling pathway. J. BUON 2019, 24, 2136–2140. [Google Scholar]
- Wierzbicki, M.; Sawosz, E.; Strojny, B.; Jaworski, S.; Grodzik, M.; Chwalibog, A. NF-kappaB-related decrease of glioma angiogenic potential by graphite nanoparticles and graphene oxide nanoplatelets. Sci. Rep. 2018, 8, 14733. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Yin, S.Y.; Wei, W.C.; Jian, F.Y.; Yang, N.S. Therapeutic applications of herbal medicines for cancer patients. Evid. Based Complement. Altern. Med. 2013, 2013, 302426. [Google Scholar] [CrossRef]
- Liu, C.A.; Chang, C.Y.; Hsueh, K.W.; Su, H.L.; Chiou, T.W.; Lin, S.Z.; Harn, H.J. Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment. Int. J. Mol. Sci. 2018, 19, 1115. [Google Scholar] [CrossRef]
- Li, C.; Han, X. Anthecotulide Sesquiterpene Lactone Exhibits Selective Anticancer Effects in Human Malignant Melanoma Cells by Activating Apoptotic and Autophagic Pathways, S-Phase Cell Cycle Arrest, Caspase Activation, and Inhibition of NF-kappaBSignalling Pathway. Med. Sci. Monit. 2019, 25, 2852–2858. [Google Scholar] [CrossRef]
- Deng, D.; Luo, K.; Liu, H.; Nie, X.; Xue, L.; Wang, R.; Xu, Y.; Cui, J.; Shao, N.; Zhi, F. p62 acts as an oncogene and is targeted by miR-124-3p in glioma. Cancer Cell Int. 2019, 19, 280. [Google Scholar] [CrossRef]
- Xu, H.; Sun, L.; Zheng, Y.; Yu, S.; Ou-Yang, J.; Han, H.; Dai, X.; Yu, X.; Li, M.; Lan, Q. GBP3 promotes glioma cell proliferation via SQSTM1/p62-ERK1/2 axis. Biochem. Biophys. Res. Commun. 2018, 495, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Narasimamurthy, R.; Xia, Y.; Myskiw, C.; Soda, Y.; Verma, I.M. Targeting NF-kappaB in glioblastoma: A therapeutic approach. Sci. Adv. 2016, 2, e1501292. [Google Scholar] [CrossRef] [PubMed]
- Hehner, S.P.; Heinrich, M.; Bork, P.M.; Vogt, M.; Ratter, F.; Lehmann, V.; Schulze-Osthoff, K.; Droge, W.; Schmitz, M.L. Sesquiterpene lactones specifically inhibit activation of NF-kappa B by preventing the degradation of I kappa B-alpha and I kappa B-beta. J. Biol. Chem. 1998, 273, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Gajkowska, B.; Orzechowski, A. Molecular basis of parthenolide-dependent proapoptotic activity in cancer cells. Folia Histochem. Cytobiol. 2008, 46, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. Willmar Schwabe Award 2006: Antiplasmodial and antitumor activity of artemisinin—From bench to bedside. Planta Med. 2007, 73, 299–309. [Google Scholar] [CrossRef]
- Su, J.; Liu, F.; Xia, M.; Xu, Y.; Li, X.; Kang, J.; Li, Y.; Sun, L. p62 participates in the inhibition of NF-kappaB signaling and apoptosis induced by sulfasalazine in human glioma U251 cells. Oncol. Rep. 2015, 34, 235–243. [Google Scholar] [CrossRef]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef]
- Izdebska, M.; Halas-Wisniewska, M.; Zielinska, W.; Klimaszewska-Wisniewska, A.; Grzanka, D.; Gagat, M. Lidocaine induces protective autophagy in rat C6 glioma cell line. Int. J. Oncol. 2018, 54, 1099–1111. [Google Scholar] [CrossRef]
- Li, S.S.; Xu, L.Z.; Zhou, W.; Yao, S.; Wang, C.L.; Xia, J.L.; Wang, H.F.; Kamran, M.; Xue, X.Y.; Dong, L.; et al. p62/SQSTM1 interacts with vimentin to enhance breast cancer metastasis. Carcinogenesis 2017, 38, 1092–1103. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotondo, R.; Oliva, M.A.; Staffieri, S.; Castaldo, S.; Giangaspero, F.; Arcella, A. Implication of Lactucopicrin in Autophagy, Cell Cycle Arrest and Oxidative Stress to Inhibit U87Mg Glioblastoma Cell Growth. Molecules 2020, 25, 5843. https://doi.org/10.3390/molecules25245843
Rotondo R, Oliva MA, Staffieri S, Castaldo S, Giangaspero F, Arcella A. Implication of Lactucopicrin in Autophagy, Cell Cycle Arrest and Oxidative Stress to Inhibit U87Mg Glioblastoma Cell Growth. Molecules. 2020; 25(24):5843. https://doi.org/10.3390/molecules25245843
Chicago/Turabian StyleRotondo, Rossella, Maria Antonietta Oliva, Sabrina Staffieri, Salvatore Castaldo, Felice Giangaspero, and Antonietta Arcella. 2020. "Implication of Lactucopicrin in Autophagy, Cell Cycle Arrest and Oxidative Stress to Inhibit U87Mg Glioblastoma Cell Growth" Molecules 25, no. 24: 5843. https://doi.org/10.3390/molecules25245843
APA StyleRotondo, R., Oliva, M. A., Staffieri, S., Castaldo, S., Giangaspero, F., & Arcella, A. (2020). Implication of Lactucopicrin in Autophagy, Cell Cycle Arrest and Oxidative Stress to Inhibit U87Mg Glioblastoma Cell Growth. Molecules, 25(24), 5843. https://doi.org/10.3390/molecules25245843