
New Advances in the Synthetic Application of Enantiomeric 1-Phenylethylamine (α-PEA): Privileged Chiral Inducer and Auxiliary



Abstract
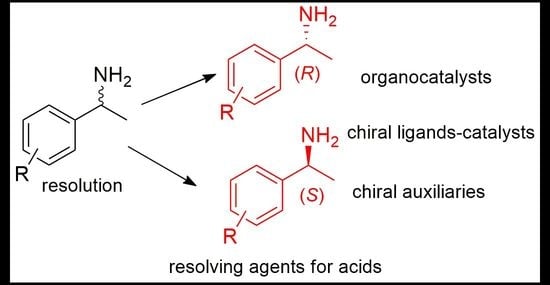
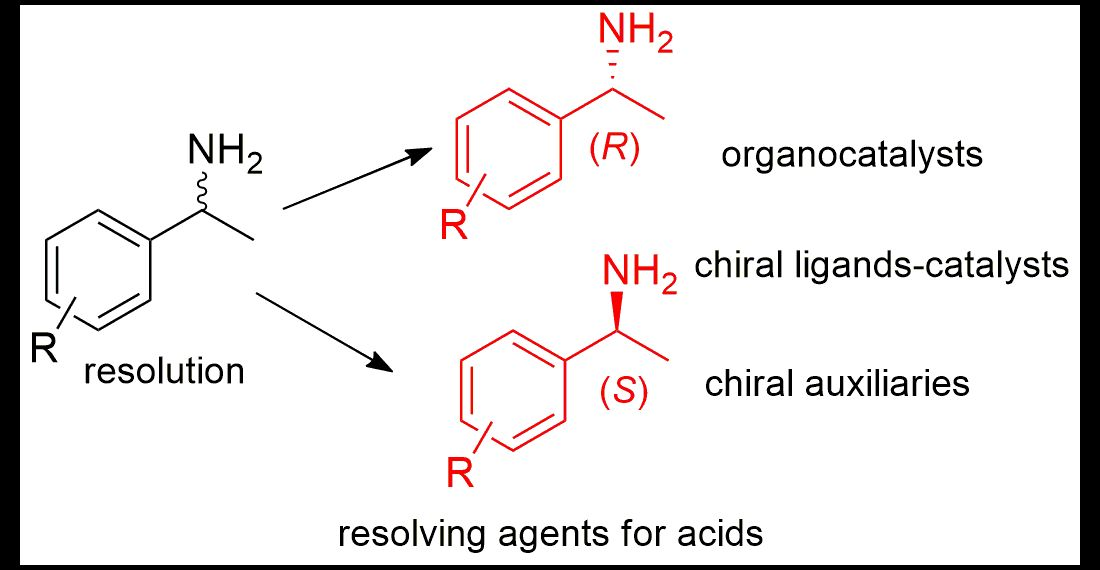{kind=link}
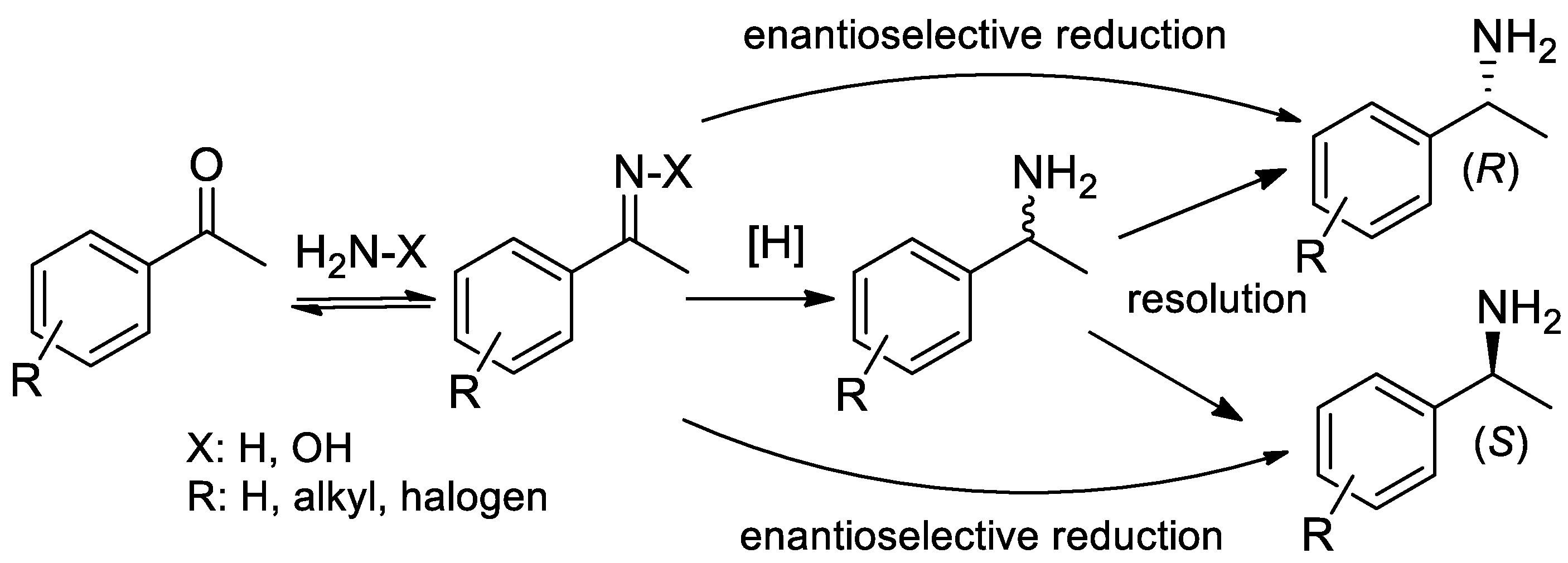{kind=link}
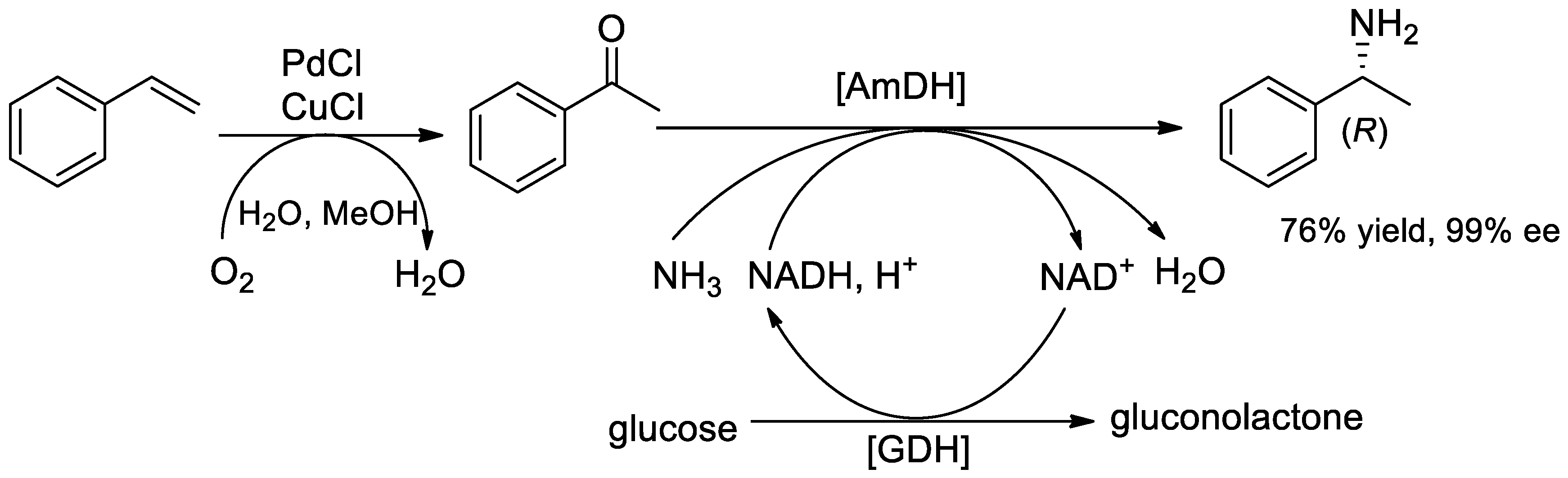{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
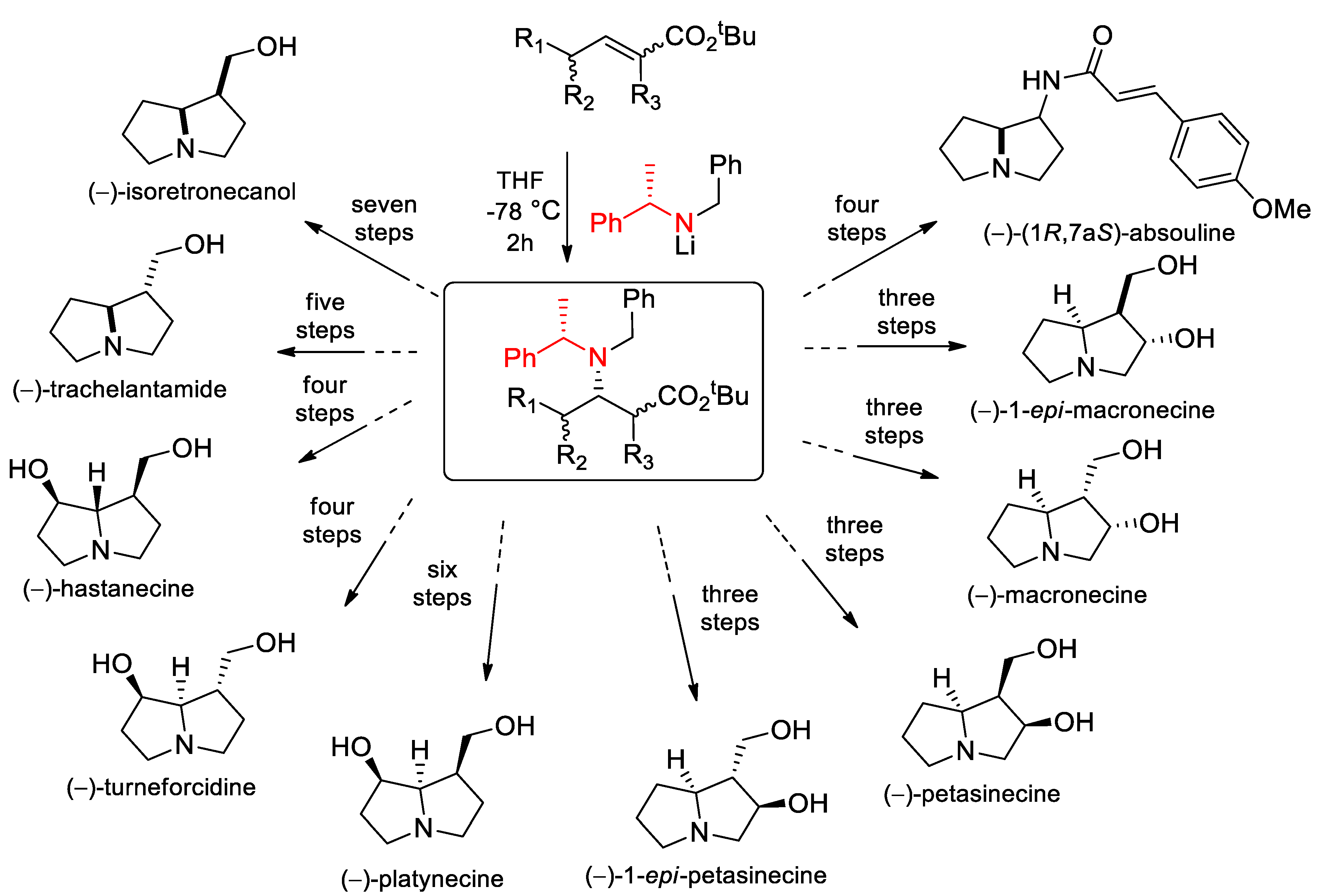{kind=link}
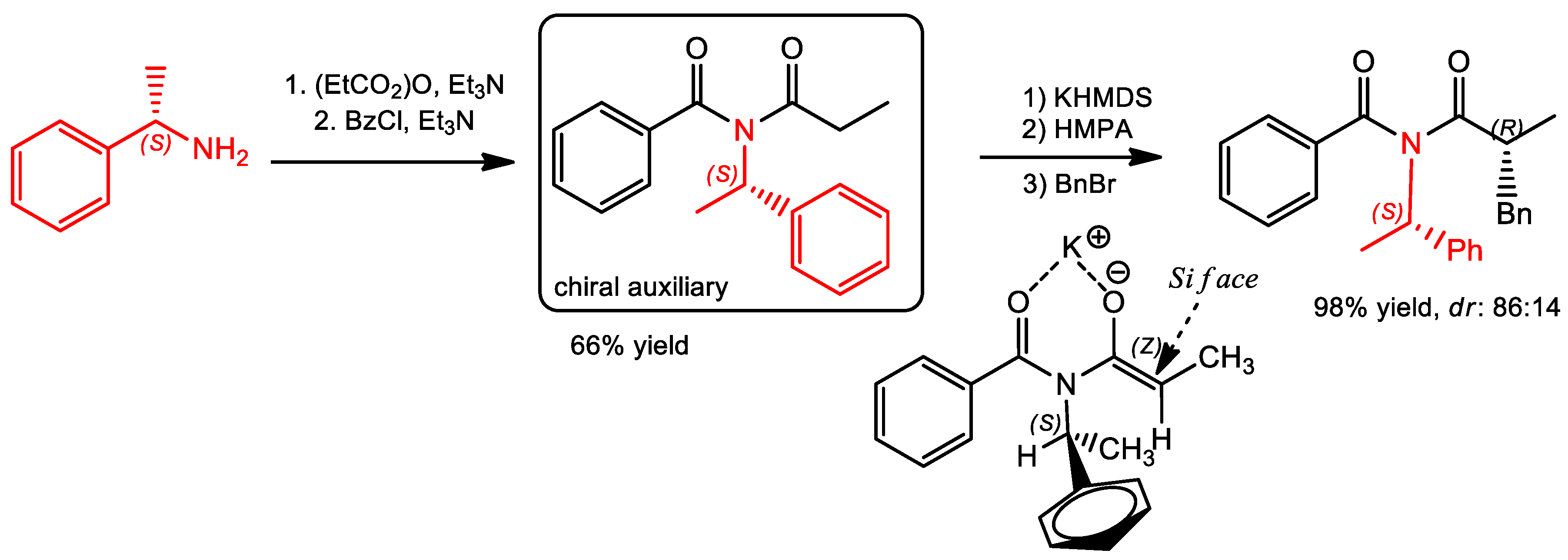{kind=link}
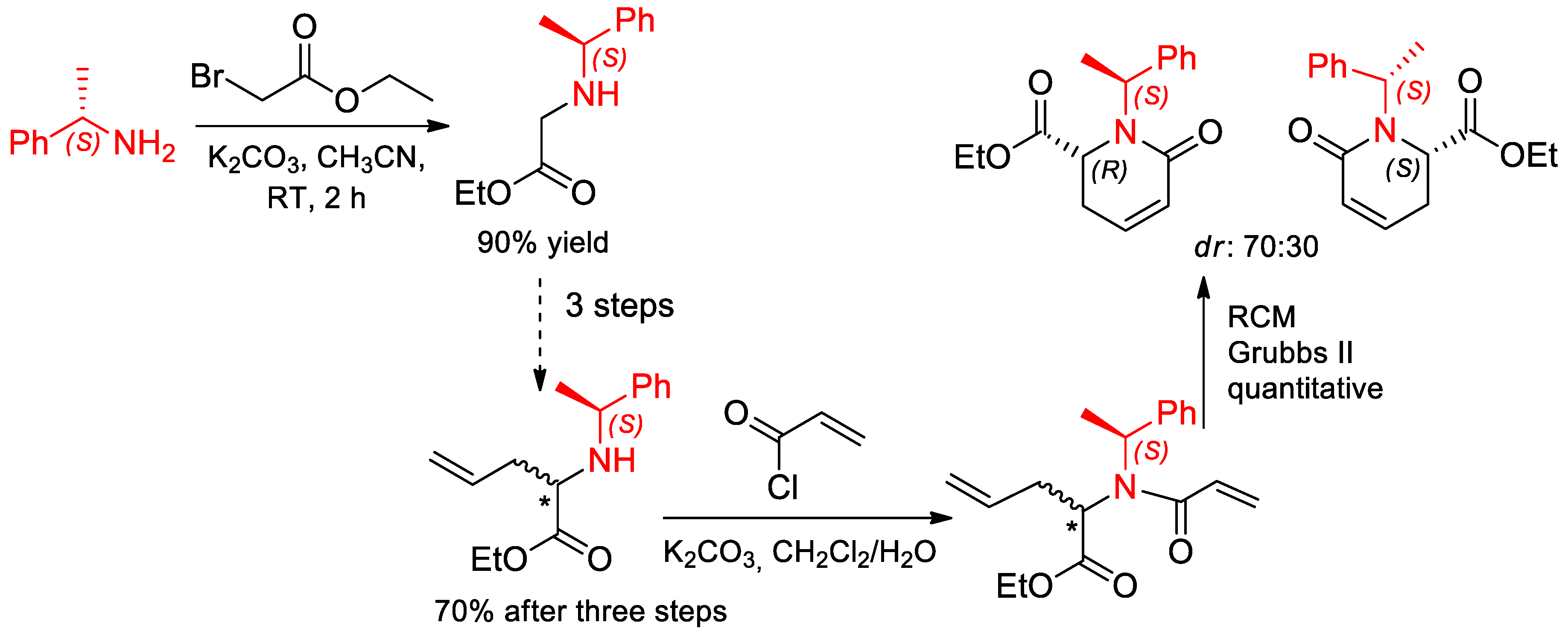{kind=link}
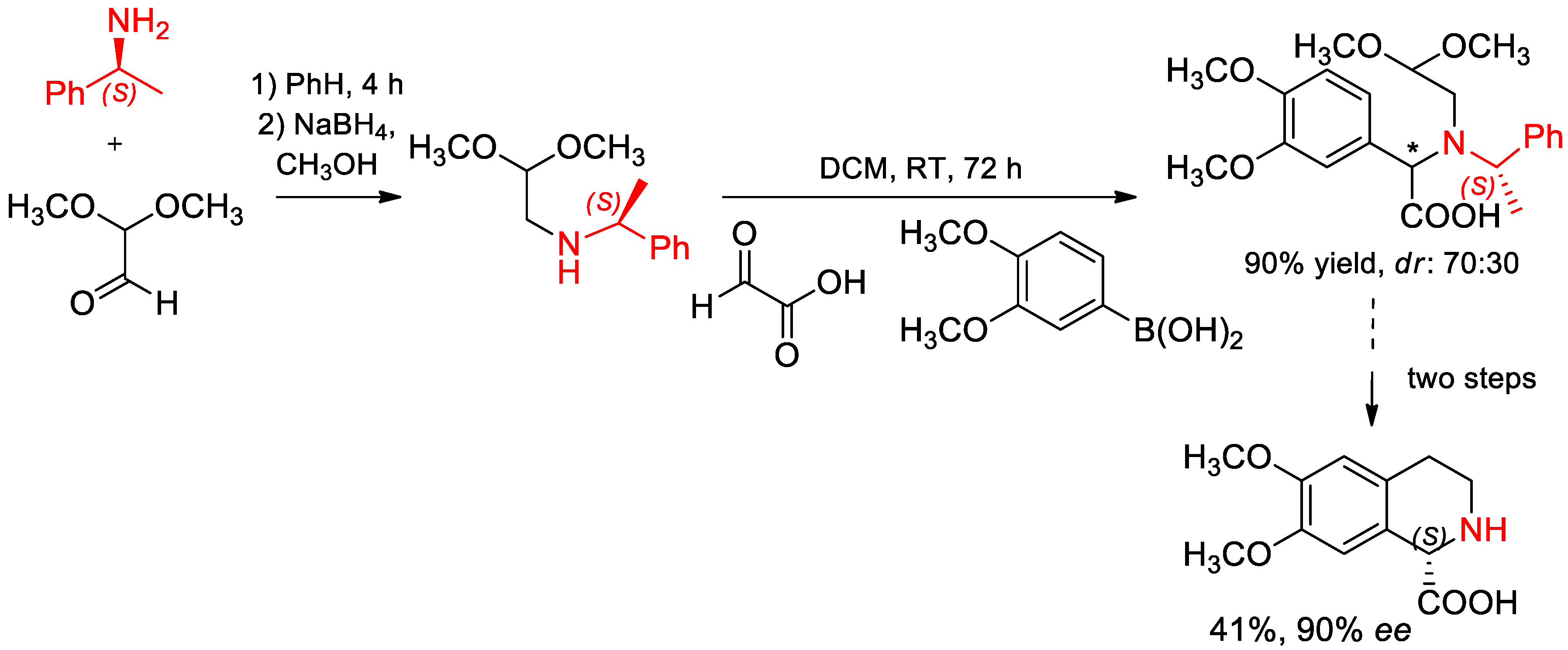{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Improvements in the Synthesis of α-PEA and Its Derivatives
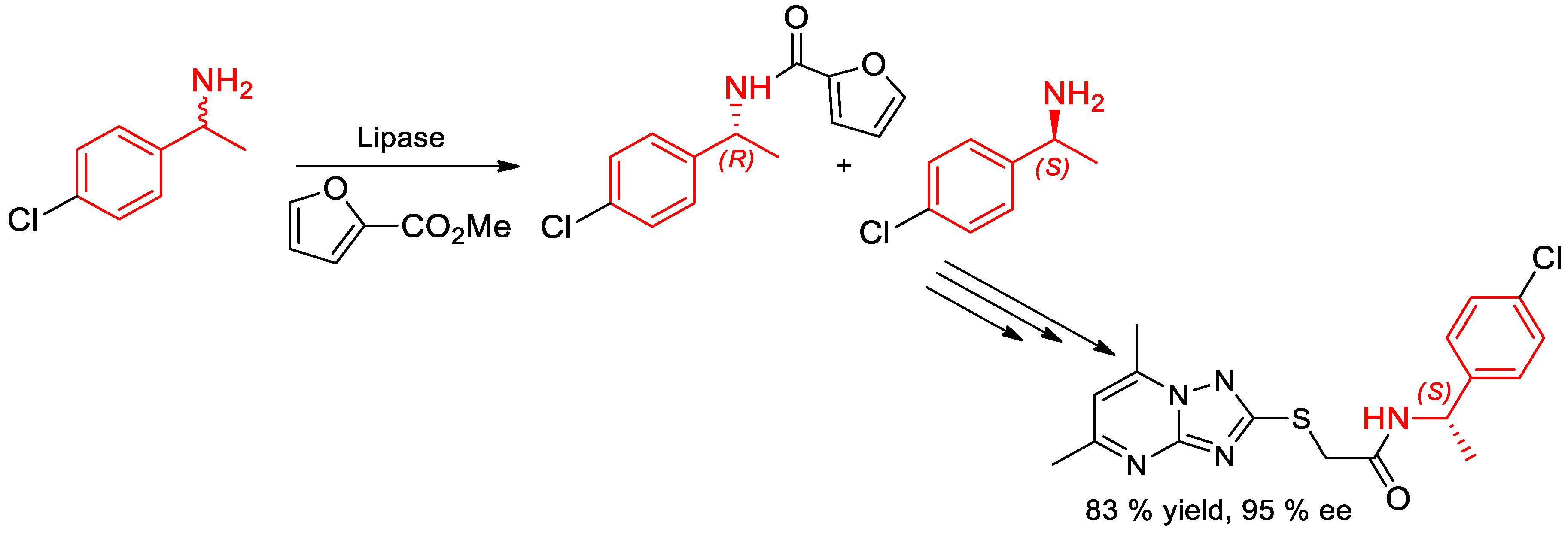
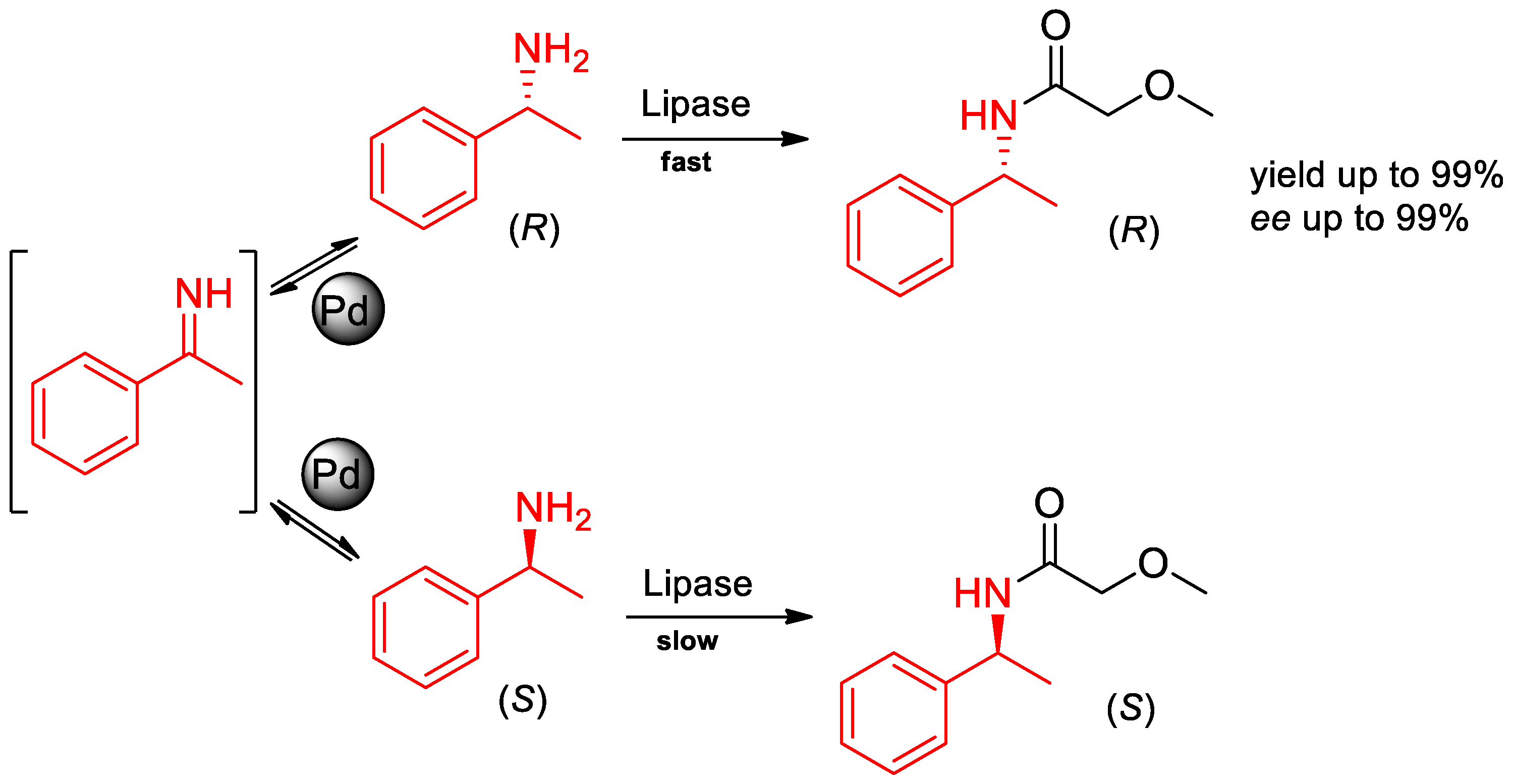
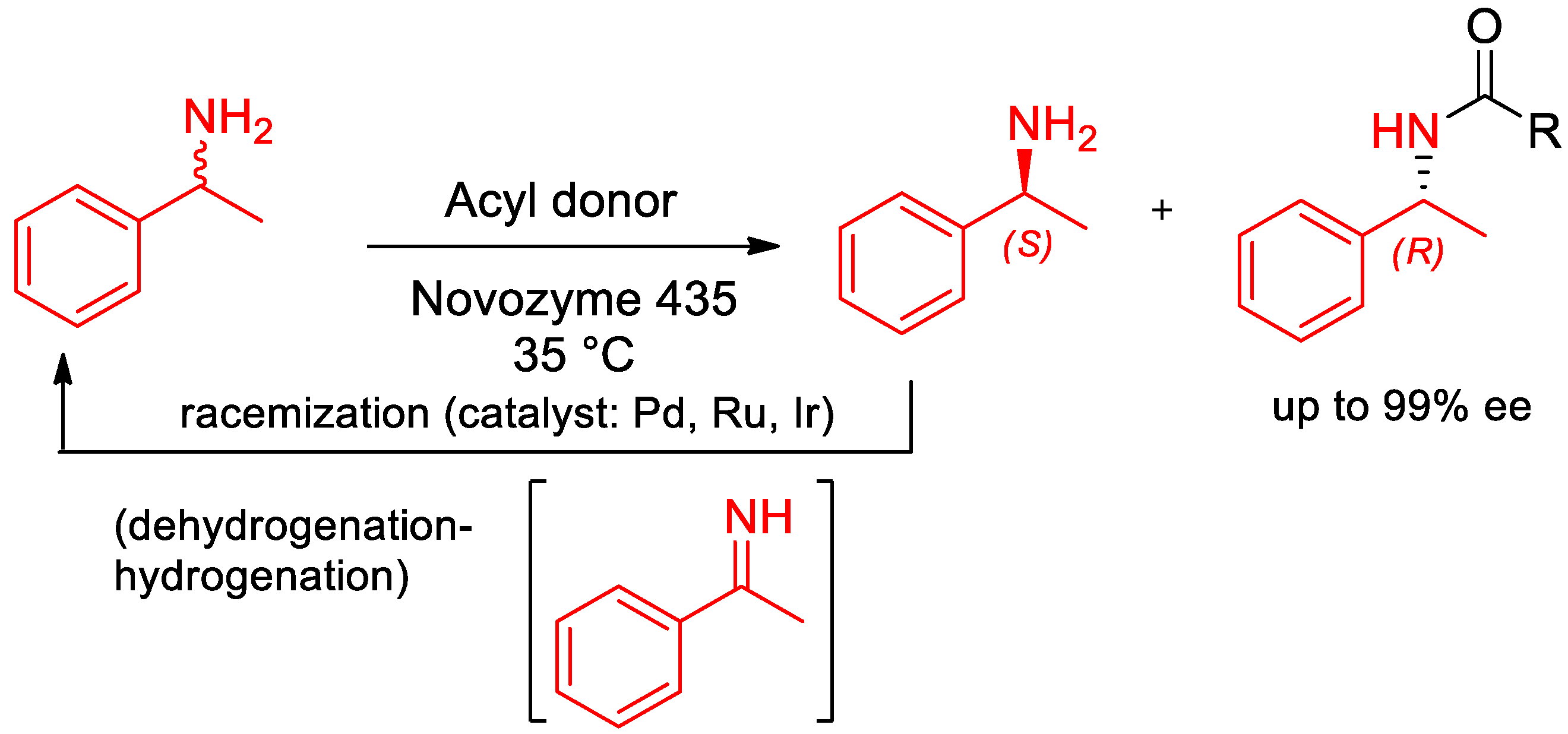
3. Chiral Resolution of α-PEA and Its Derivatives
4. Application of Enantiomeric α-PEA in Chiral Resolution
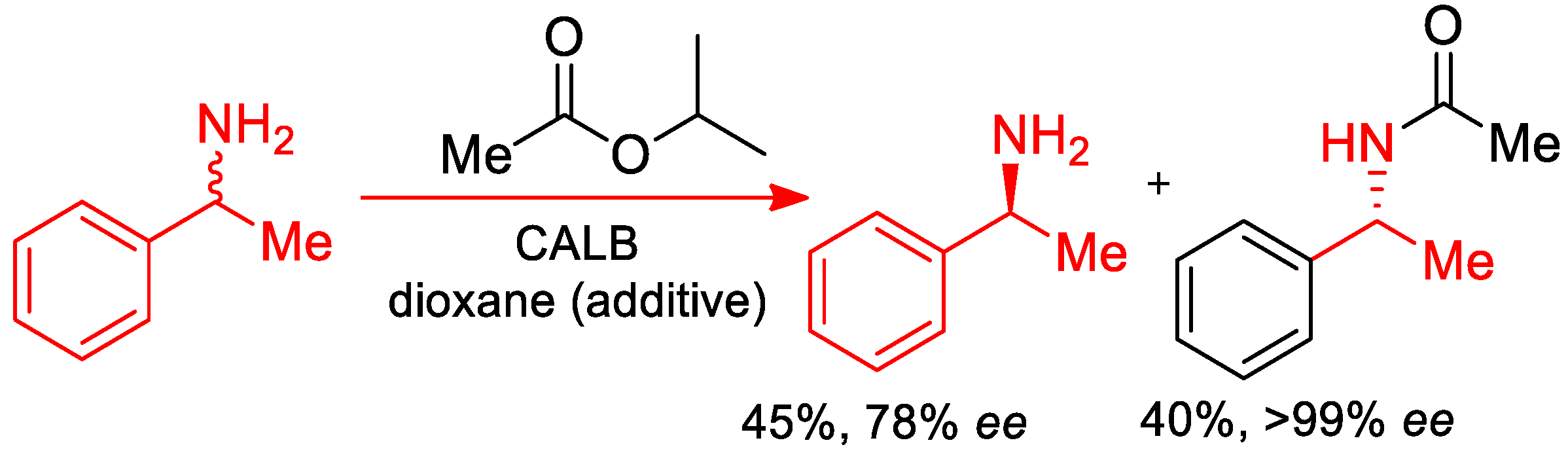
5. Enantiomeric Enrichment of α-PEA and Its Derivatives—Self-Disproportionation of Enantiomers (SDE)
6. α-PEA as a Chiral Auxiliary in Diastereoselective Synthesis
6.1. Synthesis of Compounds with Biological Activity
6.2. Synthesis of Imides
6.3. α-PEA and Its Derivatives in the Diastereoselective Cyclization Reaction
6.4. Asymmetric Aminocarbonylation of Iodoalkenes
6.5. Multicomponent Amino Alkylations
6.6. Novel Derivatives of α-PEA. Synthesis of Enantiomeric Guanidines and Guanidinium Salts
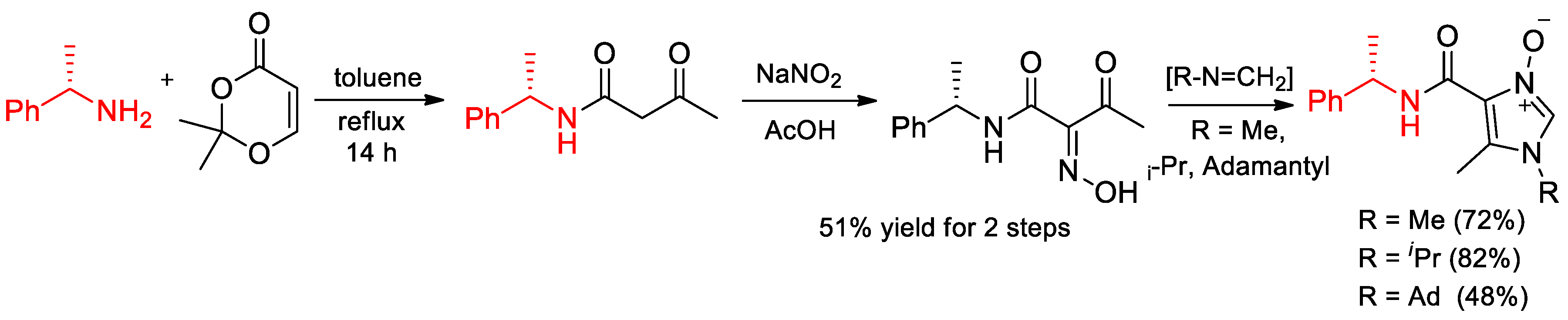
6.7. Novel Derivatives of α-PEA. Synthesis of Enantiomeric Derivative of Imidazole N-Oxides
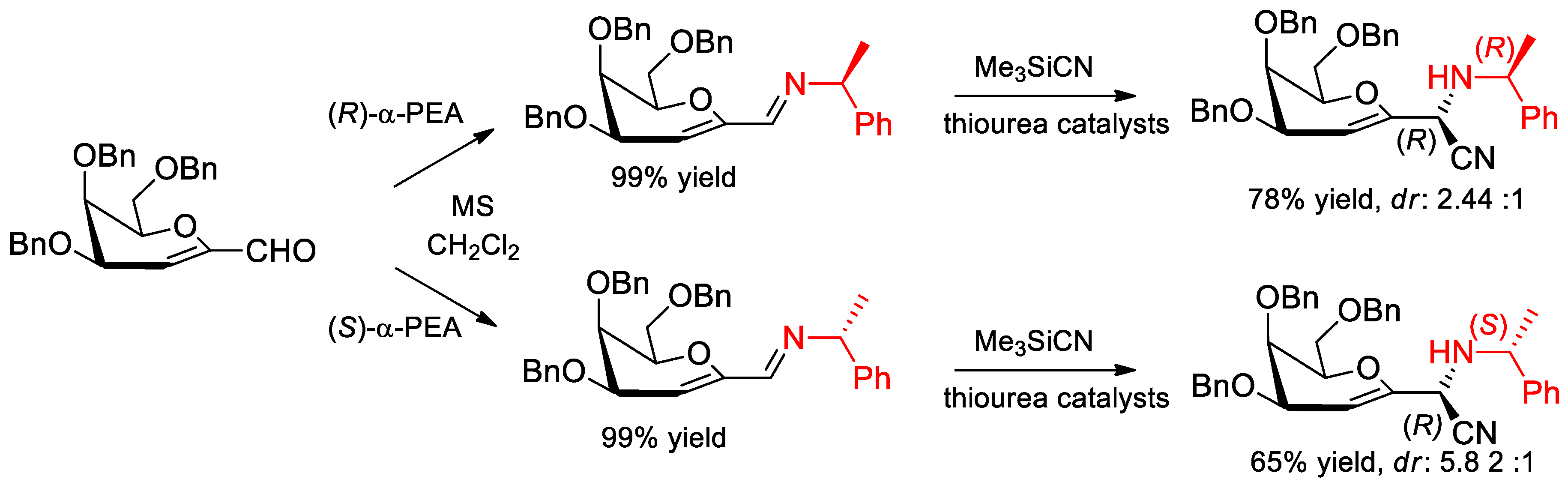
6.8. α-PEA in the Strecker Reaction
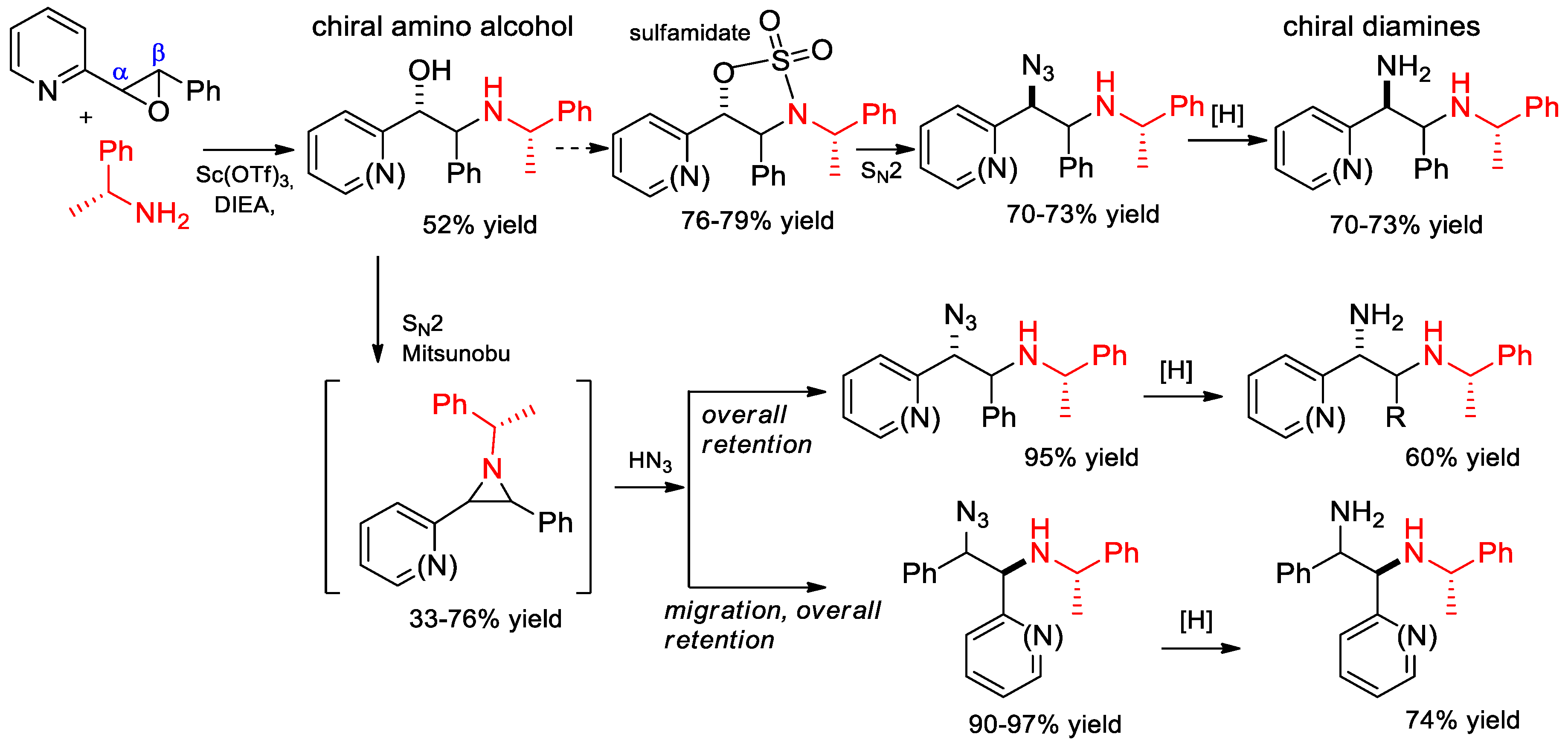
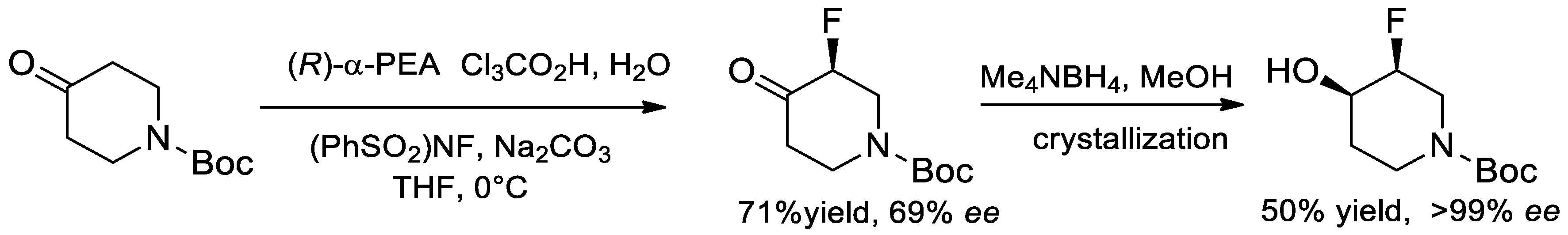
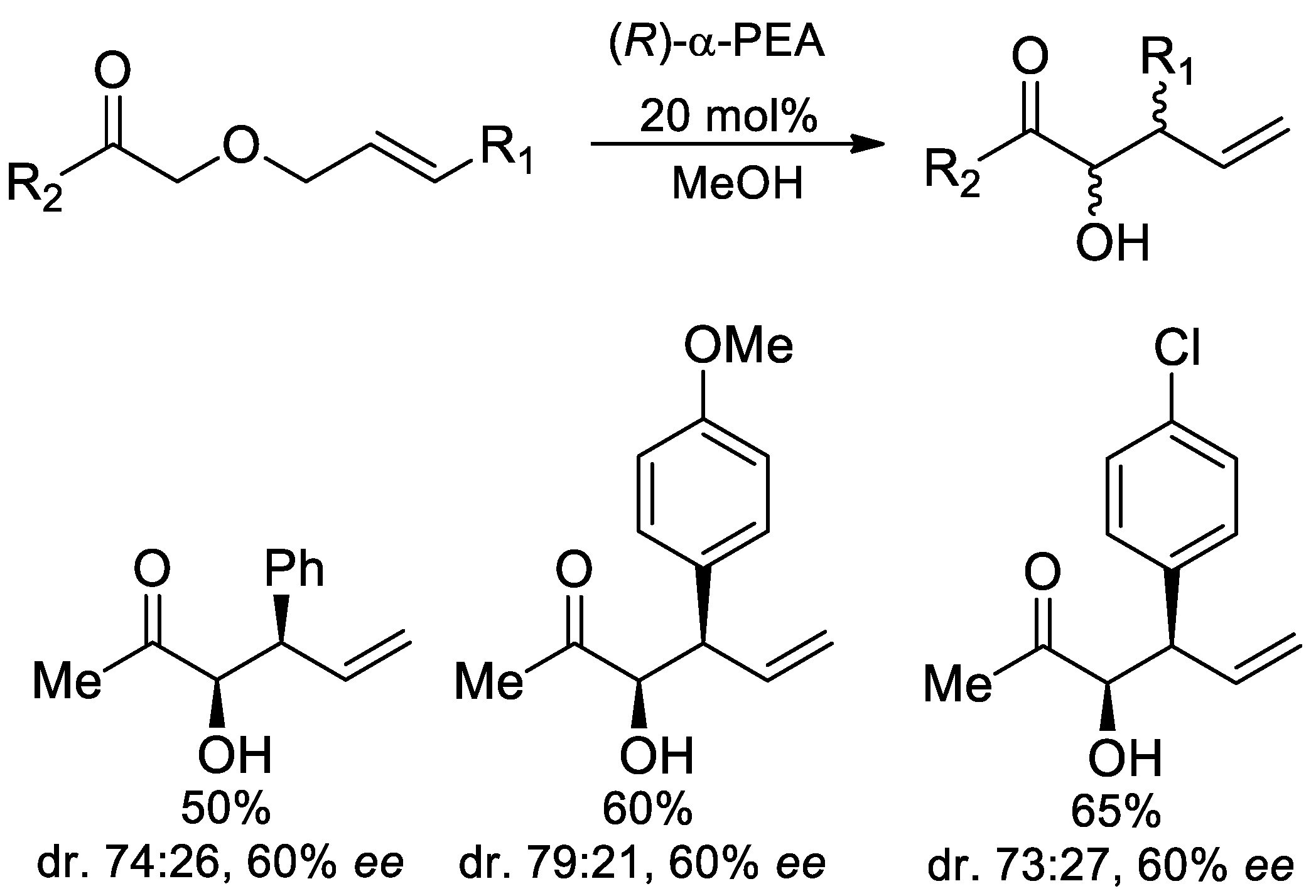
6.9. Synthesis of Chiral Amino Alcohols and Diamines from α-PEA
6.10. Removal of α-PEA Used as Chiral Auxiliary
7. Synthesis and Application of Chiral Ligands with α-PEA Fragments in Asymmetric Reactions
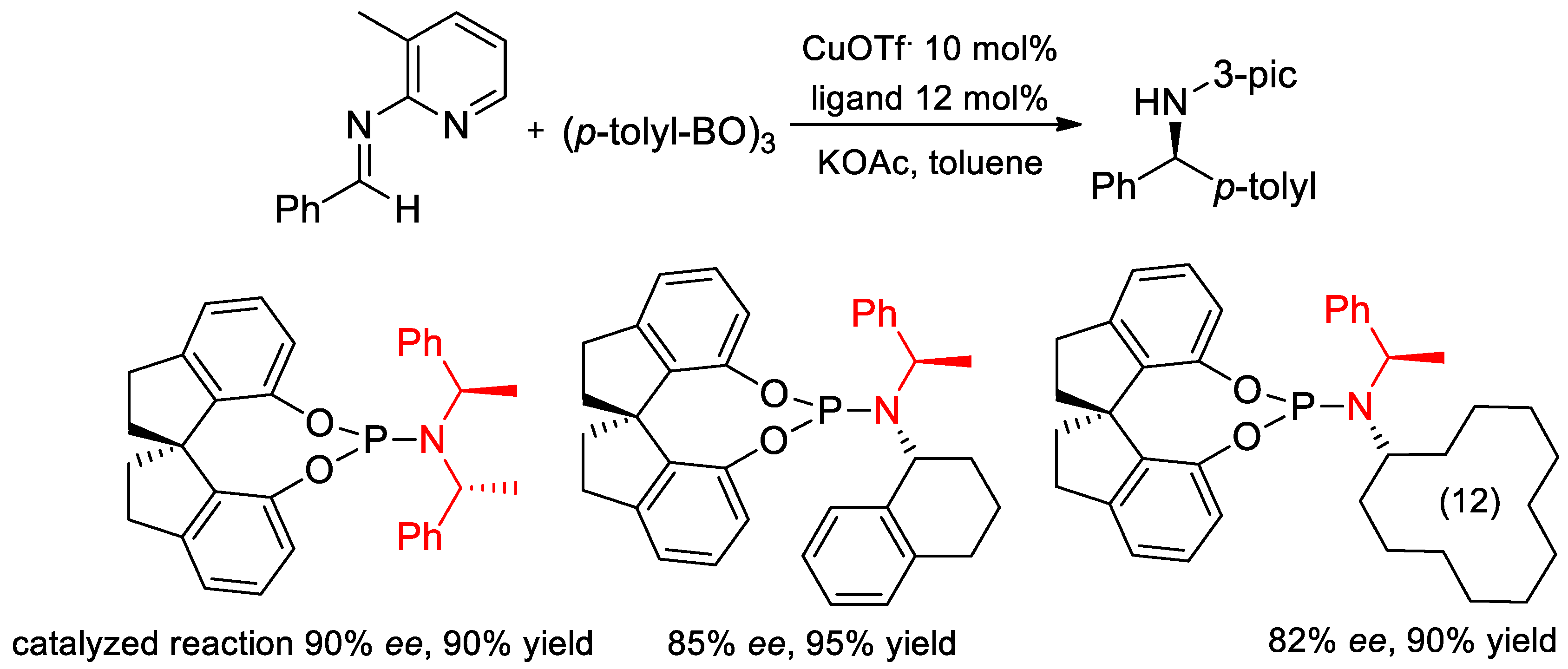
7.1. Enantioselective Arylation of N-Heteroaryl Aldimines and Hydrogenation Catalyzed by Ligands with PEA Fragments
7.2. Catalyzed Enantioselective Addition of Organo-Zinc to Aldehydes
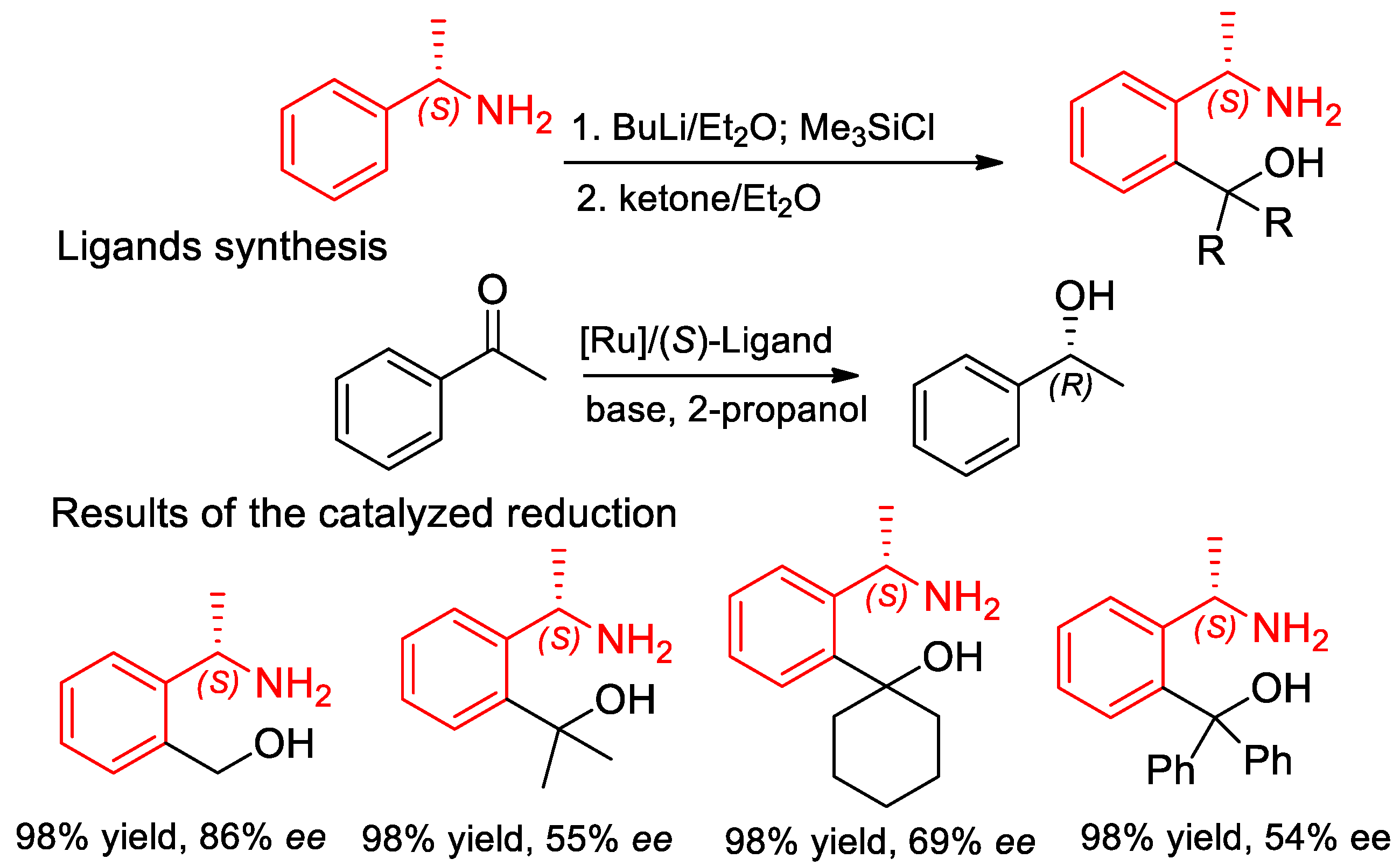
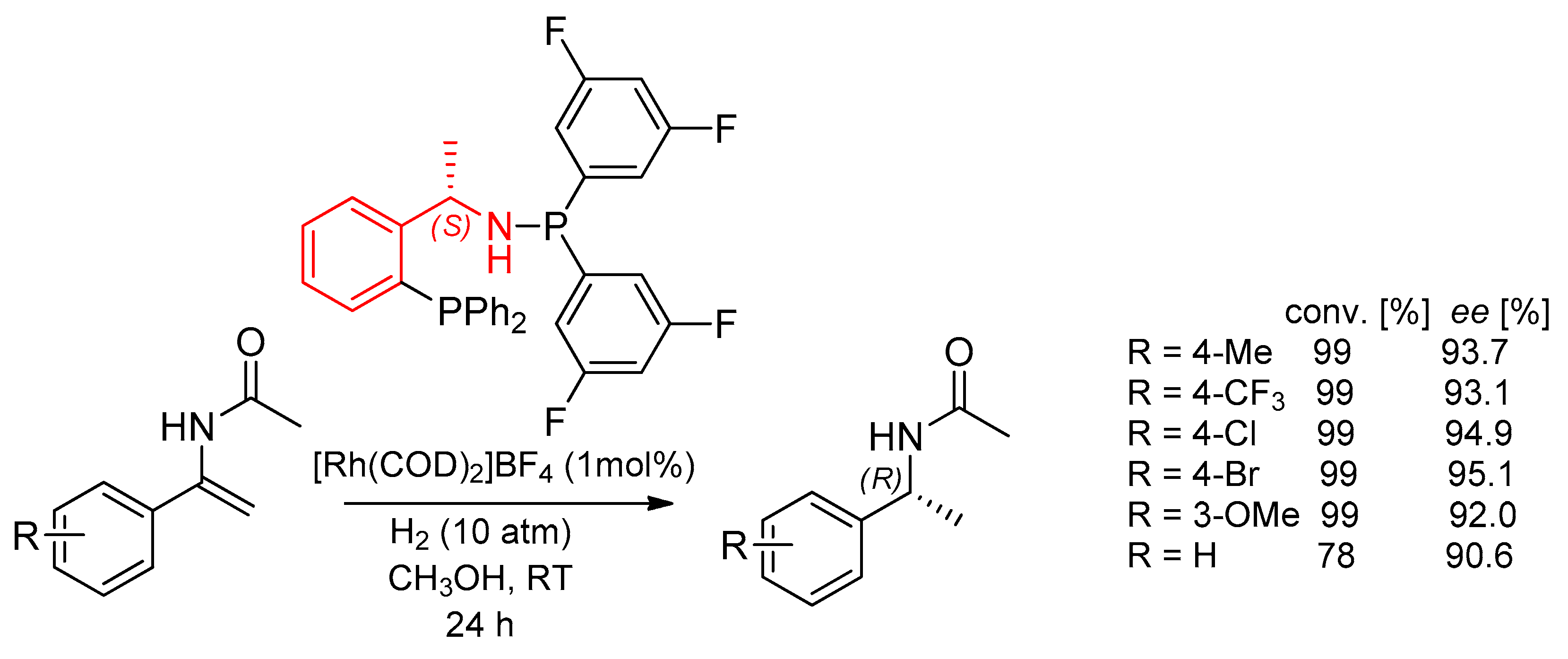
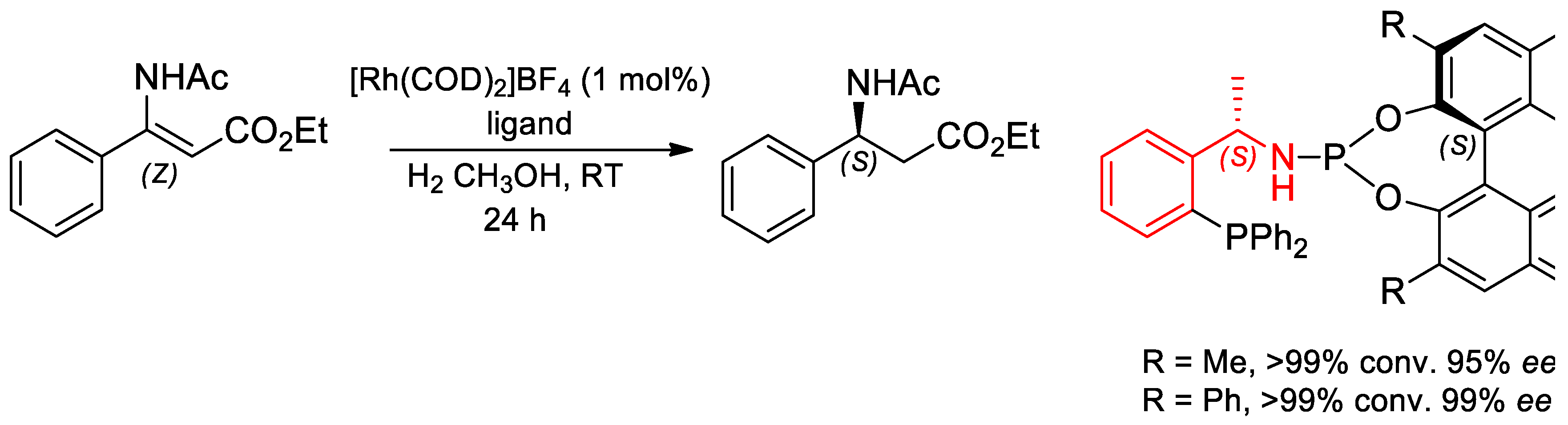
7.3. Asymmetric Hydrogenation Reactions
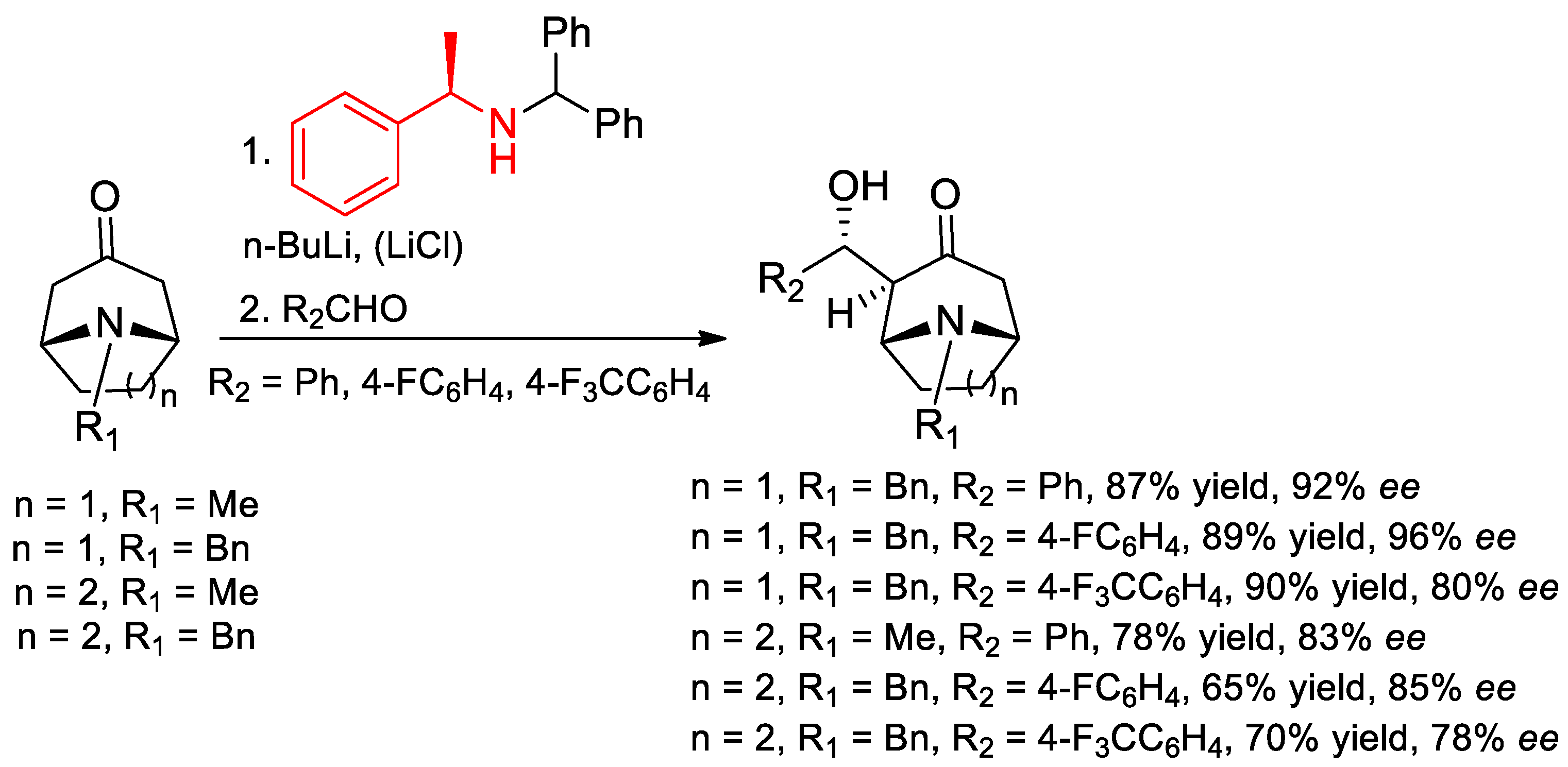
7.4. Deprotonations of Bicyclic Amino Ketones
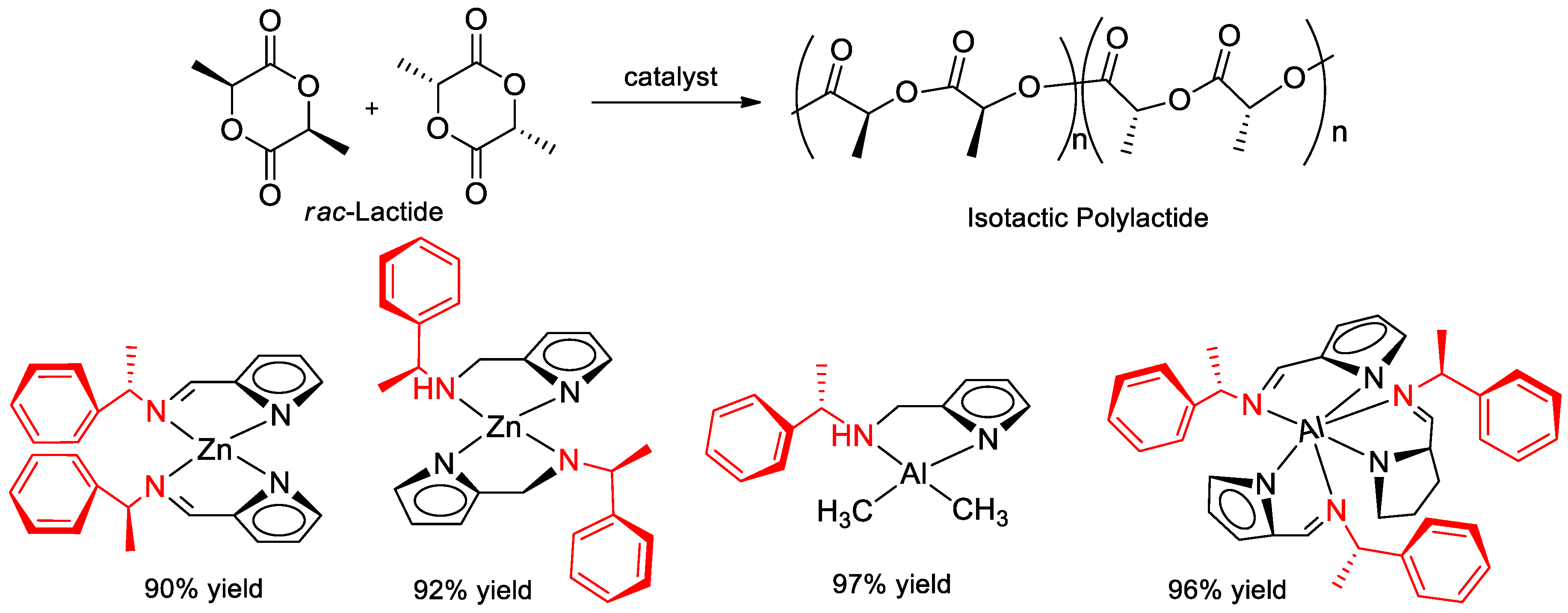
7.5. Polymerization of Rac-Lactide
8. α-PEA and Its Derivatives in Organocatalysis
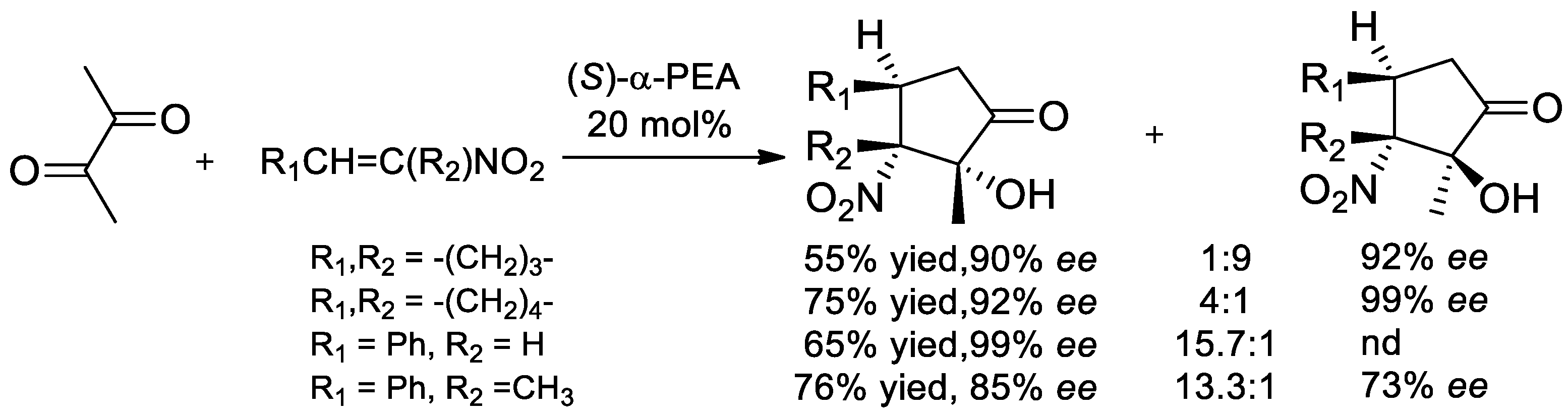
8.1. Direct Use of α-PEA as an Organocatalyst
8.2. Enantioselective Aldol Addition
8.3. Enantioselective Micheal Addition
9. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Ingersoll, A.W. α-phenylethylamine. Org. Synth. 1937, 17, 76. [Google Scholar] [CrossRef]
- Tse-Lok, H.; Fieser, M.; Fieser, L. (Eds.) Fiesers’ Reagents for Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1967; Volume 1, p. 838, 1969; Volume 2, p. 271; 1972; Volume 3, p. 199; 1975; Volume 5, p. 441; 1977, Volume 6, pp. 162, 457; 1980; Volume 8, p. 393; 1981; Volume 9, p. 363; 1982, Volume 10, p. 67; 1984, Volume 11, p. 411; 1986; Volume 12, p. 319; 1989; Volume 14, p. 257; 1994; Volume 17, p. 276. [Google Scholar]
- Juaristi, E.; Escalante, J.; León-Romo, J.L.; Reyes, A. Recent applications of α-phenylethylamine (α-PEA) in the preparation of enantiopure compounds. Part 1: Incorporation in chiral catalysts. Part 2: α-PEA and derivatives as resolving agents. Tetrahedron Asymmetry 1998, 9, 715–740. [Google Scholar] [CrossRef]
- Juaristi, E.; León-Romo, J.L.; Reyes, A.; Escalante, J. Recent applications of α-phenylethylamine (α-PEA) in the preparation of enantiopure compounds. Part 3: α-PEA as chiral auxiliary. Part 4: α-PEA as chiral reagent in the stereodifferentiation of prochiral substrates. Tetrahedron Asymmetry 1999, 10, 2441–2495. [Google Scholar] [CrossRef]
- Bandala, Y.; Juaristi, E. Recent Advances in the Application od α-Phenylethylamine (α-PEA) in the Preparation of Enantiopure Compounds. Aldrichim. Acta 2010, 43, 65–78. [Google Scholar]
- Carreira, E.M.; Yamamoto, H. (Eds.) 1-Phenylethylamine in Comprehensive Chirality, Reference Work; Elsevier: Oxford, UK, 2012; Available online: https://www.sciencedirect.com/topics/chemistry/1-phenylethylamine (accessed on 18 September 2020).
- Smirnov, Y.D.; Tomilov, A.P. Electrochemical synthesis of 1-phenylethylamine. Russ. J. Electrochem. 1996, 32, 106–109, Chem. Abstr.1996, 124, 158674. [Google Scholar]
- Uthoff, F.; Groeger, H. Asymmetric Synthesis of 1-Phenylethylamine from Styrene via Combined Wacker Oxidation and Enzymatic Reductive Amination. J. Org. Chem. 2018, 83, 9517–9521. [Google Scholar] [CrossRef]
- Kodama, K.; Kimura, Y.; Shitara, H.; Yasutake, M.; Sakurai, R.; Hirose, T. Solvent-induced chirality control in the enantioseparation of 1-phenylethylamine via diastereomeric salt formation. Chirality 2011, 23, 326–332. [Google Scholar] [CrossRef]
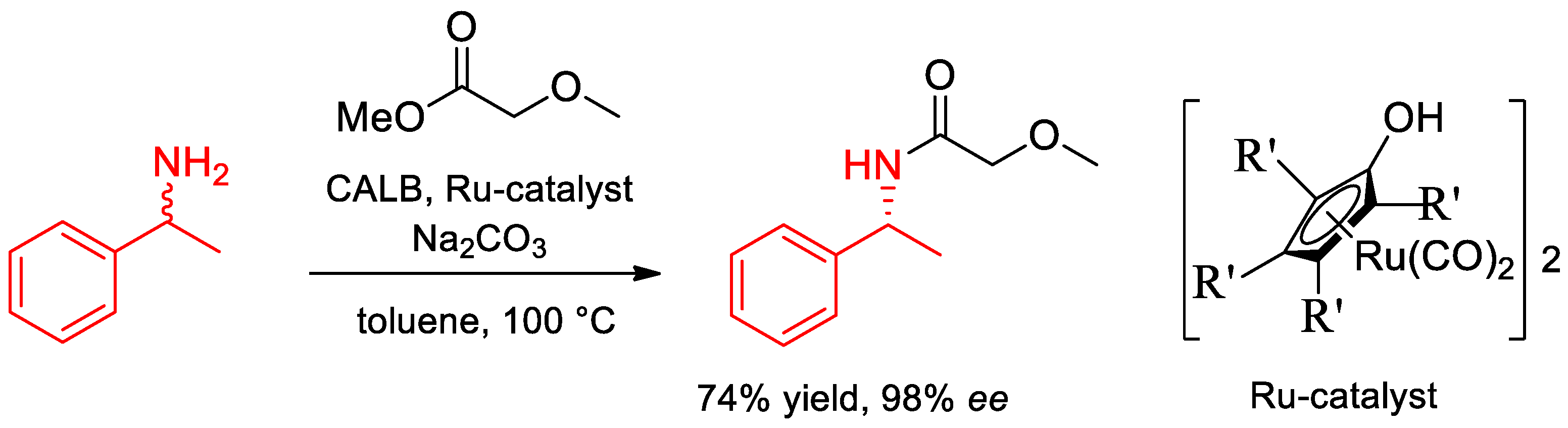
- Päiviö, M.; Perkiö, P.; Kanerva, L.T. Solvent-free kinetic resolution of primary amines catalyzed by Candida Antarctica lipase B: Effect of immobilization and recycling stability. Tetrahedron Asymmetry 2012, 23, 230–236. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, F.; Yan, H.; Zheng, J.; Wang, Z. The enzymatic resolution of 1-(4-chlorophenyl)ethylamine by Novozym 435 to prepare a novel triazolopyrimidine herbicide. Chirality 2018, 30, 1225–1232. [Google Scholar] [CrossRef]
- Gustafson, K.P.J.; Lihammar, R.; Verho, O.; Engström, K.; Bäckvall, J.-E. Chemo-enzymatic Dynamic Kinetic Resolution of Primary Amines Using a Recyclable Palladium Nanoparticle Catalyst Together with Lipases. J. Org. Chem. 2014, 79, 3747–3751. [Google Scholar] [CrossRef]
- Mirinda, A.S.; Mirinda, L.S.M.; Souza, R.O.M.A. Ethyl acetate as an acyl donor in the continuous flow kinetic resolution of (±)-1-phenylethylamine catalyzed by lipases. Org. Biomol. Chem. 2013, 11, 3332–3336. [Google Scholar] [CrossRef] [PubMed]
- Perez-Venegas, M.; Juaristi, E. Mechanoenzymatic resolution of racemic chiral amines, a green technique for the synthesis of pharmaceutical building blocks. Tetrahedron 2018, 74, 6453–6458. [Google Scholar] [CrossRef]
- Thalén, L.K.; Bäckvall, J.-E. Development of dynamic kinetic resolution on large scale for (±)-1-phenylethylamine. Beilstein J. Org. Chem. 2010, 6, 823–829. [Google Scholar] [CrossRef] [PubMed]
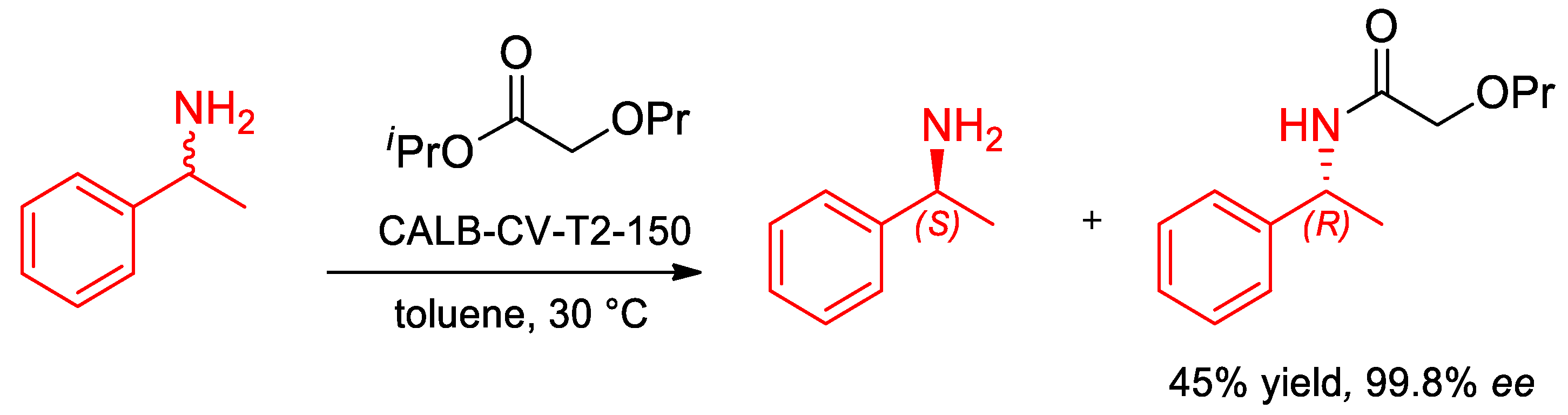
- Oláh, M.; Kovács, D.; Katona, G.; Hornyánszky, G.; Poppe, L. Optimization of 2-alkoxyacetates as acylating agent for enzymatic kinetic resolution of chiral amines. Tetrahedron 2018, 74, 3663–3670. [Google Scholar] [CrossRef]
- Peng, Y.; He, Q.; Rohani, S.; Jenkins, H. Resolution of 2-chloromandelic acid with (R)-(+)-N-benzyl-1-phenylethylamine: Chiral discrimination mechanism. Chirality 2012, 24, 349–355. [Google Scholar] [CrossRef]
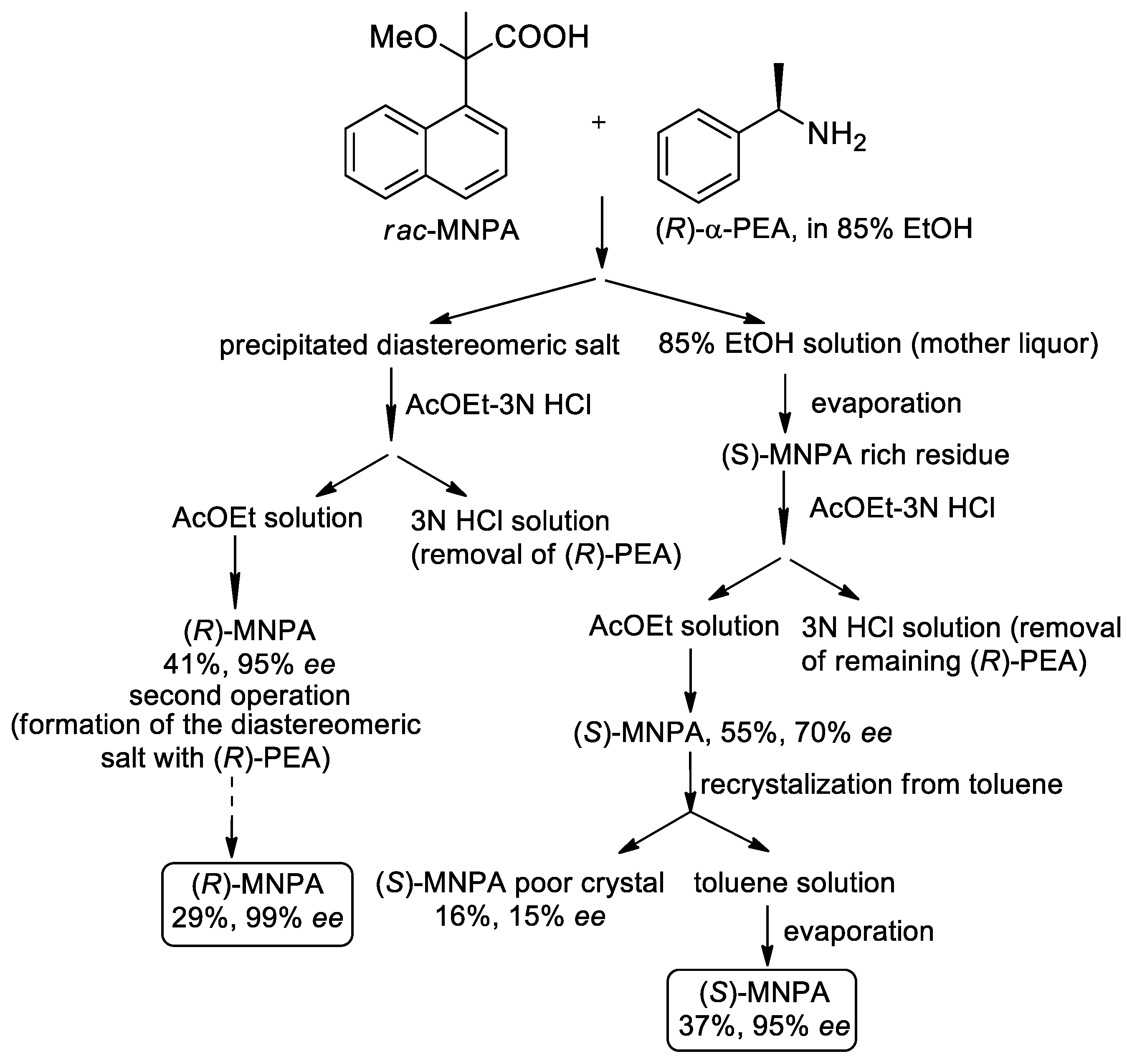
- Kusakari, M.; Ohta, Y.; Nakagawa, H.; Katagiri, H.; Kijima, T.; Murakami, S.; Matsuba, S.; Hatano, B. Enantioresolution of 2-methoxy-2-(1-naphthyl)propionic acid using diastereomeric salt formation with chiral phenylethylamine. Tetrahedron Lett. 2014, 55, 4114–4116. [Google Scholar] [CrossRef]
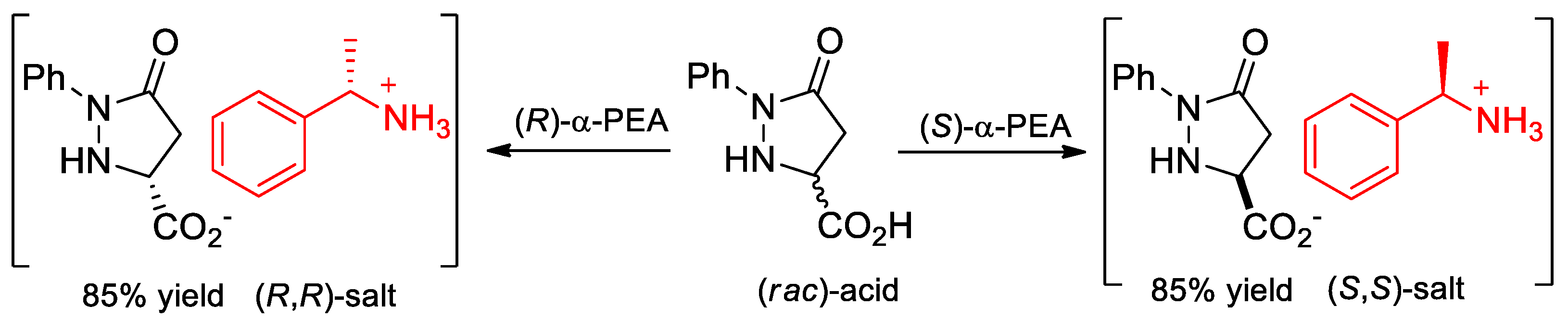
- Juaristi, E.; Melgar-Fernández, R.; González-Olvera, R.; Vargas-Caporali, J.; Pérez-Isidoro, R. Resolution of 5-oxo-1-phenylpyrazolidine-3-carboxylic acid and synthesis of novel enantiopure amide derivatives. Arkivoc 2010, 2010, 55–75. [Google Scholar] [CrossRef]
- Kaboudin, B.; Alaie, S.; Yokomatsu, T. Resolution of enantiomers of α-hydroxy-(o-chlorophenyl)methylphosphinic acid via diastereomeric salt formation with enantiopure 1-phenylethylamines. Tetrahedron Asymmetry 2011, 22, 1813–1816. [Google Scholar] [CrossRef]
- Kovalenko, V.N.; Kulinkovich, O.G. The resolution of trans-2,2-dichloro-3-methylcyclopropanecarboxylic acid via crystallization of its salts with (+)- and (−)-α-phenylethylamine, and the transformation of the resulting enantiomers into (R)- and (S)-dimethyl 2-methylsuccinates. Tetrahedron Asymmetry 2011, 22, 26–30. [Google Scholar] [CrossRef]
- Kovalenko, V.N.; Kozyrkov, Y.Y. A Simple Method for Resolution of Endo-/Exo-Monoesters of Trans-Norborn-5-Ene-2,3-Dicarboxylic Acids Into Their Enantiomers. Chirality 2015, 27, 151–155. [Google Scholar] [CrossRef]
- Gaidai, A.V.; Volochnyuk, D.M.; Shishkin, O.V.; Fokin, A.A.; Levandovskiy, I.A.; Shubina, T.E. D3-Trishomocubane-4-carboxylic Acid as a New Chiral Building Block: Synthesis and Absolute Configuration. Synthesis 2012, 44, 810–816. [Google Scholar] [CrossRef]
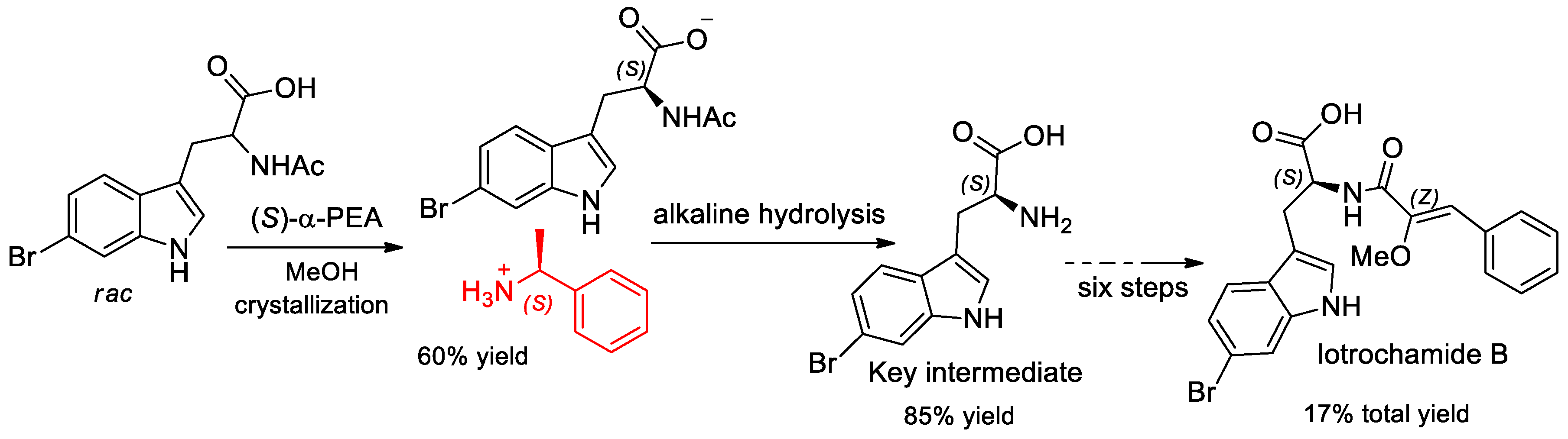
- Wang, S.Y.; Zhao, J.; Que, H.D. Synthesis of the Natural Product Iotrochamide B. Chem. Nat. Compd. 2019, 55, 499–501. [Google Scholar] [CrossRef]
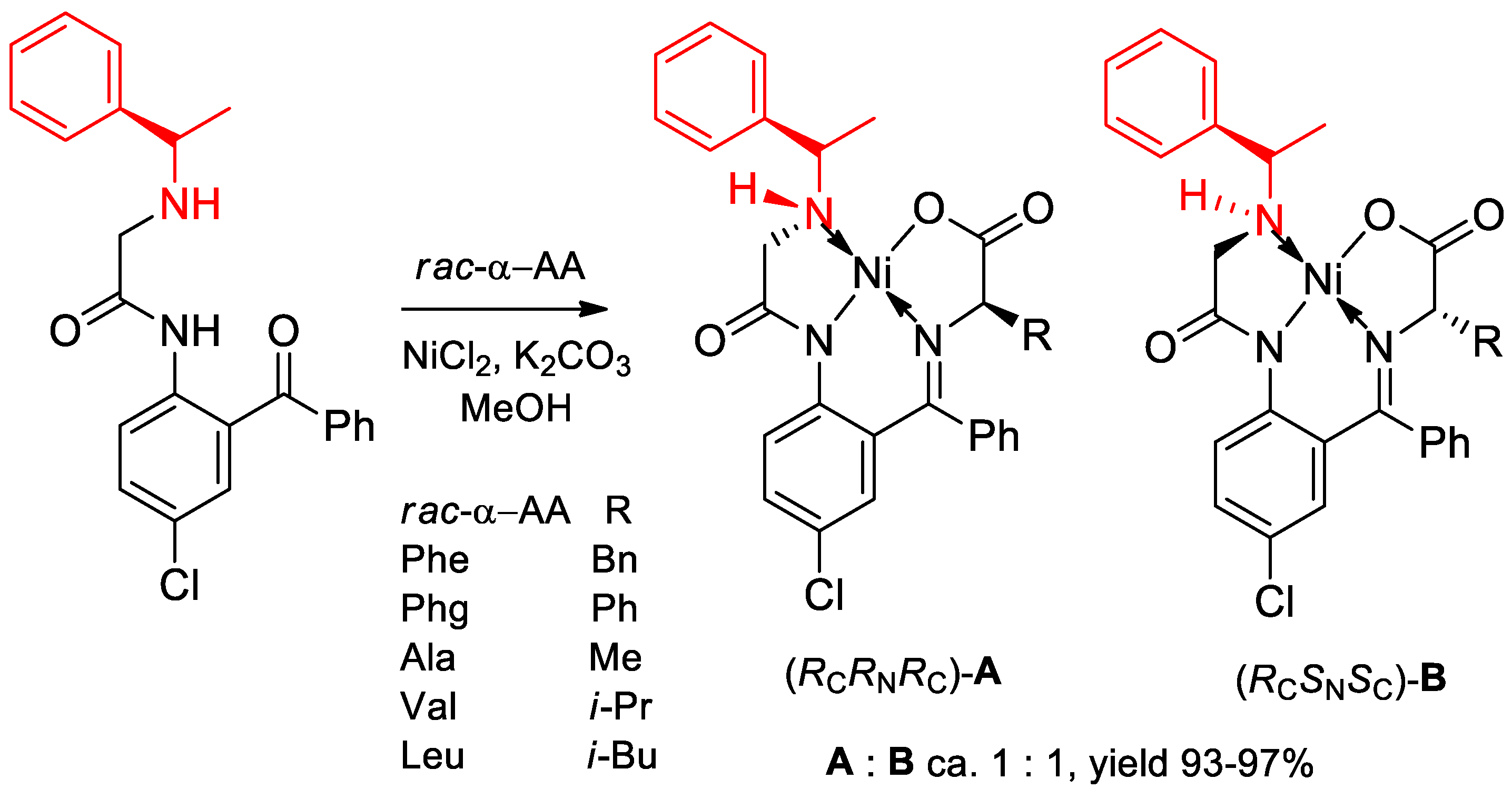
- Takeda, R.; Kawamura, A.; Kawashima, A.; Moriwaki, H.; Sato, T.; Aceña, J.L.; Soloshonok, V.A. Design and synthesis of (S)- and (R)-α-(phenyl)ethylamine-derived NH-type ligands and their application for the chemical resolution of α-amino acids. Org. Biomol. Chem. 2014, 12, 6239–6249. [Google Scholar] [CrossRef] [PubMed]
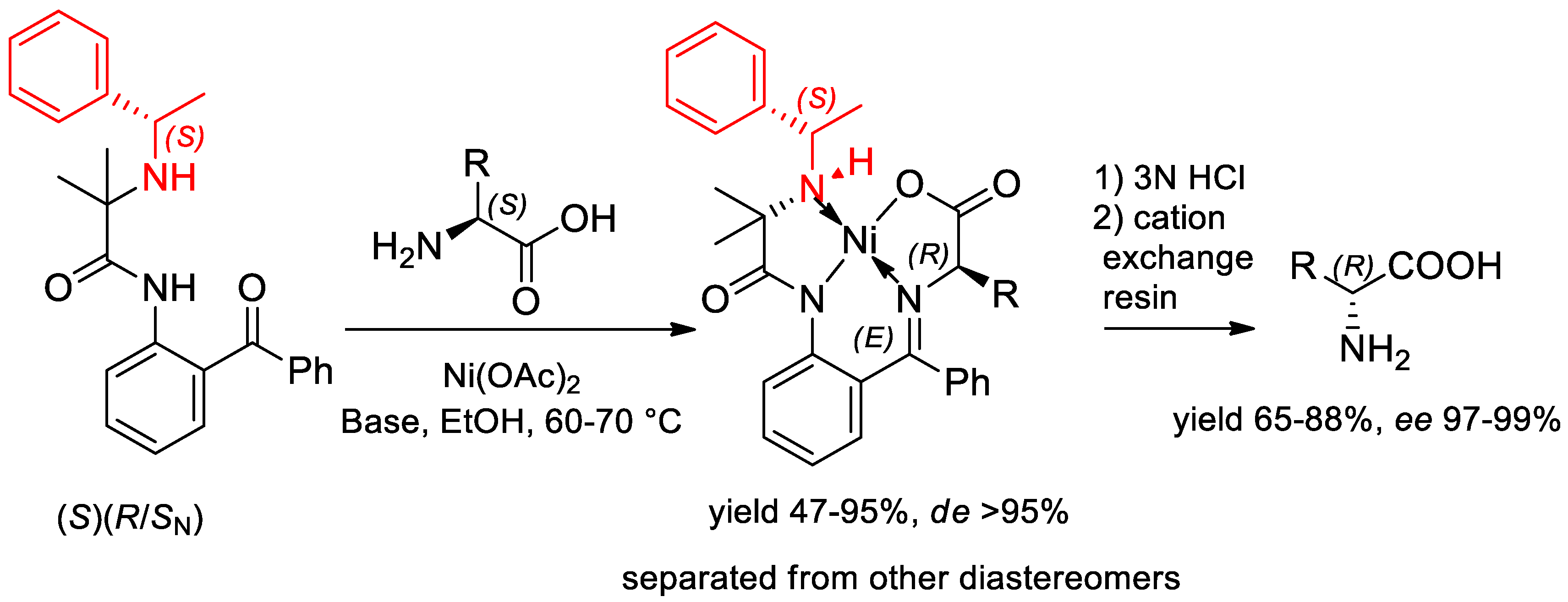
- Sorochinsky, A.E.; Ueki, H.; Aceña, J.L.; Ellis, T.K.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Chemical approach for interconversion of (S)- and (R)-α-amino acids. Org. Biomol. Chem. 2013, 11, 4503–4507. [Google Scholar] [CrossRef] [PubMed]
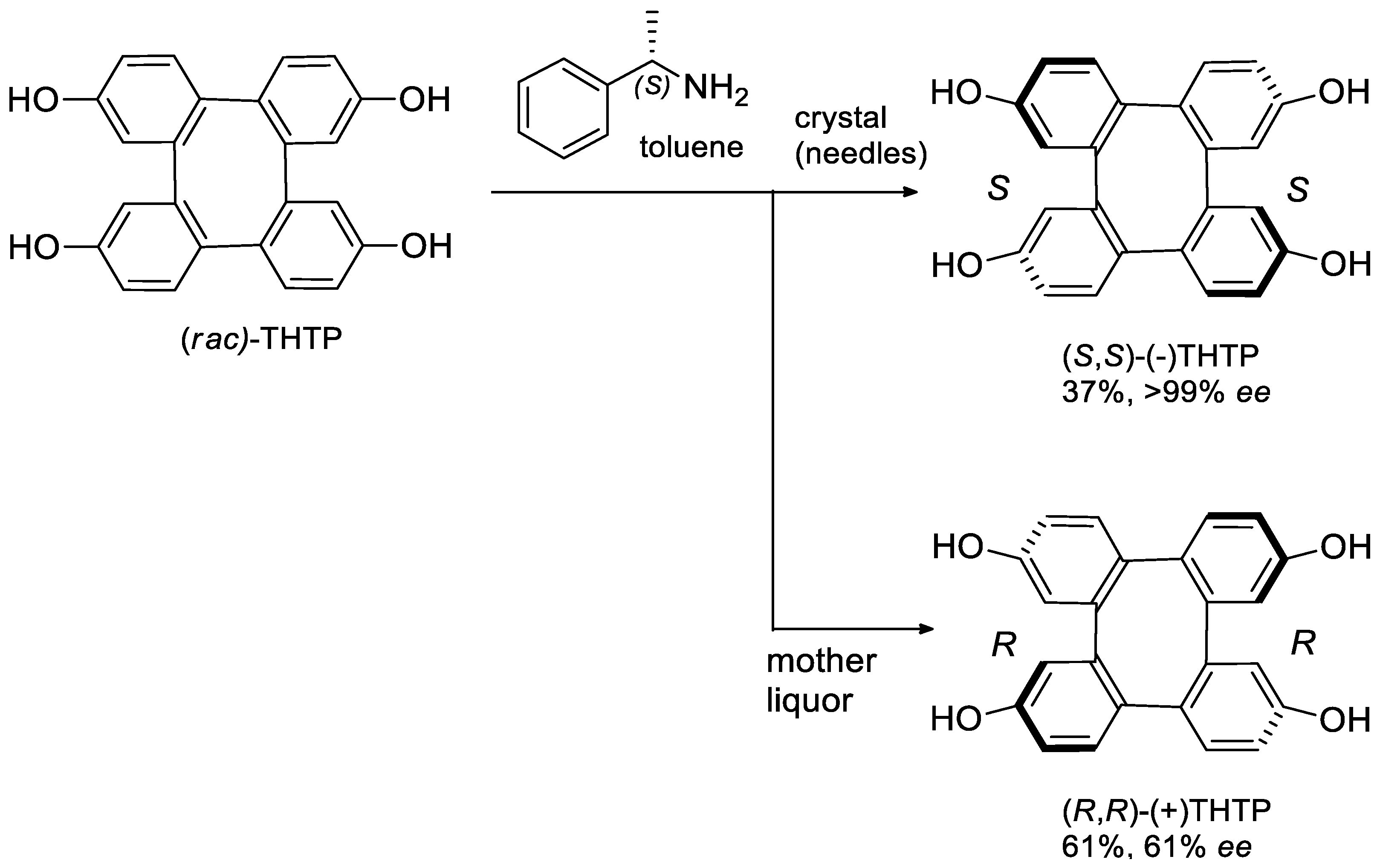
- Kaku, H.; Mitarai, A.; Okamoto, N.; Tanaka, K.; Ichikawa, S.; Yamamoto, T.; Inai, M.; Nishii, T.; Horikawa, M.; Tsunoda, T. Optically Active 2,7,10,15-Tetrahydroxytetraphenylene: Clathrates with Both Enantiomers of 1-Phenylethylamine and Their Stability. Eur. J. Org. Chem. 2018, 6991–6999. [Google Scholar] [CrossRef]
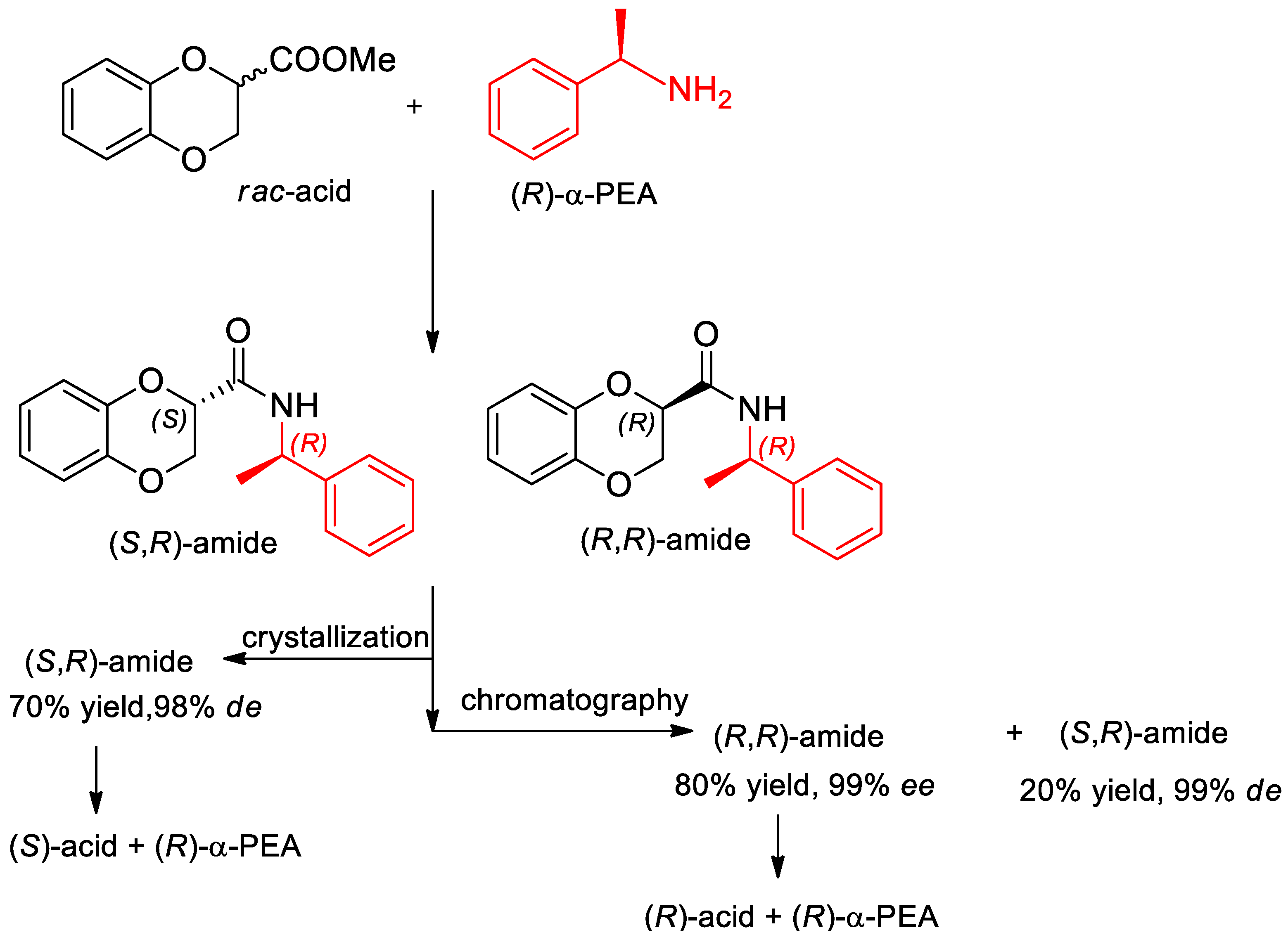
- Fumagalli, L.; Bolchi, C.; Bavo, F.; Pallavicini, M. Crystallization-based resolution of 1,4-benzodioxane-2-carboxylic acid enantiomers via diastereomeric 1-phenylethylamides. Tetrahedron Lett. 2016, 57, 2009–2011. [Google Scholar] [CrossRef]
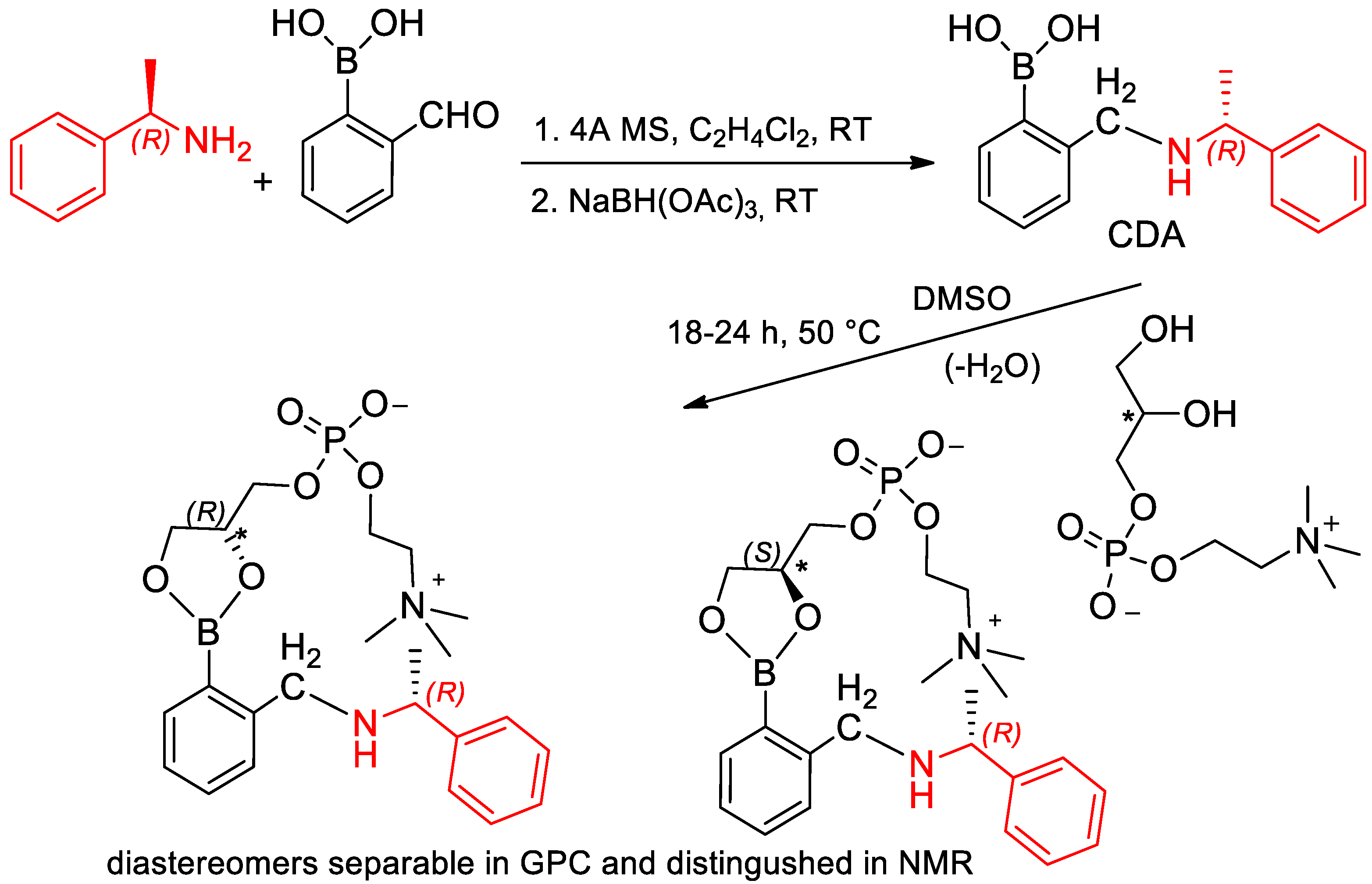
- Kelly, A.M.; Pe’rez-Fuertes, Y.; Arimori, S.; Bull, S.D.; James, T.D. Simple Protocol for NMR Analysis of the Enantiomeric Purity of Diols. Org. Lett. 2006, 8, 1971–1974. [Google Scholar] [CrossRef]
- De Ferra, L.; Massa, A.; Di Mola, A.; Diehlc, B. An effective method for the determination of the enantio-purity of L-glycerophosphocholine (L-GPC). J. Pharm. Biomed. Anal. 2020, 183, 113152. [Google Scholar] [CrossRef]
- Shabbir, S.H.; Regan, C.J.; Anslyn, E.V. A general protocol for creating high-throughput screening assays for reaction yield and enantiomeric excess applied to hydrobenzoin. Proc. Natl. Acad. Sci. USA 2009, 106, 10487–10492. [Google Scholar] [CrossRef]
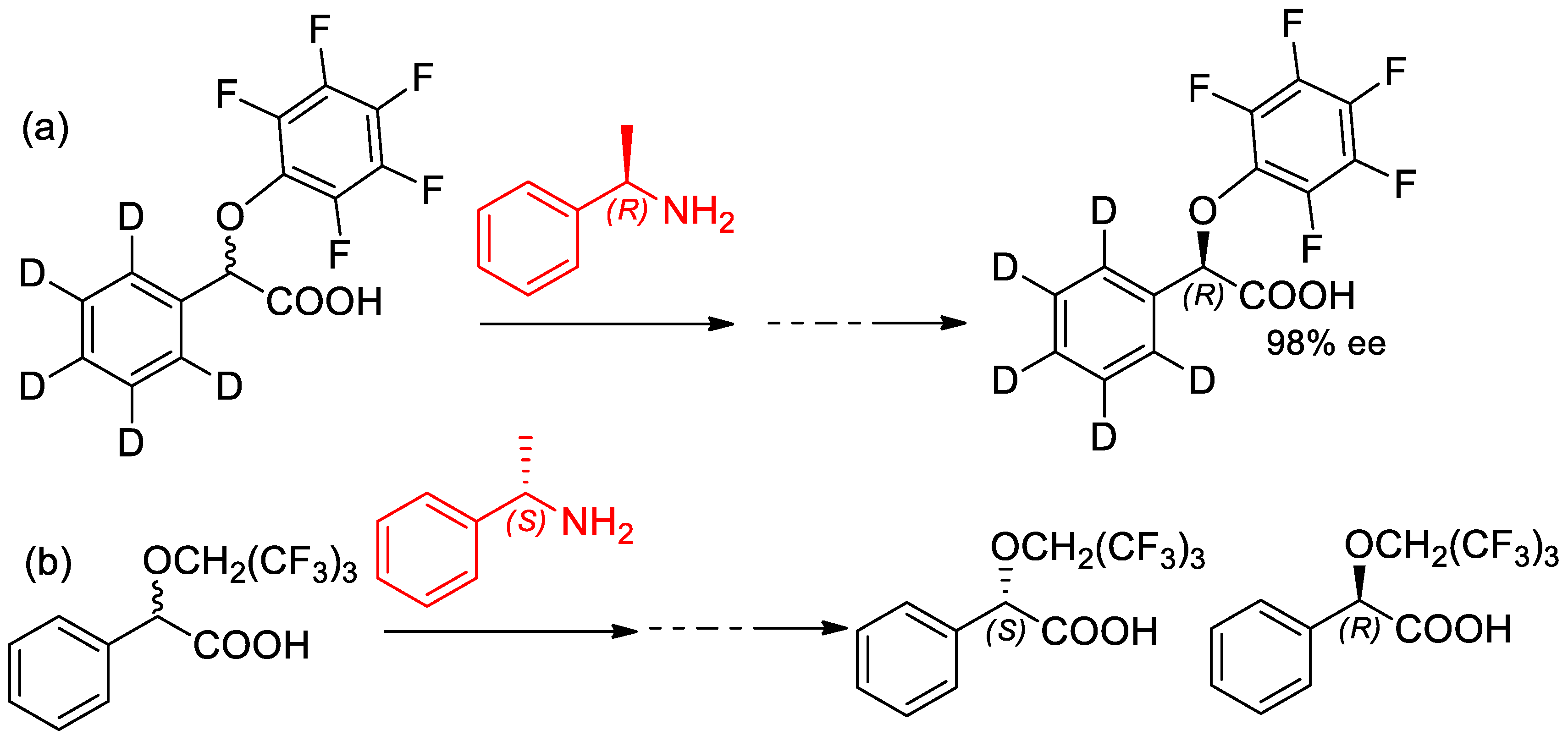
- Cavalluzzi, M.M.; Lovece, A.; Bruno, C.; Franchini, C.; Lentini, G. Preparation of (−)-(R)-2-(2,3,4,5,6-pentafluorophenoxy)-2-(phenyl-d5)acetic acid: An efficient 1H NMR chiral solvating agent for direct enantiomeric purity evaluation of quinoline-containing antimalarial drugs. Tetrahedron Asymmetry 2014, 25, 1605–1611. [Google Scholar] [CrossRef]
- Nemes, A.; Csóka, T.; Béni, S.; Farkas, V.; Rábai, J.; Szabó, D. Chiral Recognition Studies of α-(Nonafluoro-tert-butoxy)carboxylic Acids by NMR Spectroscopy. J. Org. Chem. 2015, 80, 6267–6274. [Google Scholar] [CrossRef] [PubMed]
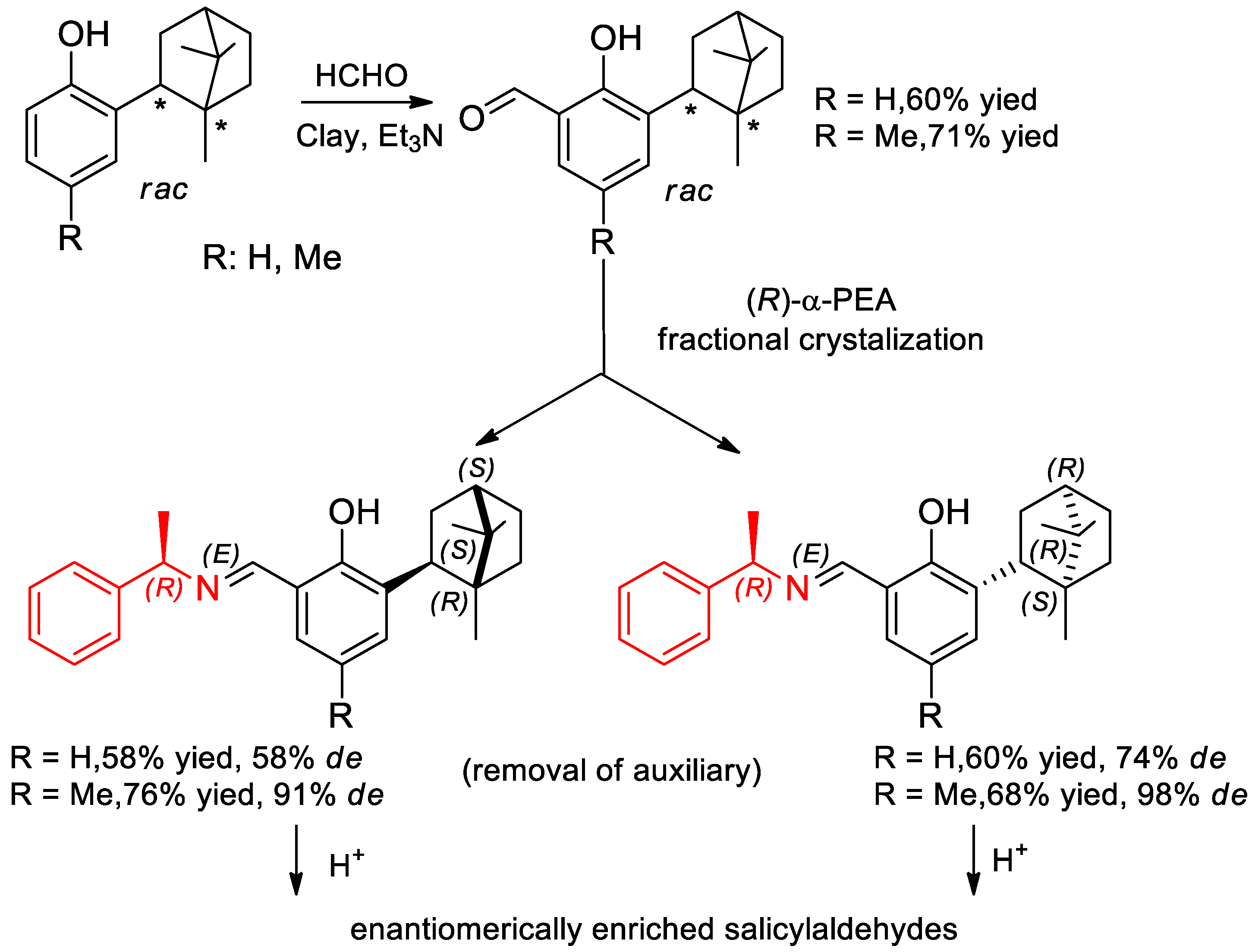
- Buravlev, E.V.; Chukicheva, I.Y.; Dolgushin, F.M.; Kuchin, A.V. Separation of racemic salicylaldehydes containing isobornyl substituent using (R)-1-phenylethylamine. Russ. J. Org. Chem. 2010, 46, 649–654. [Google Scholar] [CrossRef]
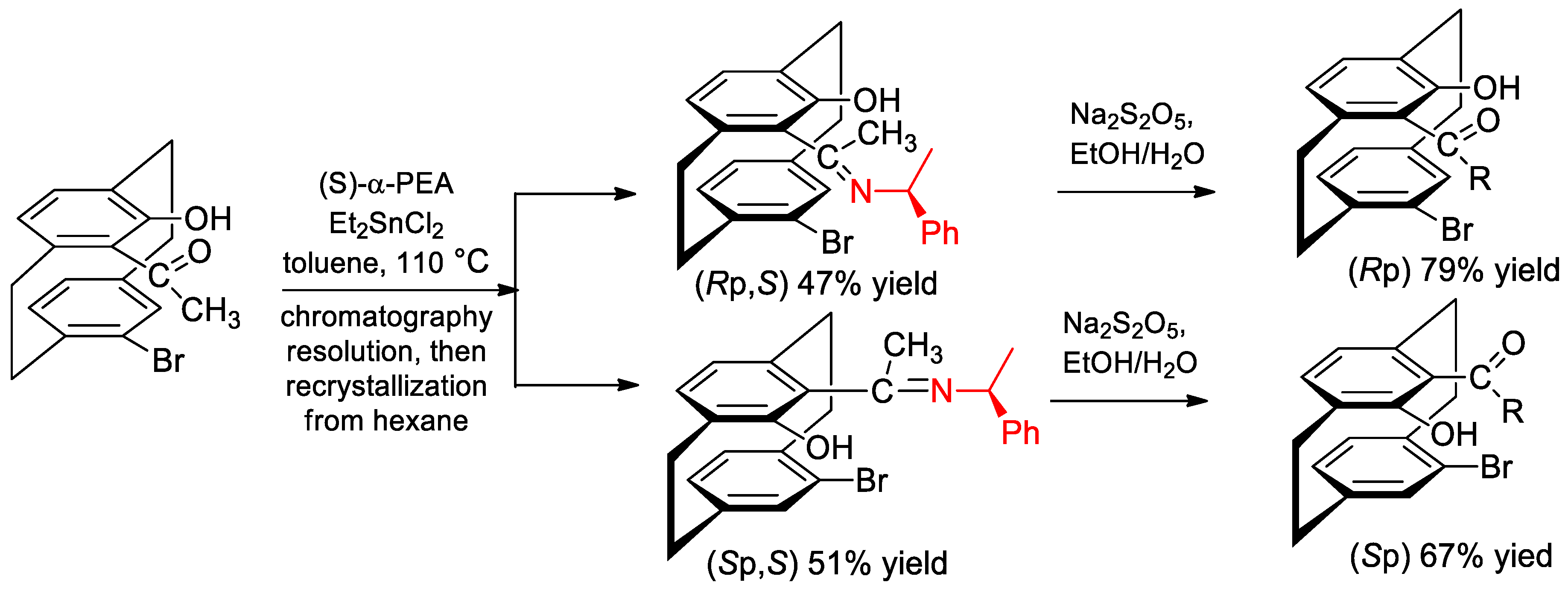
- Vorontsova, N.V.; Bystrova, G.S.; Antonov, D.Y.; Vologzhanina, A.V.; Godovikov, I.A.; Il’in, M.M. Novel ligands based on bromosubstituted hydroxycarbonyl2.2paracyclophane derivatives: Synthesis and application in asymmetric catalysis. Tetrahedron Asymmetry 2010, 21, 731–738. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Klika, K.D. Terminology Related to the Phenomenon ‘Self-Disproportionation of Enantiomers’ (SDE). Helv. Chim. Acta 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Nakamura, T.; Tateishi, K.; Tsukagoshi, S.; Hashimoto, S.; Watanabe, S.; Soloshonok, V.A.; Aceña, J.L.; Kitagawa, O. Self-disproportionation of enantiomers of non-racemic chiral amine derivatives through achiral chromatography. Tetrahedron 2012, 68, 4013–4017. [Google Scholar] [CrossRef]
- Suzuki, Y.; Han, J.; Kitagawa, O.; Aceña, J.L.; Klika, K.D.; Soloshonok, V.A. A comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography. ACS Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Enantiomeric Enrichments via the Self-Disproportionation of Enantiomers (SDE) by Achiral, Gravity-Driven Column Chromatography: A Case Study Using N-(1-Phenylethyl)acetamide for Optimizing the Enantiomerically Pure Yield and Magnitude of the SDE. Helv. Chim. Acta 2015, 98, 1147–1159. [Google Scholar] [CrossRef]
- Wzorek, A.; Kamizela, A.; Sato, A.; Soloshonok, V.A. Self-Disproportionation of Enantiomers (SDE) via achiral gravity-driven column chromatography of N-fluoroacyl-1-phenylethylamines. J. Fluor. Chem. 2017, 196, 37–43. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE): The effect of scaling down, potential problems versus prospective applications, possible new occurrences, and unrealized opportunities? Electrophoresis 2019, 40, 1869–1880. [Google Scholar] [CrossRef]
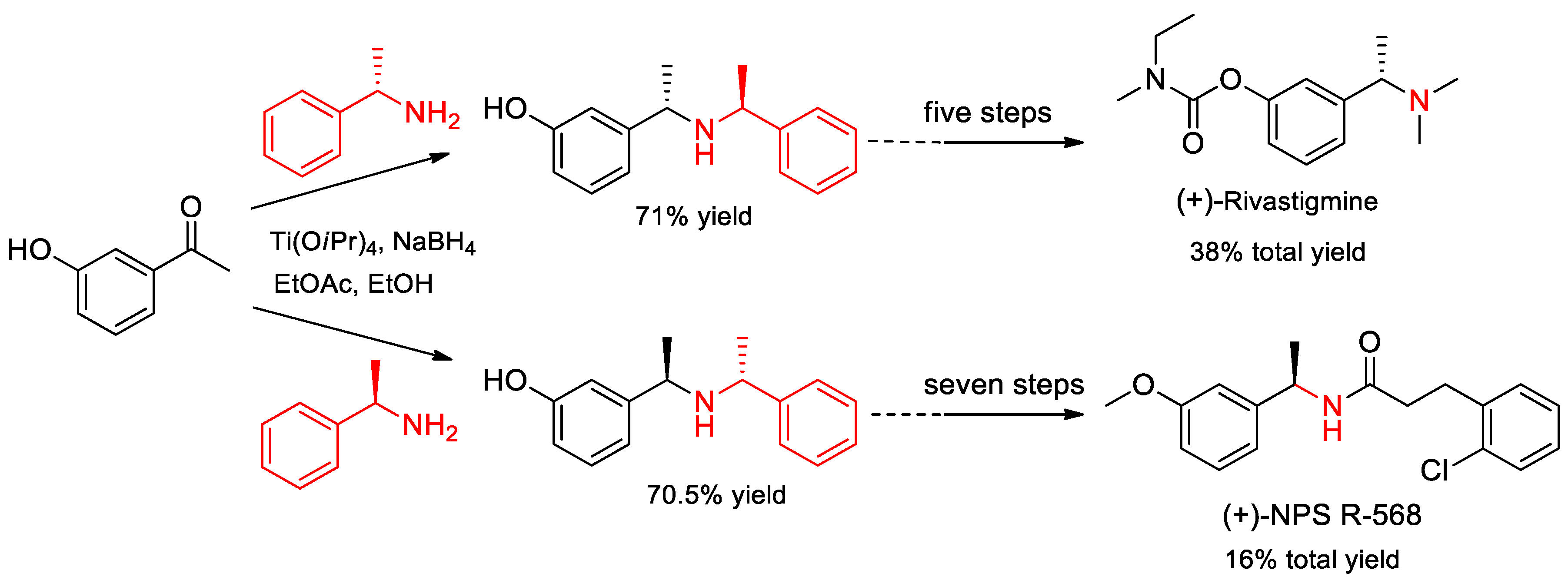
- Rao, R.; Shewalkar, M.P.; Nandipati, R.; Yadav, J.S.; Khagga, M.; Shinde, D.B. General Strategy for Large-Scale Synthesis of (+)-Rivastigmine and (+)-NPS R-568. Synth. Commun. 2012, 42, 589–598. [Google Scholar] [CrossRef]
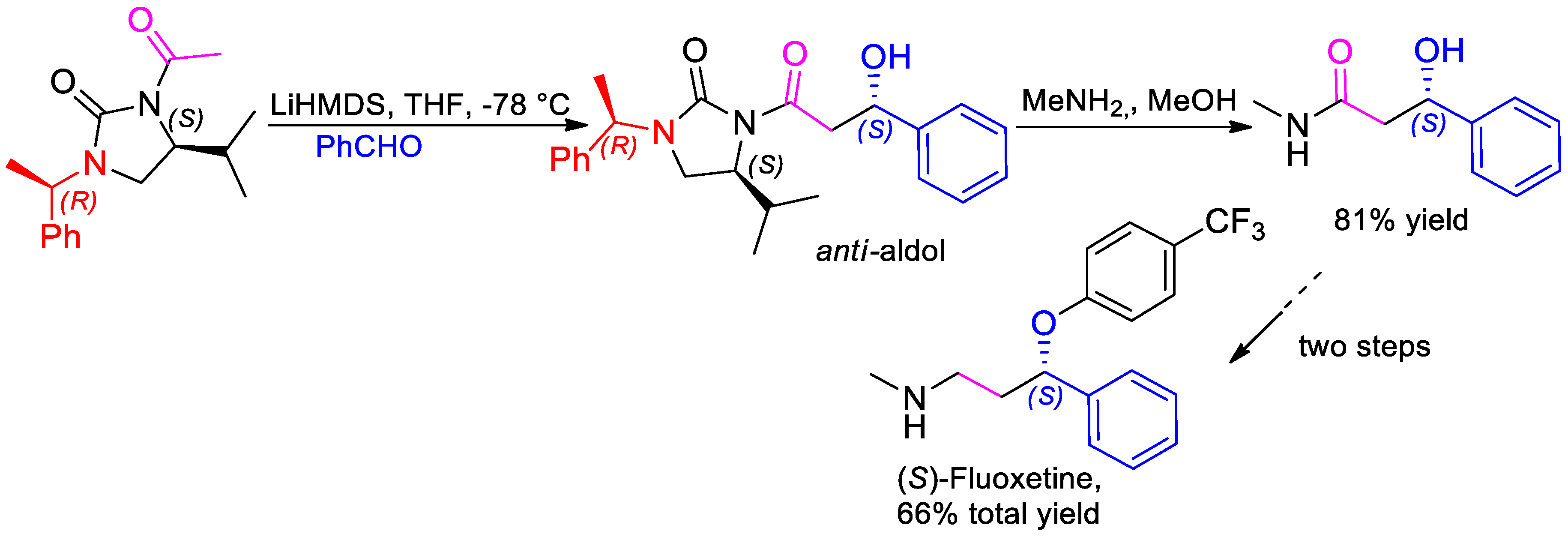
- Khatik, G.L.; Kumar, V.; Nair, V.A. Reversal of Selectivity in Acetate Aldol Reactions of N-Acetyl-(S)-4-isopropyl-1-(R)-1-phenylethylimidazolidin-2-one. Org. Lett. 2012, 14, 2442–2445. [Google Scholar] [CrossRef] [PubMed]
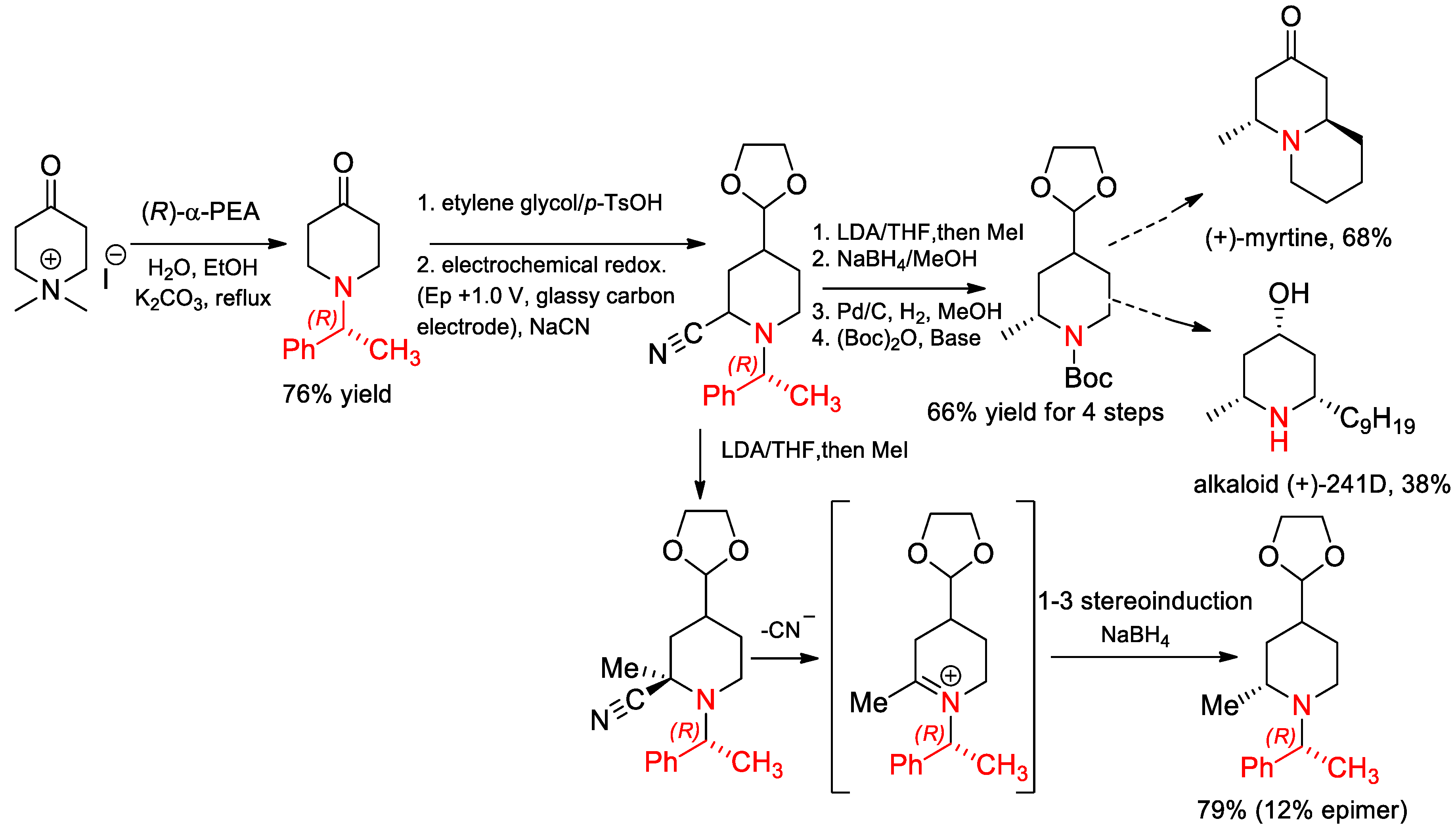
- Vu, V.H.; Louafi, F.; Girard, N.; Marion, R.; Roisne, T.; Dorcet, V.; Hurvois, J.-P. Electrochemical Access to 8-(1-Phenyl-ethyl)-1,4-dioxa-8-aza-spiro4.5decane-7-carbonitrile. Application to the Asymmetric Syntheses of (+)-Myrtine and Alkaloid (+)-241D. J. Org. Chem. 2014, 79, 3358–3373. [Google Scholar] [CrossRef] [PubMed]
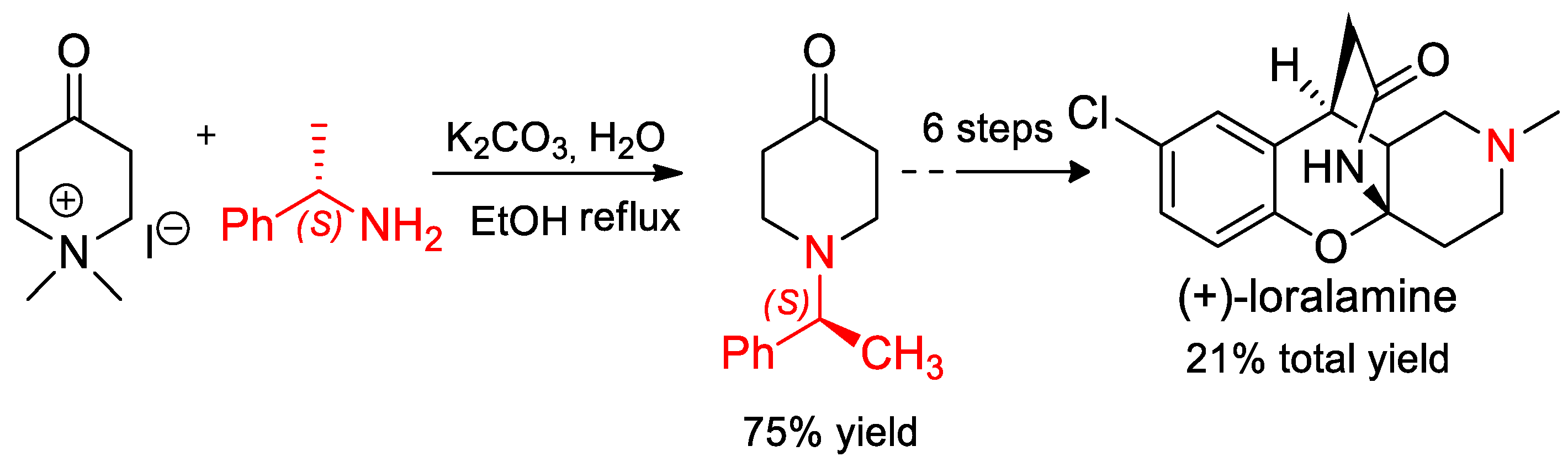
- Pawłowska, J.; Krawczyk, K.K.; Wojtasiewicz, K.; Maurin, J.K.; Czarnocki, Z. (S)-(−)-α-Methylbenzylamine as chiral auxiliary in the synthesis of (+)-lortalamine. Monatsh. Chem. 2009, 140, 83–86. [Google Scholar] [CrossRef]
- Kuehne, M.E.; Matson, P.A.; Bornmann, W.G. Enantioselective syntheses of vinblastine, leurosidine, vincovaline and 20’-epi-vincovaline. J. Org. Chem. 1991, 56, 513–528. [Google Scholar] [CrossRef]
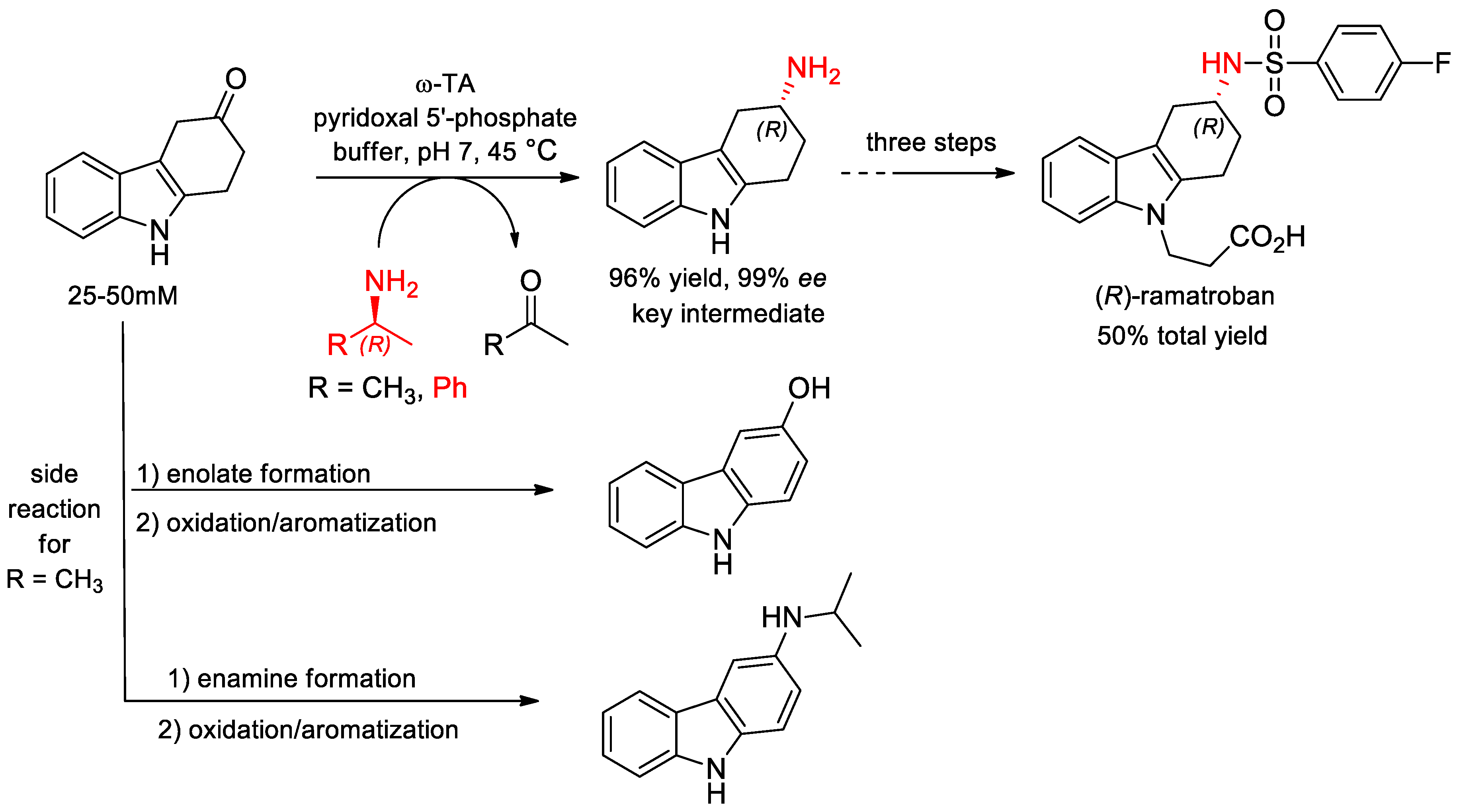
- Busto, E.; Simon, R.C.; Grischek, B.; Gotor-Fernndez, V.; Kroutil, W. Cutting Short the Asymmetric Synthesis of the Ramatroban Precursor by Employing ω-Transaminases. Adv. Synth. Catal. 2014, 356, 1937–1942. [Google Scholar] [CrossRef]
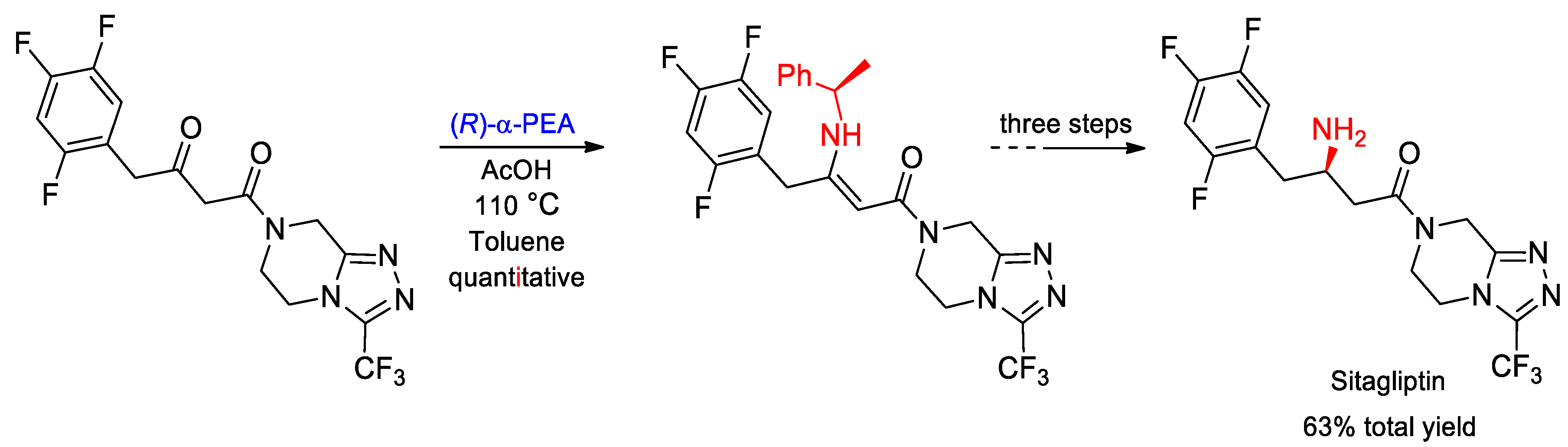
- Gutierrez, O.; Metil, D.; Dwivedi, N.; Gudimalla, N.; Chandrashekar, E.R.R.; Dahanukar, V.H.; Bhattacharya, A.; Bandichhor, R.; Kozlowski, M.C. Practical, Asymmetric Route to Sitagliptin and Derivatives: Development and Origin of Diastereoselectivity. Org. Lett. 2015, 17, 1742–1745. [Google Scholar] [CrossRef]
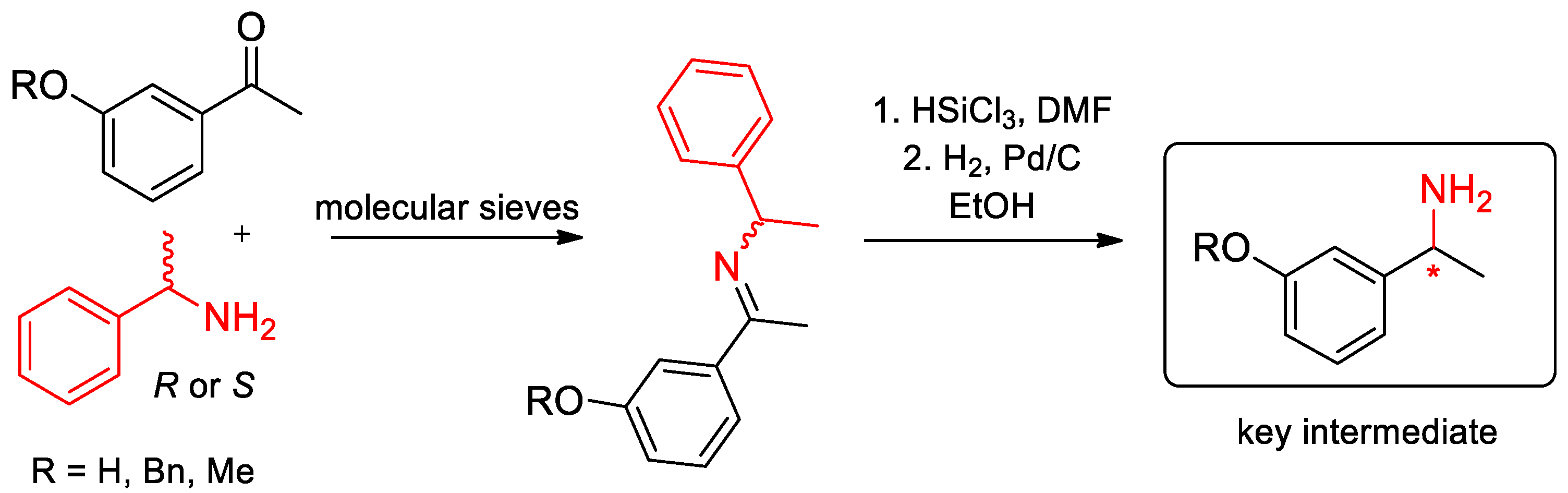
- Brenna, D.; Benaglia, M.; Porta, R.; Fernandes, S.; Burke, A.J. Stereoselective Metal-Free Reduction of Chiral Imines in Batch and Flow Mode: A Convenient Strategy for the Synthesis of Chiral Active Pharmaceutical Ingredients. Eur. J. Org. Chem. 2017, 39–44. [Google Scholar] [CrossRef]
- Brenna, D.; Rossi, S.; Cozzi, F.; Benaglia, M. Iron catalyzed diastereoselective hydrogenation of chiral imines. Org. Biomol. Chem. 2017, 15, 5685–5688. [Google Scholar] [CrossRef]
- Davies, S.G.; Fletcher, A.M.; Lebee, C.; Roberts, P.M. Asymmetric synthesis of (−)-(1R,7aS)-absouline. Tetrahedron 2013, 69, 1369–1377. [Google Scholar] [CrossRef]
- Brambilla, M.; Davies, S.G.; Fletcher, A.M.; Roberts, P.M.; Thomson, J.E. Asymmetric syntheses of (−)-isoretronecanol and (−)-trachelantamidine. Tetrahedron 2014, 70, 204–211. [Google Scholar] [CrossRef]
- Brambilla, M.; Davies, S.G.; Fletcher, A.M.; Roberts, P.M.; Thomson, J.E. Asymmetric syntheses of (−)-hastanecine, (−)-turneforcidine and (−)-platynecine. Tetrahedron 2016, 72, 4523–4535. [Google Scholar] [CrossRef]
- Brambilla, M.; Davies, S.G.; Fletcher, A.M.; Roberts, P.M.; Thomson, J.E. Asymmetric syntheses of the 1-hydroxymethyl-2-hydroxy substituted pyrrolizidines (−)-macronecine, (−)-petasinecine, (−)-1-epi-macronecine, (+)-1-epi-petasinecine and (+)-2-epi-rosmarinecine. Tetrahedron 2016, 72, 7449–7461. [Google Scholar] [CrossRef]
- Garduño-Castro, M.H.; Hernández-Rodríguez, M. Application of acyclic chiral auxiliaries on alkylation reactions. Tetrahedron Lett. 2014, 55, 193–196. [Google Scholar] [CrossRef]
- Zárate, A.; Orea, L.; Juárez, J.R.; Castro, A.; Mendoza, A.; Gnecco, D.; Terán, J.L. Diastereoselective Approach to cis-4-Methyl/thiol-Pipecolic Esters Based on RCM Reaction and Conjugate Michael Addition. Synth. Commun. 2014, 44, 2838–2847. [Google Scholar] [CrossRef]
- Bułyszko, I.; Chrzanowska, M.; Grajewska, A.; Rozwadowska, M.D. Synthesis of (+)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid, a Diastereoselective Approach. Eur. J. Org. Chem. 2015, 383–388. [Google Scholar] [CrossRef]
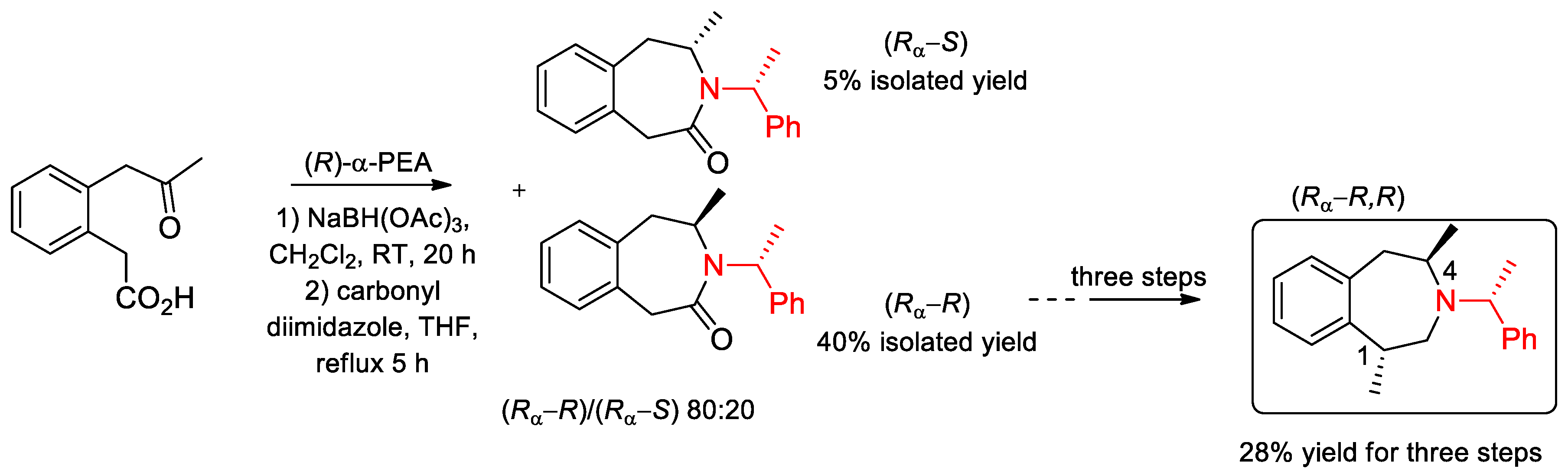
- Sarkar, S.; Schepmann, D.; Köhler, J.; Fröhlich, R.; Wünsch, B. Asymmetric Synthesis of Potent and Selective σ1 Receptor Ligands with Tetrahydro-3-benzazepine Scaffold. Eur. J. Org. Chem. 2012, 5980–5990. [Google Scholar] [CrossRef]
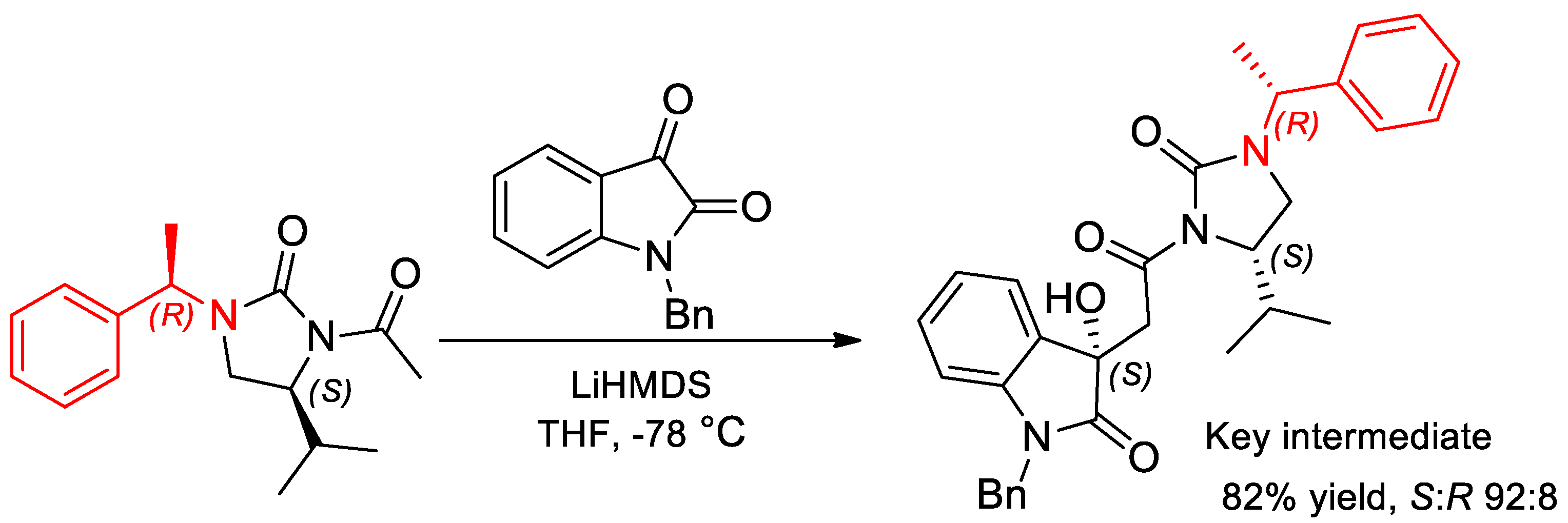
- Gangar, M.; Kashyap, N.; Kumar, K.; Goyal, S.; Nair, V.A. Imidazolidinone based chiral auxiliary mediated acetate aldol reactions of isatin derivatives and stereoselective synthesis of 3-substituted-3-hydroxy-2-oxindoles. Tetrahedron Lett. 2015, 56, 7074–7081. [Google Scholar] [CrossRef]
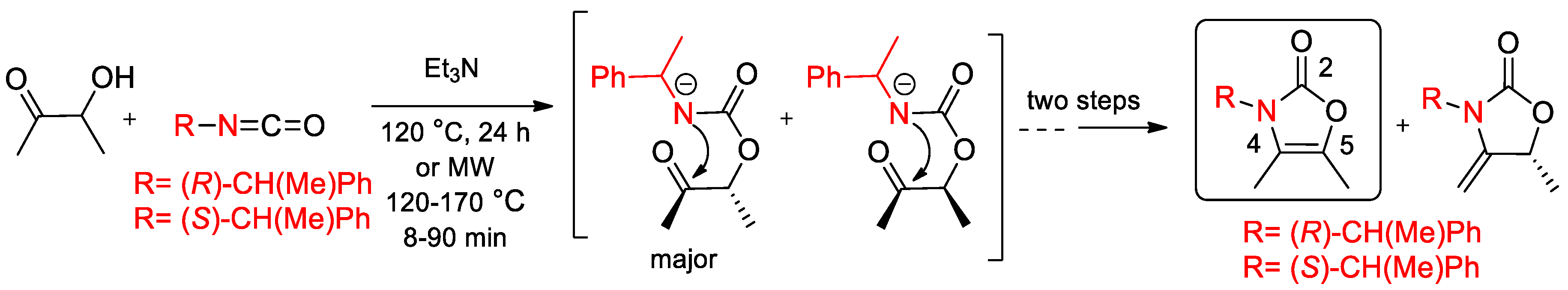
- Santoyo, B.M.; González-Romero, C.; Zárate-Zárate, D.; Hernández-Benitez, R.I.; Pelayo, V.; Barrera, E.; Escalante, C.H.; Fuentes-Benites, A.; Martínez-Morales, G.; López, J.; et al. Enantiopure 4-oxazolin-2-ones and 4-methylene-2-oxazolidinones as chiral building blocks in a divergent asymmetric synthesis of heterocycles. Chirality 2019, 31, 719–749. [Google Scholar] [CrossRef]
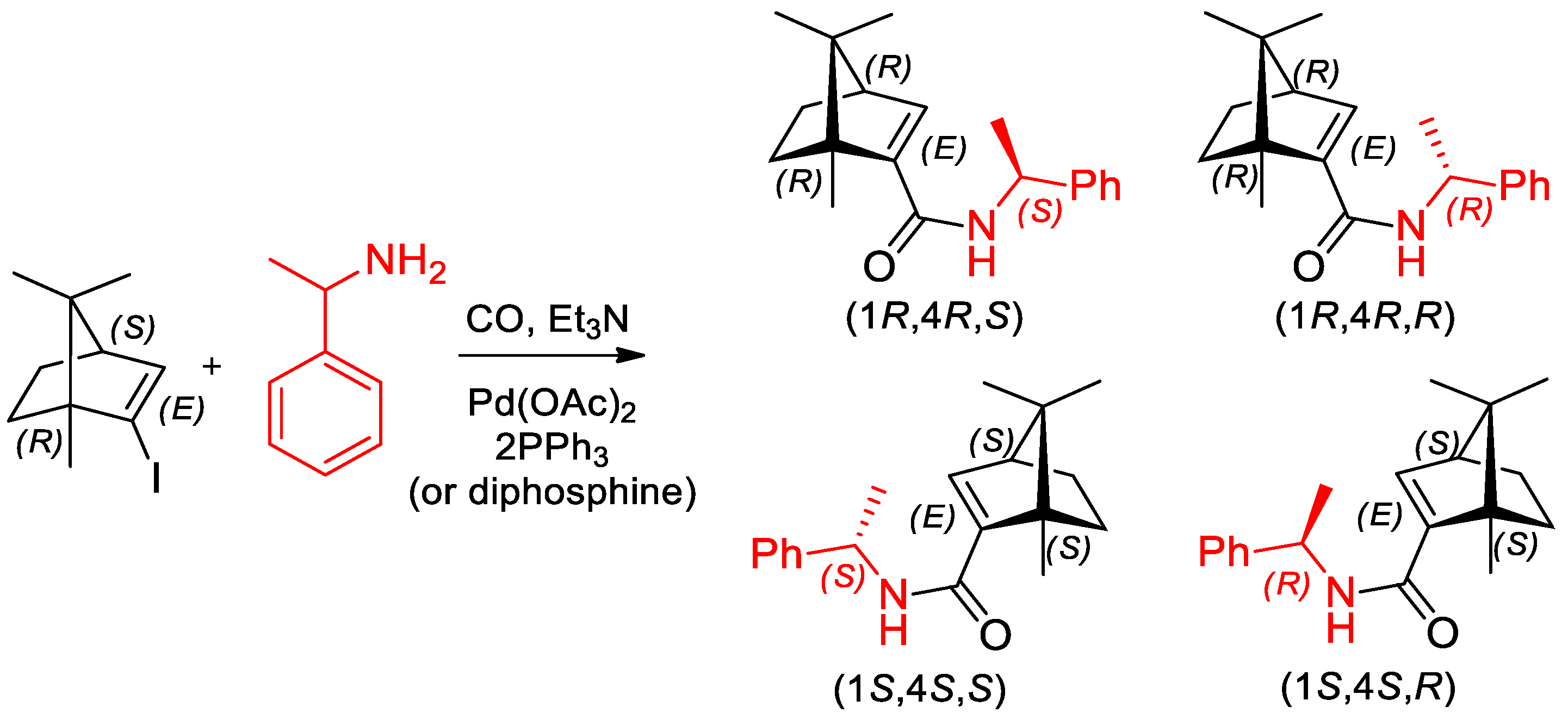
- Mikle, G.; Boros, B.; Kollár, L. Asymmetric aminocarbonylation of iodoalkenes in the presence of α-phenylethylamine as an N-nucleophile. Tetrahedron Asymmetry 2017, 28, 1733–1738. [Google Scholar] [CrossRef]
- Balint, E.; Tajti, A.; Kalocsai, D.; Matravolgy, B.; Karaghiosoff, K.; Czugler, M.; Keglevich, G. Synthesis and utilization of optically active α-aminophosphonate derivatives by Kabachnik-Fields reaction. Tetrahedron 2017, 73, 5659–5667. [Google Scholar] [CrossRef]
- Cimarelli, C.; Fratoni, D.; Palmieri, G. Novel stereoselective synthesis of 2,3-dihydro-1H-benzofchromen-3-amine derivatives through a one-pot three-component reaction. Tetrahedron Asymmetry 2011, 22, 1542–1547. [Google Scholar] [CrossRef]
- Markus, J.; Ferko, B.; Berkeš, D.; Moncol, J.; Lawson, A.M.; Othman, M.; Daïch, A. Indolylglycines Backbones in the Synthesis of Enantiopure 3,3-Spiroindolenines, Indolyl Tetracyclic Hemiaminals, and 3-Indolyl-maleimides Frameworks. Eur. J. Org. Chem. 2019, 5662–5677. [Google Scholar] [CrossRef]
- Castigliaa, A.; El Sehrawia, H.M.; Orbegozo, T.; Spitznera, D.; Claasen, B.; Frey, W.; Kantlehnera, W.; Jager, V. Synthesis and Characterization of Chiral Guanidines und Guanidinium Salts Derived from 1-Phenylethylamine. Z. Naturforsch. 2012, 67, 337–346. [Google Scholar] [CrossRef]
- Mlostoń, G.; Rygielska, D.; Jasiński, M.; Heimgartner, H. Optically active imidazoles derived from enantiomerically pure trans-1,2-diaminocyclohexane. Tetrahedron Asymmetry 2011, 22, 669–674. [Google Scholar] [CrossRef]
- Sipos, S.; Jablonkai, I.; Egyed, O.; Czugler, M. Preparation of 2-amino-2-C-glycosyl-acetonitriles from C-glycosyl aldehydes by Strecker reaction. Carbohydr. Res. 2011, 346, 2862–2871. [Google Scholar] [CrossRef]
- Wosińska-Hrydczuk, M.; Skarżewski, J. 2-Oxiranyl-pyridines: Synthesis and Regioselective Epoxide Ring Openings with Chiral Amines as a Route to Chiral Ligands. Heteroat. Chem. 2019, 2019, 2381208. [Google Scholar] [CrossRef]
- Wosińska-Hrydczuk, M.; Boratyński, P.J.; Skarżewski, J. Regioselective and Stereodivergent Synthesis of Enantiomerically Pure Vic-Diamines from Chiral β-Amino Alcohols with 2-Pyridyl and 6-(2,2′-Bipyridyl) Moieties. Molecules 2020, 25, 727. [Google Scholar] [CrossRef]
- Wu, C.; Qin, X.; Mangala, A.; Moeljadi, P.; Hirao, H.; Zhou, J.S. Copper-Catalyzed Asymmetric Arylation of N-Heteroaryl Aldimines: Elementary Step of a 1,4-Insertion. Angew. Chem. Int. Ed. 2019, 58, 2705–2709. [Google Scholar] [CrossRef]
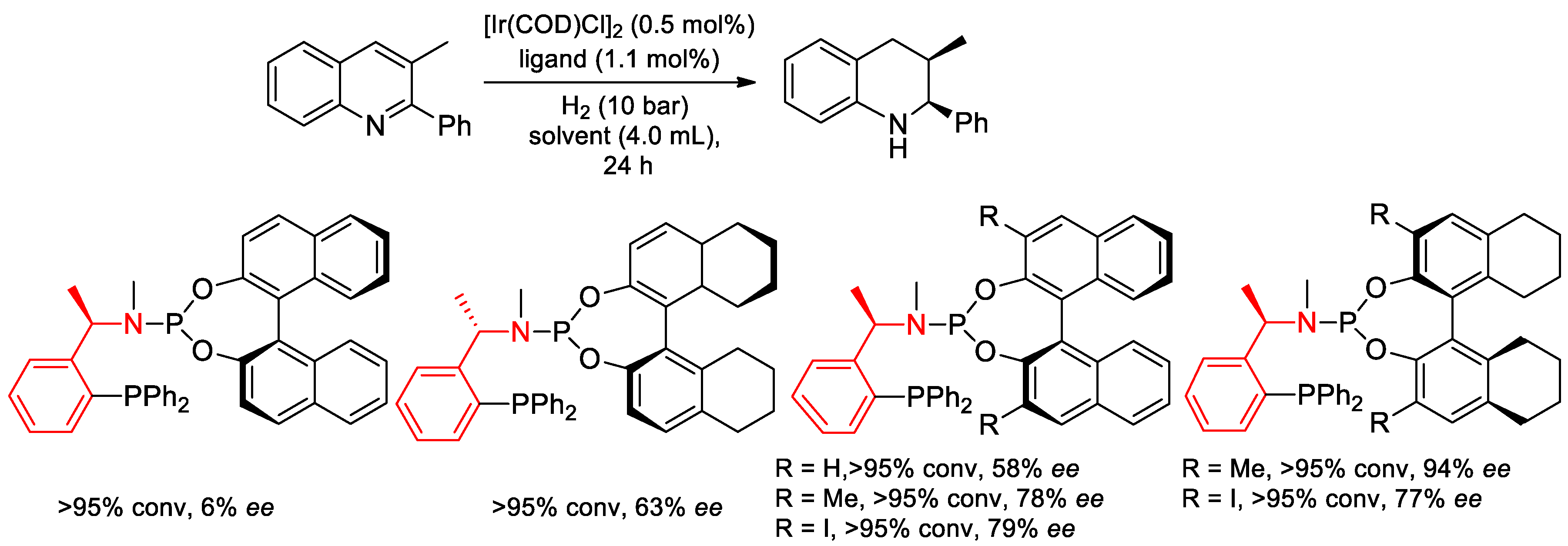
- Hu, X.-H.; Hu, X.-P. Highly Diastereo- and Enantioselective Ir-Catalyzed Hydrogenation of 2,3-Disubstituted Quinolines with Structurally Fine-Tuned Phosphine–Phosphoramidite Ligands. Org. Lett. 2019, 21, 10003–10006. [Google Scholar] [CrossRef] [PubMed]
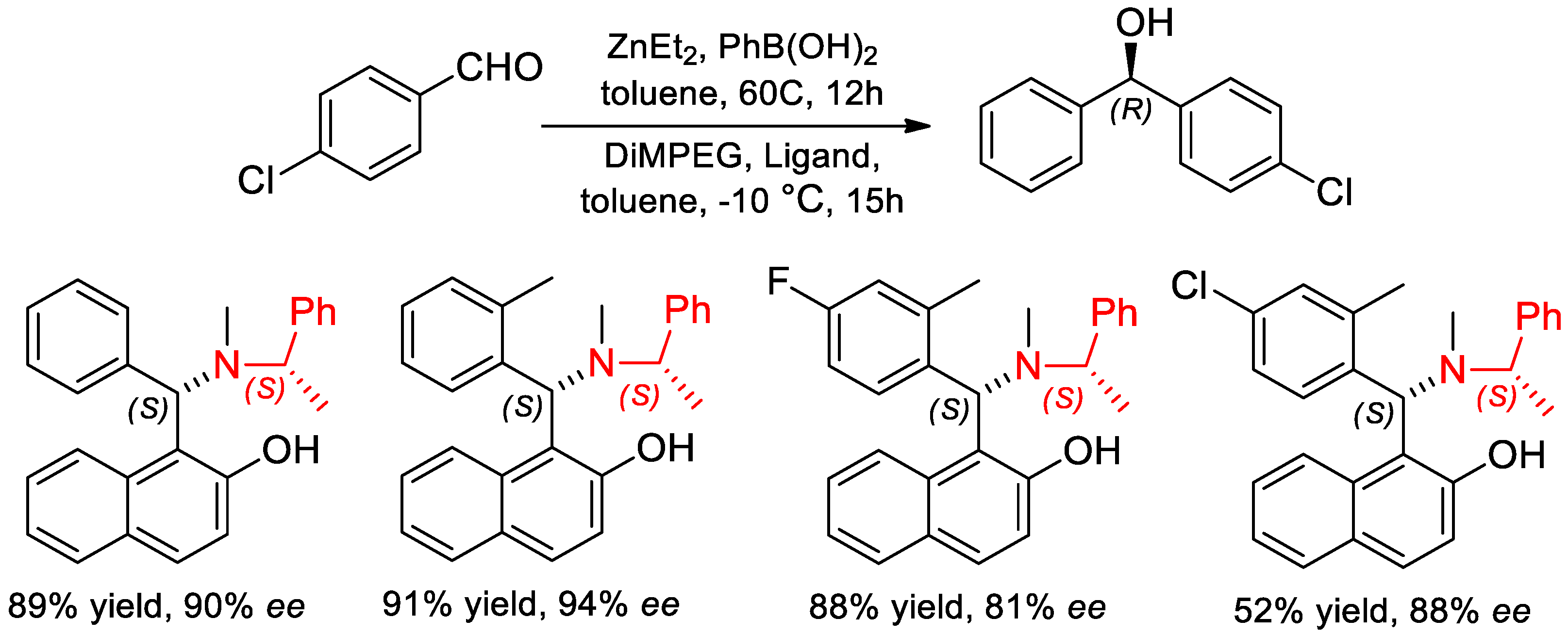
- Wei, H.; Yin, L.; Luo, H.; Li, X.; Chan, A.S.C. Structural influence of chiral tertiary aminonaphthol ligands on the asymmetric phenyl transfer to aromatic aldehydes. Chirality 2011, 23, 222–227. [Google Scholar] [CrossRef]
- Marinova, M.; Kostova, K.; Tzvetkova, P.; Tavlinova-Kirilova, M.; Chimov, A.; Nikolova, R.; Shivachev, B.; Dimitrov, V. Synthesis of 1,3-aminonaphthols by diastereoselective Betti-type aminoalkylation of dihydroxy naphthalenes; diastereoselectivity, absolute configuration, and application. Tetrahedron Asymmetry 2013, 24, 1453–1466. [Google Scholar] [CrossRef]
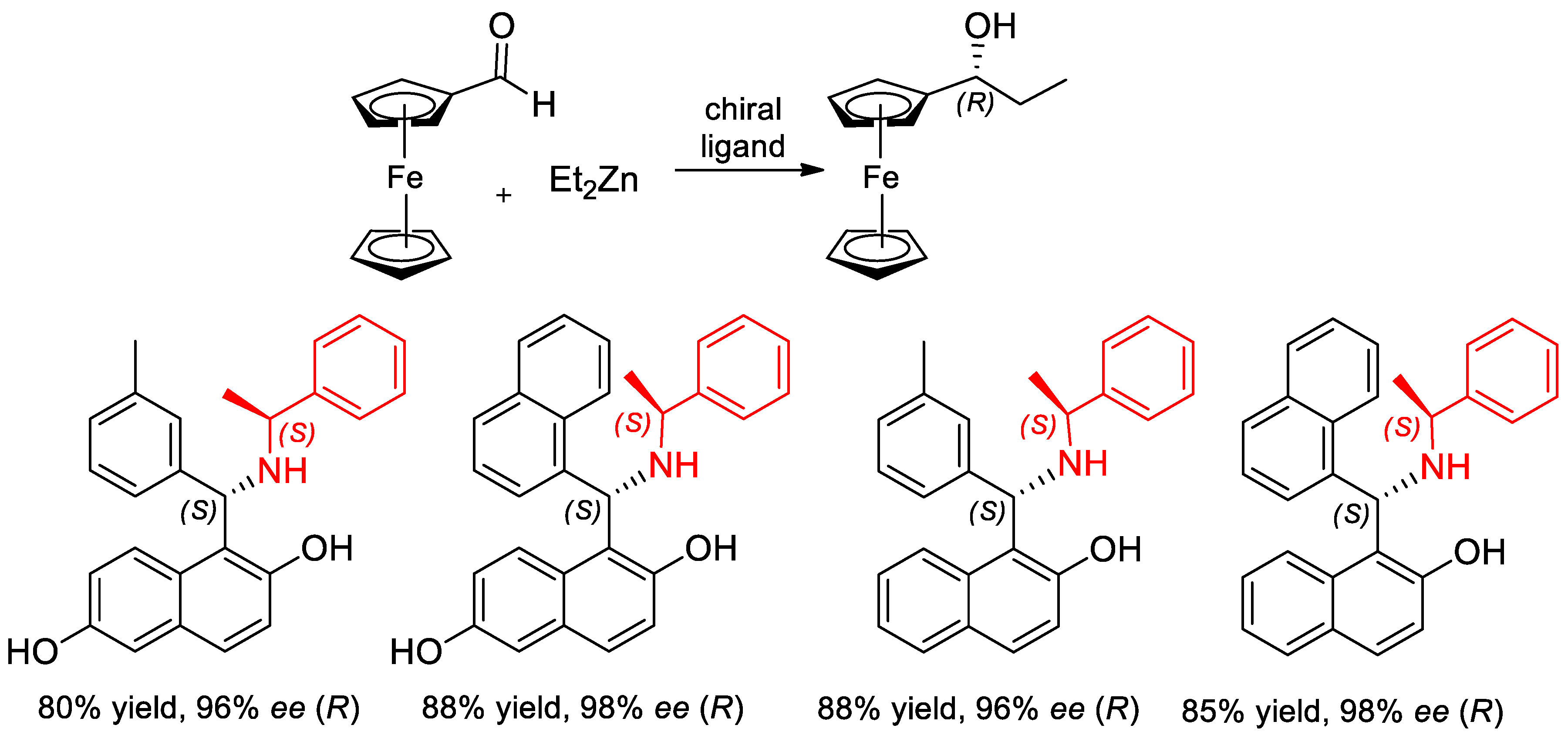
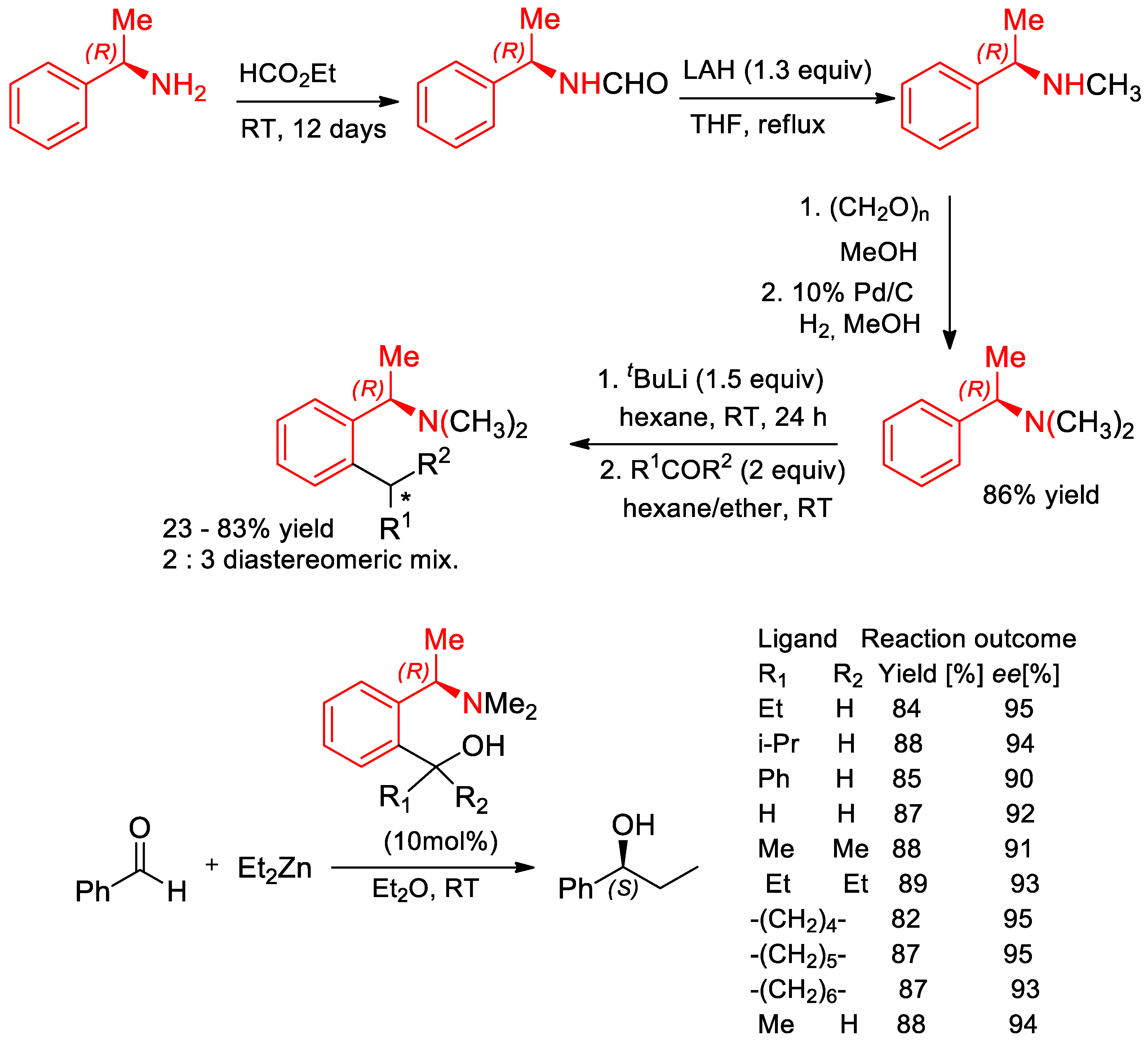
- Asami, M.; Miyairi, N.; Sasahara, Y.; Ichikawa, K.-I.; Hosoda, N.; Ito, S. Enantioselective addition of diethylzinc to aldehydes catalyzed by (R)-1-phenylethylamine-derived 1,4-amino alcohols. Tetrahedron 2015, 71, 6796–6802. [Google Scholar] [CrossRef]
- Han, M.-L.; Hu, X.-P.; Huang, J.-D.; Chen, L.-G.; Zheng, Z. New chiral amino alcohol ligands derived from 1-phenylethylamine for efficient Ru-catalyzed asymmetric transfer hydrogenation. Tetrahedron Asymmetry 2011, 22, 222–225. [Google Scholar] [CrossRef]
- Zhou, X.-M.; Huang, J.-D.; Luo, L.-B.; Zhang, C.-L.; Zheng, Z.; Hu, X.-P. Readily available chiral phosphine–aminophosphine ligands derived from 1-phenylethylamine for Rh-catalyzed enantioselective hydrogenations. Tetrahedron Asymmetry 2010, 21, 420–424. [Google Scholar] [CrossRef]
- Zhou, X.-M.; Huang, J.-D.; Luo, L.-B.; Zhang, C.-L.; Hu, X.-P.; Zheng, Z. Chiral 1-phenylethylamine-derived phosphine-phosphoramidite ligands for highly enantioselective Rh-catalyzed hydrogenation of β-(acylamino)acrylates: Significant effect of substituents on 3,3′-positions of binaphthyl moiety. Org. Biomol. Chem. 2010, 8, 2320–2322. [Google Scholar] [CrossRef]
- Nodzewska, A.; Sidorowicz, K.; Sienkiewicz, M. Solvent-Free Synthesis of a Secondary N-Benzhydrylamine as a Chiral Reagent for Asymmetric Deprotonation of Bicyclic N-Benzylamino Ketones. Synthesis 2014, 46, 1475–1480. [Google Scholar] [CrossRef]
- Jia, B.; Hao, J.; Wei, X.; Tong, H.; Zhou, M.; Liu, D. Zinc and aluminum complexes of chiral ligands: Synthesis, characterization and application to rac-lactide polymerization. J. Organomet. Chem. 2017, 831, 11–17. [Google Scholar] [CrossRef]
- Felluga, F.; Forzato, C.; Nitti, P.; Pitacco, G.; Prati, F.; Valentin, E.; Zangrando, E. Optically Active α-Phenylethylamine as Efficient Organocatalyst in the Solvent-free Reactions Between 2,3-Butanedione and Conjugated Nitroolefins. Chirality 2012, 24, 1005–1012. [Google Scholar] [CrossRef]
- Shaw, S.J.; Goff, D.A.; Boralsky, L.A.; Irving, M.; Singh, R. Enantioselective Synthesis of cis-3-Fluoropiperidin-4-ol, a Building Block for Medicinal Chemistry. J. Org. Chem. 2013, 78, 8892–8897. [Google Scholar] [CrossRef]
- Peňaška, T.; Mojzes, M.M.; Filo, J.; Jurdáková, H.; Mečiarová, M.; Šebesta, R. Organocatalysts Effect on the Stereoselectivity of 2,3-Wittig Rearrangement. Eur. J. Org. Chem. 2019, 605–610. [Google Scholar] [CrossRef]
- Yadav, G.D.; Singh, S. Direct asymmetric aldol reactions catalysed by trans-4-hydroxy-(S)-prolinamide in solvent-free conditions. Tetrahedron Asymmetry 2015, 26, 1156–1166. [Google Scholar] [CrossRef]
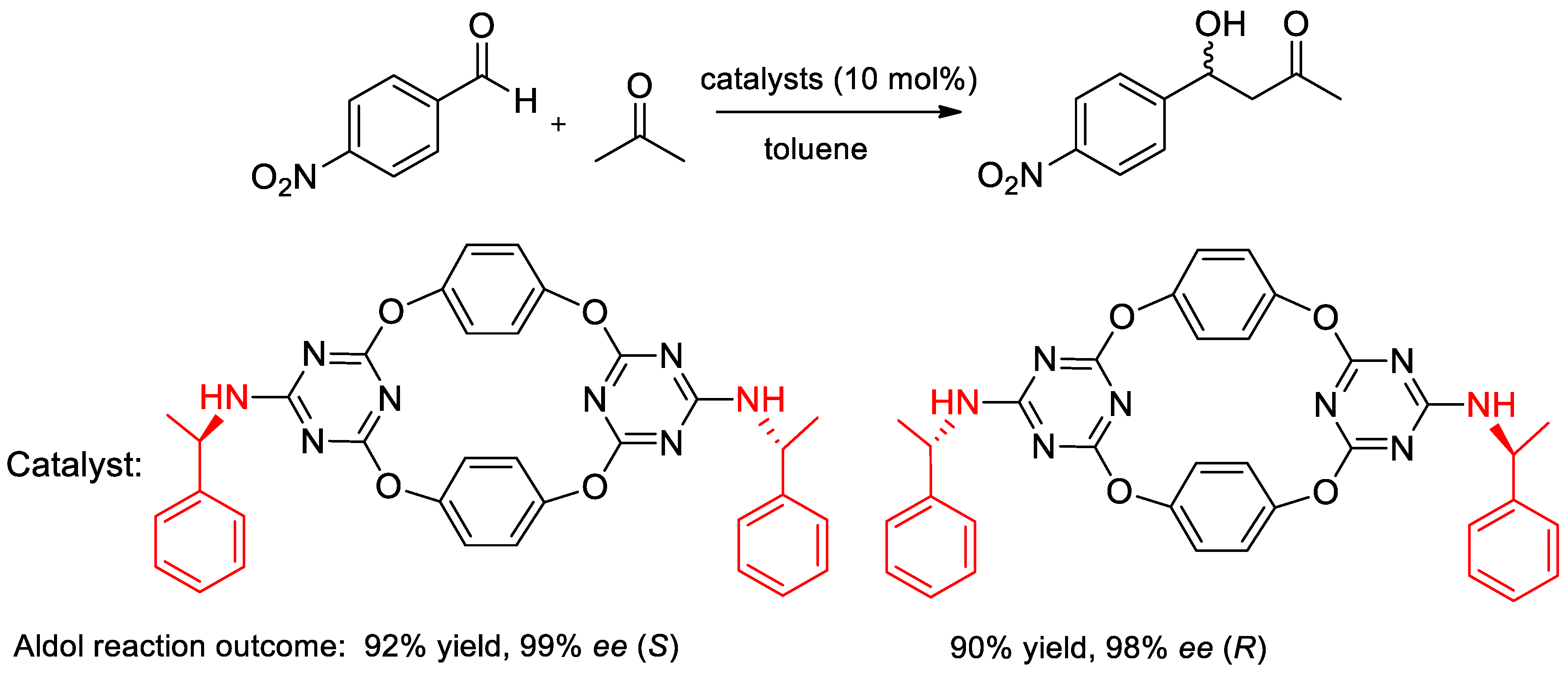
- Genc, H.N.; Ozgun, U.; Sirit, A. Chiral tetraoxacalix2arene2triazine-based organocatalysts for Enantioselective Aldol reactions. Tetrahedron Lett. 2019, 60, 1763–1768. [Google Scholar] [CrossRef]
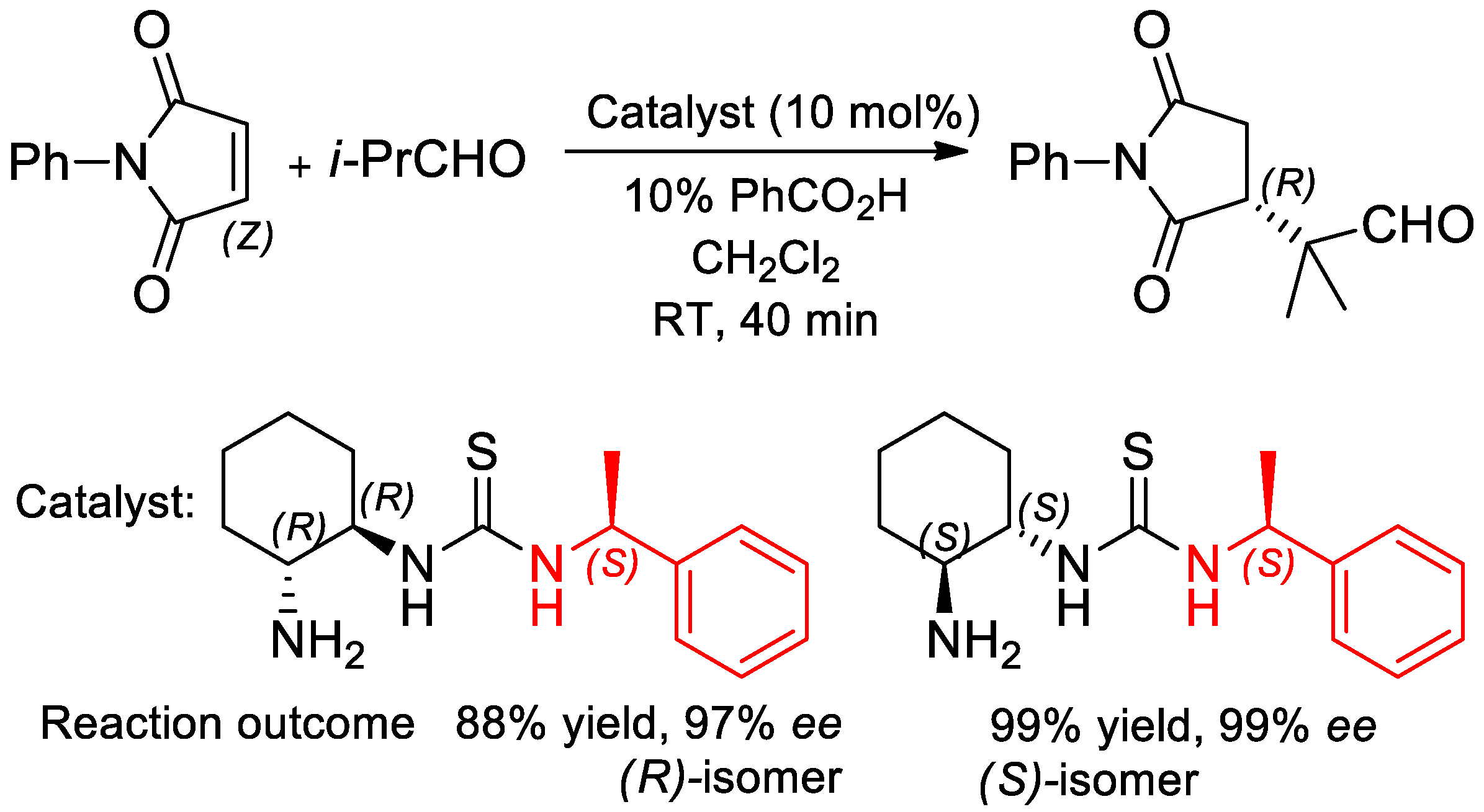
- Vazquez-Chavez, J.; Luna-Morales, S.; Cruz-Aguilar, D.A.; Díaz-Salazar, H.; Narváez, V.W.E.; Silva-Gutiérrez, R.S.; Hernández-Ortega, S.; Rocha-Rinza, T.; Hernández-Rodríguez, M. The effect of chiral N-substituents with methyl or trifluoromethyl groups on the catalytic performance of mono- and bifunctional thioureas. Org. Biomol. Chem. 2019, 17, 10045–10051. [Google Scholar] [CrossRef] [PubMed]





























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wosińska-Hrydczuk, M.; Skarżewski, J. New Advances in the Synthetic Application of Enantiomeric 1-Phenylethylamine (α-PEA): Privileged Chiral Inducer and Auxiliary. Molecules 2020, 25, 4907. https://doi.org/10.3390/molecules25214907
Wosińska-Hrydczuk M, Skarżewski J. New Advances in the Synthetic Application of Enantiomeric 1-Phenylethylamine (α-PEA): Privileged Chiral Inducer and Auxiliary. Molecules. 2020; 25(21):4907. https://doi.org/10.3390/molecules25214907
Chicago/Turabian StyleWosińska-Hrydczuk, Marzena, and Jacek Skarżewski. 2020. "New Advances in the Synthetic Application of Enantiomeric 1-Phenylethylamine (α-PEA): Privileged Chiral Inducer and Auxiliary" Molecules 25, no. 21: 4907. https://doi.org/10.3390/molecules25214907
APA StyleWosińska-Hrydczuk, M., & Skarżewski, J. (2020). New Advances in the Synthetic Application of Enantiomeric 1-Phenylethylamine (α-PEA): Privileged Chiral Inducer and Auxiliary. Molecules, 25(21), 4907. https://doi.org/10.3390/molecules25214907