Diastereoisomerically Pure, (S)-O-1,2-O-isopropyli dene-(5-O-α-d-glucofuranosyl) t-butanesulfinate: Synthesis, Crystal Structure, Absolute Configuration and Reactivity




Abstract
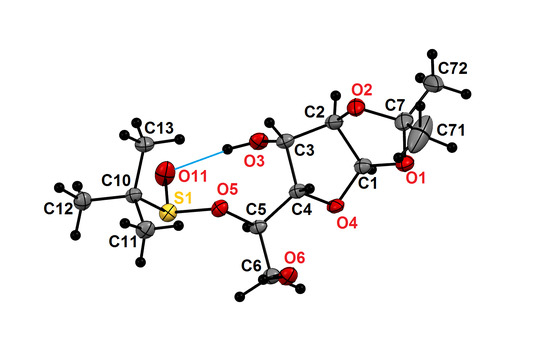
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Disscusion
3. Materials and Methods
3.1. (R)-1,2-O-Isopropylidene-3,5-O-Sulfinyl-α-d-Glucofuranose (R)-4 [28]
3.2. (S)-1,2-O-Isopropylidene-(5-O-α-d-glucofuranosyl) t-Butanesulfinate (-)-(S)-10
3.3. t-Butyl Methyl Sulfoxide (+)-(R)-11
3.4. Reaction of (R)-1,2-O-Isopropylidene-3,5-O-Sulfinyl- α-d-Glucofuranose (+)-(R)-4 with Phenylmagnesium Bromide
3.5. Reaction of (R)-1,2-O-Isopropylidene-3,5-O-Sulfinyl-α-d-Glucofuranose (+)-(R)-4 with 2,4,6-tri-tert-Butylphenylmagnesium Bromide
3.6. X-ray Crystallography of (S)-1,2-O-Isopropylidene-(5-O-α-d-glucofuranosyl)t-Butanesulfinate (-)-(S)-10
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drabowicz, J.; Kiełbasiński, P.; Mikołajczyk, M. Sulphinic acids and esters in synthesis. In The Chemistry of Sulfinic Acid, Esters and Their Derivatives; Patai, S., Ed.; John Wiley & Sons: Chichester, UK, 1990; pp. 351–429. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J.; Kiełbasiński, P. Chiral Sulfur Reagents; CRC Press: Boca Raton, FL, USA, 1997; ISBN 9780849391200. [Google Scholar]
- Mikołajczyk, M.; Drabowicz, J. Chiral organosulfur compounds. Top. Stereochem. 1982, 13, 333–468. [Google Scholar] [CrossRef]
- Nudelman, A. Sulfinic acids and their derivatives. Stereochemistry and chiroptical properties. In The Chemistry of Sulfinic Acids, Esters and Their Derivatives; Patai, S., Ed.; John Wiley & Sons: Chichester, UK, 1990; pp. 35–85. [Google Scholar] [CrossRef]
- Zoller, U. Syntheses of sulfinic esters. In The Chemistry of Sulfinic Acids, Esters and Their Derivatives; Patai, S., Ed.; John Wiley & Sons: Chichester, UK, 1990; pp. 217–237. [Google Scholar] [CrossRef]
- Nudelman, A. Chemistry of Optically Active Sulfur Compounds; Gordon and Breach Science Publishers Inc.: New York, NY, USA, 1984. [Google Scholar]
- Wenschuh, E.; Dölling, K.; Mikołajczyk, M.; Drabowicz, J. Aktuelle ergebnisse zur synthese und reaktivitat der sulfinsuaren und ihrer derivate. Z. Chem. 1980, 20, 122–132. [Google Scholar] [CrossRef]
- Drabowicz, J.; Krasowska, D.; Zając, A. Advances in the synthesis of chiral sulphinyl derivatives. Spec. Chem. Mag. 2007, 27, 34–37. [Google Scholar]
- Wei, J.; Sun, Z. Tert-butyl sulfoxides as a starting point for the synthesis of sulfinyl containing compounds. Org. Lett. 2015, 17, 5396–5399. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, J.; Liang, X.; Wei, Y. Electrochemical synthesis of sulfinic esters from alcohols and thiophenols. Tetrahedron Lett. 2020, 61. [Google Scholar] [CrossRef]
- Zhou, C.; Tan, Z.; Jiang, H.; Zhang, M. A sustainable oxidative esterification of thiols with alcohols by a cobalt nanocatalyst supported on doped carbon. Green Chem. 2018, 20, 1992–1997. [Google Scholar] [CrossRef]
- Gafur, S.H.; Waggoner, S.L.; Jacobsen, E.; Hamaker, C.G.; Hithcock, S.R. Efficient synthesis of sulfinate esters and sulfinamides via activated esters of p-toluenesulfinic acid. Synthesis 2018, 50, 4855–4866. [Google Scholar] [CrossRef]
- Tranquilino, A.; Andrade, S.R.C.P.; da Silva, A.P.M.; Menezes, P.H.; Oliveira, R.A. Non-expensive, open-flask and selective catalytic systems for the synthesis of sulfinate esters and thiosulfonates. Tetrahedron Lett 2017, 58, 1265–1268. [Google Scholar] [CrossRef]
- Fragher, R.J.; Shkoor, M.; Luska, L.; Schwan, A.L. Unexpected Reactions of Grignard Reagents with Selected β-Carboalkoxy Substituted Sulfinate Esters. Can. J. Chem. 2015, 93, 37–43. [Google Scholar] [CrossRef]
- Wojaczynska, E.; Wojaczynski, J. Modern Stereoselective Synthesis of Chiral Sulfinyl Compounds. Chem. Rev. 2020, 120, 4578–4611. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H. CCCLIV.-Investigations on the dependence of rotatory power on chemical constitution. Part XXVII. The optical properties of n-alkyl p-toluenesulphinates. J. Chem. Soc. Trans. 1925, 127, 2552–2587. [Google Scholar] [CrossRef]
- Drabowicz, J.; Krawczyk-Sójka, E.; Jasiak, A.; Krasowska, D.; Mielniczak, G.; Koprowski, M.; Owsianik, K. Process for the Preparation of Diastereomerically Pure O-(1R, 2S, 5R)-mentyl p-toluenesulfinate with Absolute Configuration (S). Polish Patent No. P.433225, 12 March 2020. [Google Scholar]
- Jasiak, A. Metody Przepływowe w Syntezie Chiralnych i Achiralnych związków Heteroorganicznych. Ph.D. Thesis, Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Lodz, Poland, 2020. [Google Scholar]
- Andersen, K.K. Synthesis of (+)-ethyl p-tolyl sulfoxide from (-)-menthyl (-)-p-toluenesulfinate. Tetrahedron Lett. 1962, 3, 93–95. [Google Scholar] [CrossRef]
- Andersen, K.K.; Gaffield, W.; Papanikolaou, N.E.; Foley, J.; Perkins, R.I. Optically active sulfoxides. The synthesis and rotatory dispersion of some diaryl sulfoxides. J. Am. Chem. Soc. 1964, 86, 5637–5646. [Google Scholar] [CrossRef]
- Andersen, K.K. Configurational relationships among some sulfoxides. J. Org. Chem. 1964, 29, 1953–1956. [Google Scholar] [CrossRef]
- Ridley, D.D.; Smal, M.A. Use of carbohydrates in the preparation of optically active sulphoxides. J. Chem. Soc. Chem. Commun. 1981, 505–506. [Google Scholar] [CrossRef]
- Ridley, D.D.; Smal, M.A. Preparation of arenesulfinic esters of 1,2:5,6-Di-O-cyclohexylidene-α-D-glucofuranose, and their conversion into optically active sulfoxides. Aust. J. Chem. 1982, 35, 495–507. [Google Scholar] [CrossRef]
- Llera, J.M.; Fernandez, I.; Alcudia, F. An efficient synthesis of both enantiomers of chiral non racemic methylsulfoxides from DAG. Tetrahedron Lett. 1991, 32, 7299–7302. [Google Scholar] [CrossRef]
- Fernandez, I.; Khiar, N.; Llera, J.M.; Alcudia, F. Asymmetric synthesis of alkane- and arenesulfinates of diacetone-D-glucose (DAG): An improved and general route to both enantiomerically pure sulfoxides. J. Org. Chem. 1992, 57, 6789–6796. [Google Scholar] [CrossRef]
- Khiar, N.; Fernandez, I.; Alcudia, F. Asymmetric synthesis of optically pure tert-butyl sulfoxides using the “DAG methodology”. Tetrahedron Lett. 1994, 35, 5719–5722. [Google Scholar] [CrossRef]
- Khiar, N. Determination of the absolute configuration of sulfinyl glycosides: The role of the exo-anomeric effect. Tetrahedron Lett. 2000, 41, 9059–9063. [Google Scholar] [CrossRef]
- Borsuk, K.; Frelek, J.; Łysek, R.; Urbańczyk-Lipkowska, Z.; Chmielewski, M. Six-membered cyclic sulfites derived from glucofuranose and 1,2,4-butanetriol. Chirality 2001, 13, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Drabowicz, J.; Mikołajczyk, M. One-step synthesis of alkyl t-alkanesulfinates. Synthesis 1974. [Google Scholar] [CrossRef]
- Drabowicz, J.; Legędź, S.; Mikołajczyk, M. Organosulphur compounds LXIX: Optically active sulphinates: A new type of enantioselective asymmetric synthesis and kinetic resolution. Tetrahedron 1988, 44, 5243–5251. [Google Scholar] [CrossRef]
- Pitchen, P.; Dunsch, E.; Desmukh, M.N.; Kagan, H.B. An efficient asymmetric oxidation of sulfides to sulfoxides. J. Am. Chem. Soc. 1984, 106, 8188–8193. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Nardelli, M. Ring asymmetry parameters from out-of-plane atomic displacements. Acta Cryst. 1983, 39, 1141–1142. [Google Scholar] [CrossRef]
- Bujnicki, B.; Drabowicz, J.; Mikolajczyk, M. Acid catalyzed alcoholysis of sulfinamides: Unusual stereochemistry, kinetics and a question of mechanism involving sulfurane intermediates and their pseudorotation. Molecules 2015, 20, 2949–2972. [Google Scholar] [CrossRef]
- Drabowicz, J.; Łyżwa, P.; Popielarczyk, M.; Mikołajczyk, M. A convenient procedure for oxidation of sterically hindered sulfides to sulfoxides. Synthesis 1990, 937–938. [Google Scholar] [CrossRef]
- Safaiee, M.; Moeinimehr, M.; Zolfigol, M.A. Pyridiniumporphyrazinato oxo-vanadium tribromomethanide as a newsource of Br+ catalyst for the chemo and homoselective oxidation of sulfides and benzylic alcohols. Polyhedron 2019, 170, 138–150. [Google Scholar] [CrossRef]
- He, Y.; Ma, X.; Ji, H.F.; Zha, X.B.; Jiang, H.; Lu, M. Selective Oxidation of Sulfides to Sulfoxides/Sulfones by 30% Hydrogen Peroxide. Phosphorus Sulfur Silicon Relat. Elem. 2012, 187, 822–830. [Google Scholar] [CrossRef]
- Reddy, A.B.; Swamy, B.K.; Yadav, J.S. A concise synthesis of cleistenolide. Tetrahedron Asymmetry 2016, 27, 788–790. [Google Scholar] [CrossRef]
- Laali, K.K.; Nagvekar, D.S. Reaction of simple arenes with FSO3H·SbF5/SO2: One-pot synthesis of aromatic sulfoxides. Mechanistic aspects and synthetic utility. J. Org. Chem. 1991, 56, 1867–1874. [Google Scholar] [CrossRef]
- Davis, F.A.; Jenkins Jr., R.H.; Rizvi, S.Q.A.; Yocklovich, S.G. Chemistry of sulfenic acids. 3. Studies of sterically hindered sulfenic acids using flash vacuum pyrolysis. J. Org. Chem. 1981, 46, 3467–3474. [Google Scholar] [CrossRef]
- Bresien, J.; Hinz, A.; Schulz, A.; Suhrbier, T.; Thomas, M.; Villinger, A. Dichloro-cycloazatriphosphane: The missing link between N2P2 and P4 ring systems in the systematic development of NP chemistry. Chem. Eur. J. 2017, 23, 14738–14742. [Google Scholar] [CrossRef]
- KM4CCD CrysAlis CCD, Data Collection Program. Version 1.163. Unilic & Kuma Diffraction Instruments: GmbH, Wroclaw, Poland, 2000.
- KM4CCD CrysAlis RED, Data Reduction Program. Version 1.163. Unilic & Kuma Diffraction Instruments: GmbH, Wroclaw, Poland, 2000.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, C. Features and development of Coot. Acta Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Cryst. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Cryst. 2013, B69, 249–259. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound 10 are available from the authors. |










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bujnicki, B.; Błaszczyk, J.; Chmielewski, M.; Drabowicz, J. Diastereoisomerically Pure, (S)-O-1,2-O-isopropyli dene-(5-O-α-d-glucofuranosyl) t-butanesulfinate: Synthesis, Crystal Structure, Absolute Configuration and Reactivity. Molecules 2020, 25, 3392. https://doi.org/10.3390/molecules25153392
Bujnicki B, Błaszczyk J, Chmielewski M, Drabowicz J. Diastereoisomerically Pure, (S)-O-1,2-O-isopropyli dene-(5-O-α-d-glucofuranosyl) t-butanesulfinate: Synthesis, Crystal Structure, Absolute Configuration and Reactivity. Molecules. 2020; 25(15):3392. https://doi.org/10.3390/molecules25153392
Chicago/Turabian StyleBujnicki, Bogdan, Jarosław Błaszczyk, Marek Chmielewski, and Józef Drabowicz. 2020. "Diastereoisomerically Pure, (S)-O-1,2-O-isopropyli dene-(5-O-α-d-glucofuranosyl) t-butanesulfinate: Synthesis, Crystal Structure, Absolute Configuration and Reactivity" Molecules 25, no. 15: 3392. https://doi.org/10.3390/molecules25153392
APA StyleBujnicki, B., Błaszczyk, J., Chmielewski, M., & Drabowicz, J. (2020). Diastereoisomerically Pure, (S)-O-1,2-O-isopropyli dene-(5-O-α-d-glucofuranosyl) t-butanesulfinate: Synthesis, Crystal Structure, Absolute Configuration and Reactivity. Molecules, 25(15), 3392. https://doi.org/10.3390/molecules25153392