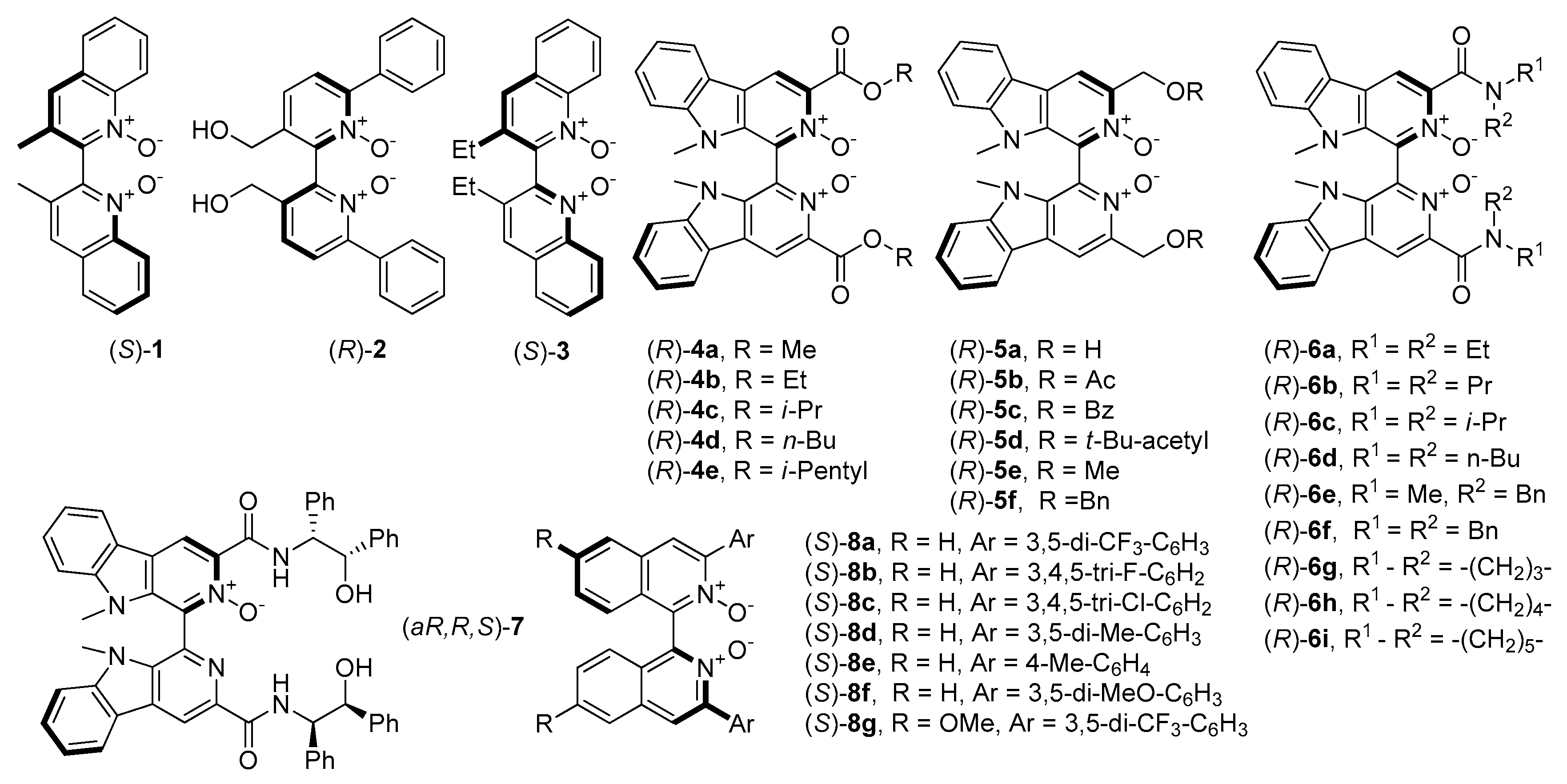
2.1. Axially Chiral N-Oxides and N,N′-Dioxides
The first mention of
N-oxides and their catalytic asymmetric applications were related to those, containing axial chirality and were developed by Nakajima et al. [
4]. Initially, compounds were based on 4,4’-biquinoline and 2,2’-bipyridine
N,
N′-dioxide backbones (
1 and
2, see
Figure 1) which, apart from Nakajima’s work, were also investigated by Feng and coworkers [
28]. A little later, Kotora et al. synthesized
N-oxides with tetrahydroisoquinoline [
29] and bis(tetrahydroisoquinoline) frameworks [
30,
31]. It is worth to highlight that organocatalysts
1 and
2 give, so far, one of the highest results (both in terms of yield and enantioselectivity) in the allylation of benzaldehyde with allyltrichlorosilane (85%, 88% ee (R) and 95%, 84% ee (S) respectively). Chang et al. received an analog of
1, containing ethyl instead of methyl groups (
3, see
Figure 1) and describe their application as organocatalyst (10 mol%) in allylation of 4- metoxybenzaldehyde with allyltrichlorosilane. The chiral product was obtained with high enantiomeric purity (92% ee) with satisfactory yield 66% [
32], which slightly exceeds the result obtained with the application of
1.
Reports concerning chiral biscarboline dioxide derivatives
4 and
5 were developed by Zhu et al. [
33,
34]. Executed screening of these compounds as benzaldehyde allylation catalysts showed, that the reaction yield for all types of catalysts was quantitative. In the case of use
4 also enantioselectivity was high (95%) while in the case of
5, results were moderate (up to 82% ee). This implies that ester groups are involved in catalyzing this reaction. Catalysts
4a and
4c were chosen to test them in allylation of substituted benzaldehydes and aliphatic aldehydes. Independently from a substrate, using
4a or
4c (1 mol%) gave high enantioselectivity (91–97% ee or 53–90% ee respectively) and moderate to high yields (up to 90%). That makes these catalysts very versatile and applicable to different types of substrates, both these with electron-withdrawing and electron-donating groups. Furnished homoallylic alcohols (except for these ones from 2,6-dichlorobenzaldehyde and 3-phenylpropanal) had configuration
S, so opposite to configuration of used catalysts (see
Figure 1).
Results of allylation using
4a and
4c are shown in
Table 1. Catalysts
4a and
4c were also tested in allylation of benzaldehyde with different, substituted allyltrichlorosilanes, including crotylation (R
1 = Me, R
2 = R
3 = H,
Scheme 1). Generally, high ee values were achieved (up to 96%) but low to moderate yields (19–88%). The ratio of syn/anti products was the same as the ratio of
Z/E isomers in the used substrate. Except for crotylation, in all other reactions slightly higher yields were obtained using catalyst
4c but the ee values, obtained for both catalysts were similar. For catalyst
4c, the impact of solvent was also tested. Enantioselectivity was satisfactory for all used solvents (85–95% ee) but the complete conversion was obtained only with solvents such as CH
2Cl
2 and CH
3CN. For THF, toluene, EtOAc and Et
2O conversion did not exceed 10%. It is also worth to mention that the syntheses of
4 and
5, although multistep, were performed using simple transformations. Overall yield was about 40–50% for
4 and about 80% for
5.
Zhu and coworkers have synthesized the chiral biscarboline
N,
N’-dioxide derivatives possessing secondary amide groups
6 (see
Figure 1) [
26,
35]. Checking the effectiveness of
6a–
b and
6d–
h as catalysts in allylation of benzaldehyde with trichlorosilane (CH
2Cl
2, −80° C, 20 h), the authors focused on optimization the catalyst loading and, choosing the most effective catalyst, on examination of its application range [
35]. The best results were observed for catalysts
6g (84% ee) and
6h (87% ee), which contained 4- and 5-membered cyclic amides, respectively. What is interesting, the highest enantioselectivity for
6h has been obtained using 1 mol% of the catalyst. With other amounts: 0.1, 0.5, 5 or 10 mol%, the enantioselectivity was lower. The
6h was applied in a series of reactions with different substrates. Generally, the resulted yield and enantioselectivity were good to excellent (12 examples, 85–97%, 67–96% ee). The influence of the position of substitution in benzaldehyde on the course of the reaction was tested. Mostly,
m- and
p-substituted benzaldehydes were compared. For electron-withdrawing groups, higher enantioselectivity was observed when the substituent was in position 3 (
Table 1, entries 7, 10). The different situations took place for the electron-donating methoxy group. In this case, also
o-substituted benzaldehyde was applied and, in all cases the yield was good (86–90%) but enantioselectivity increased in order: orto-, meta-, para-methoxybenzaldehyde (67, 83, 94% ee respectively,
Table 1, entries 2, 6, 11). All obtained homoallylic alcohols had the configuration
R, the same as the used catalyst. A comparison of the results of
6h application with the described results of the use of structurally similar catalysts
4 and also with bisquinoline derivatives
8 are presented in
Table 1. The catalyst
6h was not very effective for aliphatic aldehydes (entry 32,
Table 1). It was found that dichloromethane (DCM) was the suitable solvent, and as it was expected, the enantiomeric excess was strongly dependent on the reaction temperature.
Axially-chiral symmetrically substituted 2,2′-biquinoline
N,
N’-dioxide derivatives were developed by Takenaka and Peverati. Simple transformations allowed to receive the new Lewis base catalysts 8 (see
Figure 1) with yields up to 99% [
36]. Their catalytic utility was tested in allylation of 4-metoxybenzaldehyde with allyltrichlorosilane. Loading of 0.1 mol% was sufficient to get 83–96% conversion with enantioselectivity up to 96%. The best catalysts
8a,
8b, and
8d were also used in the allylation of cinnamaldehyde. In this case, the most efficient was
8a (having two bis 3,5-trifluoromethylphenyl substituents), which gave respective homoallylic alcohol with 84% yield and 92% ee. The obtained results were excellent, even at loading lowered to 0.05 mol%, for substituted aromatic, heteroaromatic and aliphatic aldehydes, but only if substrates were electron-rich aldehydes. The authors noticed that
8a is less reactive and less selective for the halogen-substituted aldehydes. Considering the fact, that electron-rich Lewis base catalysts are usually more reactive in reaction with halosilane compounds, they decided to prepare compound
8g, having two phenyl rings double substituted with CF
3 groups, suitable for aldehydes with halogen groups [
36]. Application of
8g increased the yield, unfortunately with unchanged or only slightly increased enantioselectivity. The catalytic efficiency of
8a and
8g, compared with previously mentioned axial-chiral catalysts is listed in
Table 1.
Good activity and selectivity in the reductive aldol reaction of chalcone and benzaldehyde with trichlorosilane (
Scheme 2) were obtained by employing bisquinoline
N,
N’-dioxide (
R)-1 as a catalyst. It resulted in up to 80% ee of the product syn [
25].
Zhu et al. have examined the catalysts
6b–
e and
6h–
i in the enantioselective hydrosilylation of ketoimines (
Table 2) [
26]. All tested
N,
N’-dioxides were effective; after 16 h (with 10 mol% of the catalyst) all the reactions were completed, but the enantioselectivity was moderate (6 examples, 42–77% ee). After optimization of the reaction conditions (1 mol% of catalyst in CH
2Cl
2 at 0 °C), catalyst
6i, as the most effective, was used in the reduction of different ketoimines (11 examples). Obtained yield, in all cases, was very high (95–99%) however the enantioselectivity was up to 85% ee. Higher enantioselectivity was observed when R
1 was an aromatic ring, furnished with an electron-donating group (reaction above
Table 2). If phenyl in R
1 had electron-withdrawing substituents, the asymmetric induction was lower. Except for three examples (
Table 2, entries 7, 11, 13), obtained products had configuration
S, opposite to used catalyst.
The next approach presented by Zhu’s research group was the mono
N-oxide
7, with amide possessing additional stereogenic center [
27], in order to improve the previously studied hydrosilylation of ketoimines. In fact, obtained results were excellent in many cases (14 examples, 94–98%, 75–96% ee), which makes the catalyst versatile and slightly more effective than 6i. A rough comparison of the catalytic efficiency of
6i and
7 are given in
Table 2. Application of
7 gave comparable yields and better enantioselectivity but requires higher catalyst loading.
2.2. N-Oxides Possessing Central Chirality
Much more attention through the past decade has been given to compounds with central chirality. The report of Mlostoń and Jurczak from 2009 deserves to be mentioned [
38]. Authors presented novel chiral
C2-symmetric bisimidazole-
N-oxides
9 (see
Figure 2), derived from trans-1,2-diaminocyclohexane, thereby breaking the tendency of catalysts based on pyridine or bipyridine
N-oxides. Screening of catalysts in the reaction of benzaldehyde with allyltrichlorosilane showed that the presence of phenyl group in the imidazole ring (R
2 in
9b) has a positive effect on the reaction efficiency (3 examples, 84–86%). Unfortunately, the introduction of two phenyl substituents (
9d), only slightly increased enantioselectivity (from 43% ee to 53% ee). However, the presence of the second phenyl substituent in
9d caused the inversion of the absolute configuration of obtained homoallyl alcohol. To improve the catalytic efficiency, different options of catalyst loading for (
R,
R)-
9d, as well as reaction temperature was checked. The best result (90%, 64% ee) has been received for a reaction carried out at 0 °C with 10 mol% of (
R,
R)-
9d. Also, estimation for a scope of aldehyde substrates was done. Higher enantioselectivity for
m-substituted, than for
o-substituted benzaldehydes was noticed. The highest asymmetric induction was observed for heteroaromatic aldehydes—furfural (76% ee) and thiophene-2-carboxyaldehyde (80% ee).
Boyd presented the synthetic pathway to obtain bipyridine
N-oxide derivatives
10 and corresponding
N,
N’-dioxides
11 [
39], showed in
Figure 2. It was found that the allylation reaction is slower when using the mono
N-oxides, and thus, the reactions applying them were carried out at higher temperatures (0 °C or −40 °C, 24 h), compared with those using the corresponding
N,
N’- dioxides (−78 °C, 12 h). In that case, although the mono- and dioxides were applied to allylation of the same aldehydes with allyltrichlorosilane, it is difficult to compare the results unambiguously. The optimal enantioselectivity was observed in the allylation of 4-methoxybenzaldehyde as a substrate, using either
N-oxides
10 (56–86% ee) or corresponding
N,
N’-dioxides 11 (59–80% ee), compared to allylation of benzaldehyde (24–35%
ee and 14–26% ee). The highest enantioselectivity (86% ee) was observed in allylation of 4-methoxybenzaldehyde using 10b as a catalyst. Despite the clear difference in the enantioselectivity of these reactions, their yields did not differ much and were mostly in the range of 30–40% (except
10a–60–72%). Better yields were observed when using
11 as the catalyst, and the compound 11a gave the highest induction (80% ee).
Conformationally rigid chiral backbone, with strong steric requirements, possessing an
N-oxide unit was developed in 2012 by Ramanathan et al. [
37]. Cycloaddition of anthracene and (E)-ethyl 3-(2-pyridyl)-propenoate, followed by the resolution of the enantiomers using
l-(+)-tartaric acid, and then oxidation with
m-chloroperoxybenzoic acid (
m-CPBA) gave nine derivatives with usually good overall yields (up to 96%). All the chiral pyridine
N-oxides
12 were evaluated in enantioselective allylation of 4-methoxybenzaldehyde (20 mol% of catalyst at −40 °C).
It was stated that an electron-rich catalyst causes greater enantioselectivity of the reaction using electron-rich aldehydes. The presence of alkyl or aryl groups at positions 2 and/or 6 of the pyridine ring reduces the nucleophilicity of the corresponding
N-oxide. Therefore, the methoxy-substituted benzene rings (electron-rich aryl groups) have been attached at position 6 of the pyridine ring to afford the catalyst which can enhance both reactivity and selectivity. After optimizing the conditions, the reactions were carried out in a solvent mixture (CHCl
3 and 1,1,2,2-tetrachloroethane in a 1:1 ratio—which enhances the yield and enantioselectivity), and the temperature was lowered from −40 °C to −78 °C.
12i was determined as the most effective catalyst, which gave 84% yield and 87% ee in the case of 4-methoxybenzaldehyde and 81%, 94% ee for 3,4-dimethoxybenzaldehyde (see
Table 1). In comparison to previously described catalysts
4a,
c and
8a,
g, for
12i higher loading (20 mol%) was needed (results compared in
Table 1). The advantage of 12i can be, that it can be used for unusual substrates. High enantioselectivity has been achieved in the allylation of heterocyclic and polycyclic aldehydes with
12i (
Table 1, entries 19, 21, 25). For others, results were rather moderate. Catalyst
12i was also examined in the crotylation of 4-metoxybenaldehyde with crotyltrichlorosilane (
E/Z = 82/12). The corresponding
anti/syn alcohols were obtained in 81:19 ratio with 83% ee and 58% ee, respectively.
Working with various enantiopure hydroxymethyl-substituted pyridine derivatives, Reissig and Eidamshaus received compounds
16 (see
Figure 2), but using them as catalysts gave rather poor results [
40]. Using 5 mol% of
16a benzaldehyde was converted into homoallylic alcohol in 65% with 24% ee after 10 days at rt. Application of desilylated compound
16b caused an increase in enantioselectivity to 47% ee but together with a simultaneous drastic decrease of yield to 12%.
Interesting attempts to prepare polysaccharide (amylose and cellulose) derivatives, bearing pyridine
N-oxide substituents, were made by the Ikai group [
7]. A controlled number of 3- or 4-pyridine
N-oxide groups have been attached to polysaccharide units by ester bonds. The great advantage of this type of compound is their non-toxicity to the environment. Unfortunately, the test applications in the reaction of asymmetric allyltrichlorosilane with benzaldehyde do not seem competitive with other described catalysts. The best results were received using amylose derivatives shown in
Figure 3 (47–62%, 13–32% ee). It has been found that the amount and the position of
N-oxide groups affected the reaction yield and enantioselectivity. Amylose not substituted with
N-oxide groups do not show any catalytic activity. The increase of the number of
N-oxide units in the catalyst from 19% to 23% caused improvement in both yield (from 47% to 62%) and enantioselectivity (from 13% to 32%). Further increase in the amount of pyridine
N-oxide had a negative effect. Also, the location of the
N-oxide groups was significant.
Ramanathan group has tested the versatility of prepared derivatives as Lewis basic activators in the desymmetrization reaction of meso-epoxides with silicon tetrachloride [
18]. Catalysts
12 applied in the reaction with
cis-stilbene epoxide were not very selective (12a gave 32% ee,
12b-5% ee and
12i-42% ee). In contrast, the
C2-symmetric bipyridine dioxide
13 with similarly conformationally rigid, chiral bicyclic skeleton gave 89% ee already in the initial trials. In subsequent experiments, the reaction conditions have been optimized in terms of the solvent used, the catalyst loading, reaction time and temperature, and the quantity of DIPEA used as a base. The best conditions for ring-opening of meso-epoxides were determined as 0.5 mol% of catalyst at −30 °C for 70 min, in CHCl
3, with 15 equivalents of DIPEA (see, reaction above
Table 3). The results of the evaluation of (–)-
13 for the enantioselective desymmetrization of meso-epoxides of various structural classes are shown in
Table 3. The best effectiveness was observed for
cis-stilbene epoxide opening (94% yield, 93% ee, entry 1). One of the challenging substrates, cyclooctene oxide (
Table 3, entry 7) successfully furnished the corresponding chlorohydrin in 84% yield with 69% ee, which is better result compare to obtained with bis(tetrahydroisoquinoline)
N,
N′-dioxides as catalysts (56% ee) [
19].
Stončius and Neniškis presented a completely different approach. The Lewis bases were invented, in which the pyridine
N-oxide unit has been attached to a chiral bicyclo [3.3.1] nonane backbone [
18]. Authors designed the structures, that contain two 2,4-diaryl-substituted pyridine
N-oxide moieties
14 and corresponding monofunctional congeners
15 (see
Figure 2). Catalyst precursors were prepared in two steps: initial Michael addition was followed by the subsequent cyclization reactions to furnish corresponding bis(pyridines). Oxidation of them with
m-CPBA produced expected
N-oxides. Another synthetic strategy was taken for
14e, which the authors briefly explained in the published work [9a]. The obtained catalysts were examined in the allylation of benzaldehyde with allyltrichlorosilane, but the results were mediocre, so they were tested also in the enantioselective ring-opening of meso-epoxides with silicon tetrachloride. Cyclohexene oxide ring-opening reaction was used as a model. The resulting yields were good, but enantioselectivity was far below expectations (7 examples, 72–85% yield, 3–47% ee). Better results obtained with the use of
14a (72%, 32% ee), compared to
14d (73%, 3% ee) suggest a beneficial effect of electron-rich 2,4,6-trimethoxyphenyl substituents on enantioselectivity. Catalysts
14a and
14e were also used in ring-opening of other epoxides, both cyclic and aromatic. Noteworthy is the result of opening the cyclopentene oxide with
14e (85%, 88% ee)–the highest reported to date for the Lewis base-catalyzed desymmetrization of cyclic substrates. Any increase in ring size resulted in decreasing in selectivity (e.g., for cyclohexene oxide 77%, 47% ee). Interesting results have been obtained in the catalytic desymmetrization reaction of an epoxy norbornene derivative, known for that furnish syn, exo-chloroalcohol
17 as a major product, rather than vicinal chlorohydrin
18 (
Scheme 3). For catalysts
14a–
c enantioselectivities of
p-nitrobenzoate (PNB)
17 were excellent (90–96% ee) but yields were not higher than 50%. In other cases, the yield of reaction and asymmetric induction were low. Scale-up (for
14b) or increase of catalyst loading (for
15b) did not affect the yield or ee values but it was possible to isolate
p-nitrobenzoate
19. In turn, the reduction of catalyst loading (for
14b) had a negative effect on reaction yield. Reduction from 10 mol% to 5 mol% caused a decrease in yield from 50% to 30%.
N-Oxides Having the Terpene Unit
Naturally occurring and easily available terpenes and alkaloids are established chiral scaffolds for asymmetric organocatalysts. They were applied for the construction of chiral pyridine
N-oxides, designed as a powerful Lewis-basic catalyst (
Figure 4) already in the first years of the 21st century [
41,
42,
43]. The latest reports about such catalysts come from 2008. Malkov and Kočovský [
44] have prepared derivatives, lacking the
C2 symmetry and having the chirality ‘‘concentrated’’ on one side of the molecule, such as dioxide
20-a quinoline analog with an isomeric terpene unit, the benzoquinoline analog
21 with similar chiral motif, and a series of bipyridine
N,
N’-dioxides with phenyl group
22 or pyridine
N-oxide having (
α-pyridyl-
N-oxide)phenyl group
23. All these compounds (in 10 mol% loading) were tested in standard benzaldehyde allylation reaction.
Chelucci et al. [
45] presented pyridine
N-oxide derivatives as a polycyclic structure with terpene fragments (
24–
28) and dipyridine monoxide with spacer (
29,
30), showed in
Figure 4. Compounds
24–
26 possess
C2-symmetry. The catalytic activity and stereo-differentiating ability of the new compounds, tested in the allyltrichlorosilane addition to benzaldehyde, appeared to be poor. The yield did not exceed 65% and enantiomeric excesses were not higher than 48%. Application of compound
24 resulted in 65% yield, but isolated homoallylic alcohol was a racemate, while in the case of
25, the obtained ee was 48%, but benzaldehyde was converted into alcohol in 31% yield. Epimers
27 and
28 behave as pseudo enantiomers and gave the opposite configuration of an allylic alcohol. Similar results of asymmetric induction have been achieved for both organocatalysts,
27 and its methyl analog
27a. Due to the previously observed relationship in similar
C2-symmetric structures, where enantioselectivity for mono
N-oxide was higher than for
N,
N’-dioxide, compounds
29 and
30 were designed. Next to
29 and
30, analogous
N,
N’-dioxides were also reported [
46]. However, in these cases, the advantage of mono-oxides has not been observed. Compounds
24–
26 and
29–
30 were used also in the enantioselective opening of
cis-stilbene. The yield achieved for all examined compounds was moderate to high (47–95%). Unfortunately, obtained halohydrin in most experiments was a racemate. Only for catalyst
25 asymmetric induction occurred (37% ee).
METHOX (
27b), developed and successfully used in allylation of aromatic aldehydes with allyltrichlorosilane a few years earlier [
47], in 2011 was tested in similar reaction but using allyldisilane (
Scheme 4) [
48]. Catalytic reactions of allyldisilane with aldehydes proceeded at very low rates and required a week or more to reach completion (10–15 mol% of (+)-
27b, at −35 °C, in CH
3CN). Nevertheless, METHOX exhibited excellent diastereo- and enantioselectivities for benzaldehyde and its derivatives (up to 97% ee). Further, this methodology was extended to the group of
α,
β-unsaturated aldehydes [
48,
49]. Different
α,
β-unsaturated aldehydes
31, incorporating aromatic, aliphatic substituents and cyclic ones were examined, and the results are shown in
Figure 5. All of the aldehydes, with or without the α-branching, were found to react with good conversion within 3–5 days, providing high enantioselectivity, up to 93% ee. However, slightly better effectiveness was observed for
31a–
c and
31f. In other cases, obtained yields varied between 37–60% and measured asymmetric induction from 66% ee to 89% ee. Received products were levorotary (except for (
S)-
31j use) when (+)-METHOX was employed. Aldehyde
31d, crotylated under the same conditions, gave rise to the expected anti-diastereoisomer as practically the only product, with the highest enantiomeric excess (96% ee). METHOX has been found to exhibit a high tolerance to aldehyde electronics, and the reactions require low catalyst loading (even 5 mol%).
The observed difference in the reactivity and selectivity of dioxide and monoxide catalysts suggests that these two catalyst types can operate
via different mechanisms [
44]. In the case of bidentate
N,
N’-dioxides, cationic transition state
A can be envisioned (see
Figure 6). On this basis, a significant impact on the observed enantioselectivity of both, the unsymmetrical catalyst substitution and the huge effect of axial chirality of the catalyst can be explained (determining the absolute configuration of the product). Alternatively, the reaction proceeds via an associative pathway, involving the neutral octahedral silicon complex
B. Transition state
B provides high enantiocontrol in the allylation of aromatic aldehydes, however, it can be sensitive to the electronic effects of substituents in a substrate and any variation in the catalyst structure proximal to the coordinating center.
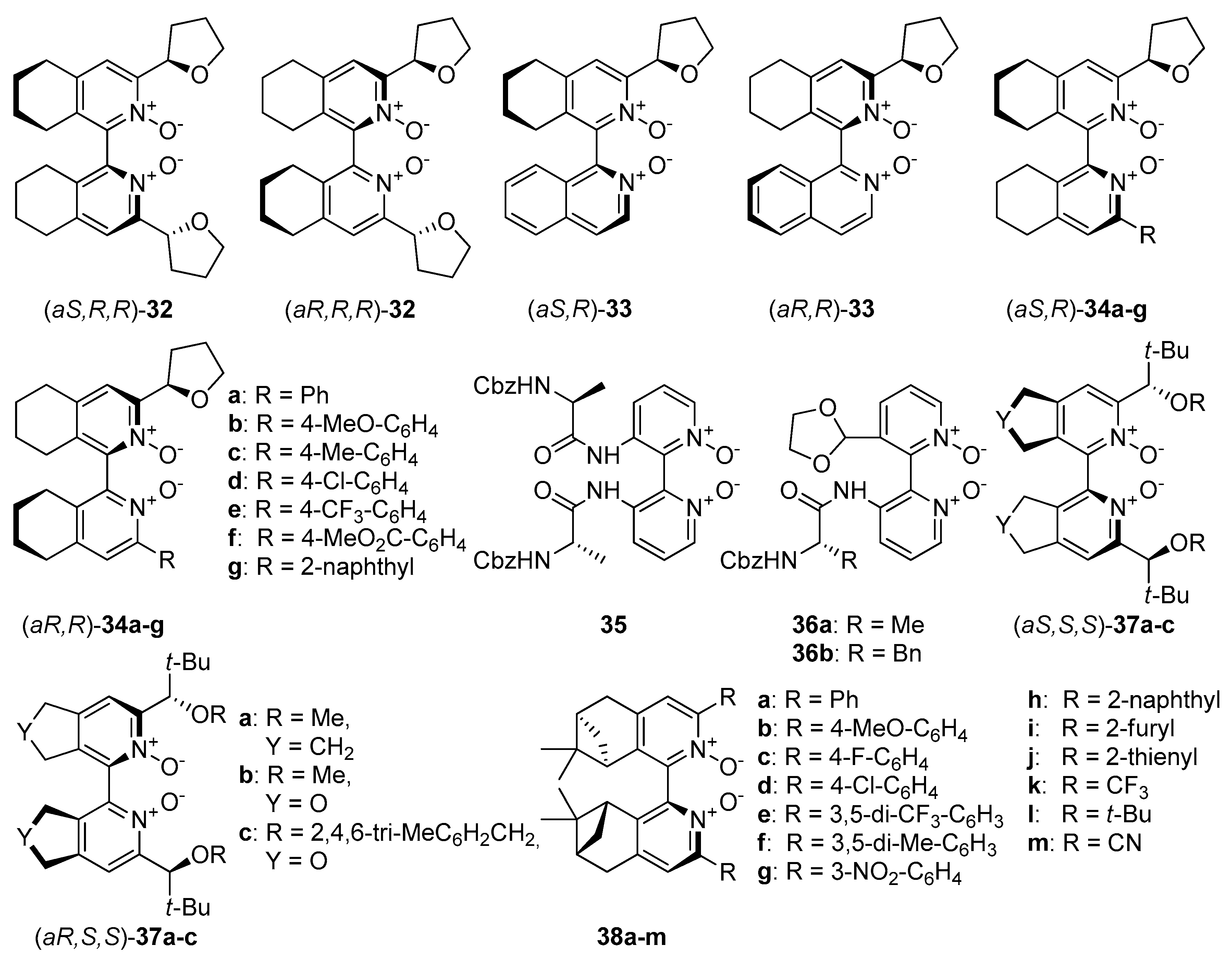
2.3. N-Oxides with Central and Axial Chirality
An interesting direction in the exploration of catalytically useful
N-oxides is the combination of two types of chirality. Researches mainly concentrate on symmetrically or unsymmetrically substituted chiral bis(tetrahydroisoquinoline)
N,
N’-dioxides (
Figure 7), which with their structure resemble compound
8. In 2008 Kotora et al. synthesized symmetrically substituted by (
R)-tetrahydrofuran-2-yl dioxides
32 (see
Figure 7), which were tested in the allylation of aromatic [
50] as well as aliphatic aldehydes [
30]. Catalysts synthesis was based on cyclotrimerization of tetrayne with (
R)-tetrahydrofuran-2-carbonitrile, followed by oxidation of received bipyridines by
m-CPBA. At the end separation of a resulted mixture of diastereoisomers was necessary. A simple column chromatography on alumina, gave isolated yields 48% for (
aR,
R,
R)-32 and 28% for (
aS,
R,
R)-
32. The configuration was assigned by X-ray crystallographic analysis. In allylation of benzaldehyde, 4-trifluoromethylbenzaldehyde or 4-methoxybenzaldehyde performed in MeCN, at −40 °C for 1 h, with 1 mol% catalyst loading, both diastereomeric catalysts were effective [
50]. Aldehydes were converted into corresponding homoallylic alcohols almost quantitatively (
Table 4, entries 1, 4, 10). Only for allylation of 4-trifluoromethylbenzaldehyde using (
aR,
R,
R)-
32 the yield was slightly lower (82%, entry 10). Applying the same catalyst, the enantioselectivity was lower for aldehyde with an electron-withdrawing group (15% ee in comparison to 48% ee for benzaldehyde,
Table 4, entry 10) and higher for aldehyde substituted with an electron-donating group (60% ee,
Table 4, entry 4). When (
aS,
R,
R)-
32 was used decrease of asymmetric induction for 4-trifluoromethylbenzaldehyde was observed again (
Table 4, entry 10), but for 4-methoxybenzaldehyde obtained alcohol was a racemate (
Table 4, entry 4).
In addition, a strong solvent effect on stereoselectivity has been observed. For (
aR,
R,
R)-
32 used in chlorobenzene no reaction was observed, but using (
aS,
R,
R)-
32 the enantioselectivity was significantly higher (
Table 4, entries 2, 5, 11), for all three substrates. For benzaldehyde yield did not change, while for substituted substrates it decreases by half. Inversion of product configuration was observed in dependence of solvent used–
R in MeCN into
S in PhCl. Kotora also mentioned in the same publication about isoquinolyl-tetrahydroisoquinoline
33 unsymmetrically substituted with tetrahydrofuran-2-yl group. The catalysts themselves were obtained in low yields (4–17%), and their use for allylation of benzaldehyde gave rather mediocre results (86% with 49% ee using (
R,
R)-
33 and 84% with 48% ee using (
S,
R)-
33). However, configurations of received homoallylic alcohols (
S with (
R,
R)-
33 and
R with (
S,
R)-
33) indicates that configuration of the product is controlled by the axial chirality of the catalyst (the opposite is obtained). The catalytic activity of
32 was also studied in the allylation of aliphatic aldehydes. Different solvents e.g., acetonitrile, dichloromethane, chloroform, and acetone have been tested in allylation of cyclohexanecarboxaldehyde as the model substrate and there is no doubt that the solvent effect controls the reaction mechanism. Using (
aR,
R,
R)-
32 enantioselectivity was low, regardless of the solvent used (10–19% ee,
Table 5, entries 1–4), but the yield proved to be highly dependent on the solvent. The highest yield was obtained when MeCN was applied (85%,
Table 5, entry 1). For the remaining solvents, the yields were similar and amounted to 34–40%. Application of (
aS,
R,
R)-
32 resulted in an increase of both the yield (52–79%,
Table 5, entries 5–8) and the reaction enantioselectivity (39–68% ee). Configuration of obtained alcohol was inverse to those obtained with (
aR,
R,
R)-
32, which confirms prior observation about the decisive influence of the axial chirality of the catalyst. (
aS,
R,
R)-
32 employed toward other aliphatic substrates gave generally high yield (79–91%,
Table 5, entries 9–12) except for pivaloyl aldehyde (10%,
Table 5, entry 13) but enantioselectivity was rather moderate (22–68% ee). For cyclic substrates, products have configuration
R (
Table 5, entries 11–12), while straight-chain aldehydes resulted in alcohols with configuration
S (
Table 5, entries 9, 10, 13).
The unsymmetrically substituted bis(tetrahydroisoquinoline)
N,
N’-dioxides
34a were also synthesized and employed in catalytic allylation of aromatic aldehydes (see
Table 4) [
31]. Again, the huge influence of the solvent on the stereochemical result of the reaction was observed. The catalyst (1 mol% loading) with
S-axial chirality gave
R-product in MeCN (
Table 4, entry 1) and
S-product in PhCl and THF (
Table 4, entries 2,3). Also, in MeCN the enantioselectivity was about two times lower than in PhCl and THF, for which results were similar. Therefore, reactions with other substrates were carried in THF. Generally, for both isomers,
34a results were very good, but (
aS,
R)-
34a worked better as a catalyst for highly enantioselective allylation of benzaldehydes, bearing electron-withdrawing as well as electron-donating groups. Slightly better yield can be observed in the case of
p-substituted benzaldehydes than for
m-substituted (
Table 4, entries 5–9, 11–14). A drastic decrease in the yield and asymmetric induction was observed for
o-chlorobenzaldehyde (
Table 4, entry 15). Results depicted in
Table 4 clearly show, that unsymmetrically substituted bis(tetrahydroisoquinoline)
N,
N’-dioxides
34a exhibit higher catalytic activity, than symmetrically substituted derivatives
32. Yields of obtained products are comparable, but much better enantioselectivity gives the use of
34 (both epimers). The enantioselectivity was highly solvent-dependent. It seems, that THF enables the reaction mechanism to proceed through the sterically more crowded neutral six-coordinate silicon species, leading to higher enantioselectivity [
30]. The scope of application of
34a for aliphatic aldehydes was tested in the reactions with
n-octanal or cyclohexylcarbaldehyde (
Table 5, entries 14–17) [
31]. The reaction in both cases was characterized by good to excellent yields (88–95%) and moderate enantioselectivity (38–67% ee).
Higher results were detected for (
aR,
R)-
34a. (
aR,
R)-
34a was also applied in allylation of benzaldehyde with
E- crotyltrichlorosilane (6:1
E/Z mixture in PhCl, for 24 h, at −40 °C with 1 mol% of catalyst), giving 2.4/1 anti/syn diastereoisomer mixture, with good yield 82% and satisfactory enantioselectivity 91% and 87% ee, respectively. The scope of application in the asymmetric allylation reaction of both, symmetric catalysts
32 was studied [
51]. Under optimized conditions (1 mol% of the catalyst in THF at −40 °C) a number of different, aromatic and aliphatic
α,
β-unsaturated aldehydes were checked, testing both epimers of
32 (
Scheme 5). Better enantioselectivity was achieved using (
aS,
R,
R) diastereomer but slightly higher yields were obtained for (
aR,
R,
R) diastereomer–respectively, 52–82% yield, 63–96% ee and 40–95% yield, 4–68% ee. The best enantioselectivity was achieved using (
aS,
R,
R)-
32 with substrates having both groups: R
1 (as phenyl) and R
2 (as methyl group or chlorine atom)-96% ee in both cases. By using (
aR,
R,
R)-
32 it was found that, that higher enantioselectivity (62–68% ee) was obtained when α-substitution (R
2) was present independently of whether R
1 was aromatic or aliphatic. Better yields for (
aR,
R,
R)-
32 (82–97%) were observed when R
1 was phenyl substituted with an electron-withdrawing group or unsubstituted phenyl group, but then the presence of electron-donating group as R
2 was necessary. For both catalysts employed, the configuration of obtained aliphatic alcohols was
R, whereas it was opposite for products having an aromatic substituent. In most applications, both catalysts appear to be similarly effective to the METHOX described earlier.
Interesting comparative studies have been done by Kotora’s research group for different enantioselective allylation procedures and different catalysts applied
N-oxides
34a-Lewis basic catalyst were compared with (
S)-BINOL-Lewis acid catalyst (Keck protocol) and (
R)-TRIP PA-Brønsted acid catalyst in enantioselective allylation of
o-substituted benzaldehydes (
Scheme 6) [
52].
The reactions performed needed different catalyst loadings and different reaction times. Despite this, an attempt to compare the results was made. Chiral catalytic systems have rarely been used for allylation of
o-substituted benzaldehydes, so the effect of substitution in the ortho position of the aromatic aldehydes was tested using the three procedures shown in
Scheme 6. Definitely, the best yields were obtained using phosphorous catalyst (
R)-TRIP PA (93–99% in comparison to results up to 70% for
34a and up to 80% for (
S)-BINOL), but achieving high enantioselectivity was problematic in all cases. In some cases the use of
N,
N’-dioxide catalysts gives better optical purity, in others, Keck or Brønsted acid allylation seems to be more effective. Also, none of the catalysts were versatile. Among tested
N,
N’-dioxides, again better yields gave (
aR,
R)-
34a and higher asymmetric induction was observed using (
aR,
S)-
34a. The best results were obtained for
o- fluorobenzaldehyde (48%, 82% ee for (
aR,
R)-dioxide and 34%, 66% ee for (
aR,
S)-dioxide),
o-vinylbenzaldehyde (40%, 72% ee and 40%, 76% ee, written in the same order) and
m- methoxybenzaldehyde (60%, 85% ee and 60%, 88% ee), which are rather moderate. The effect of the solvent was examined in allylation of 2-iodo-5-methoxybenzaldehyde. For (
R,
R)-epimer reaction proceeded better in dichloromethane than THF—70%, 41% ee in comparison to 5%, 56% ee and reversely, in case (
R,
S)-enantiomer-40%, 4% ee versus 53%, 80% ee, whereas in toluene reaction did not proceed at all, in both cases. When the reaction was performed in THF, the configuration of all products was the same as the axial configuration of the catalyst. Inversion of product configuration was observed when dichloromethane was applied.
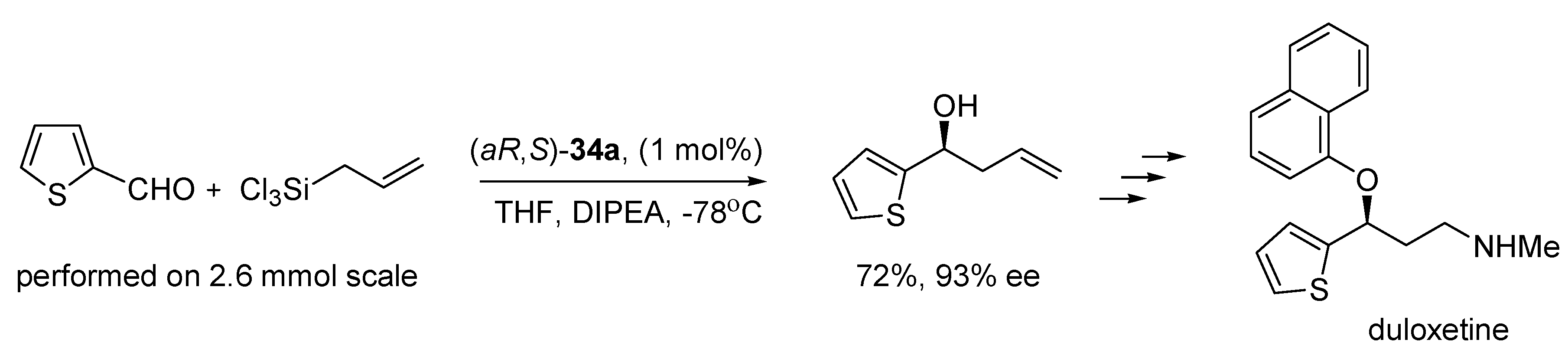
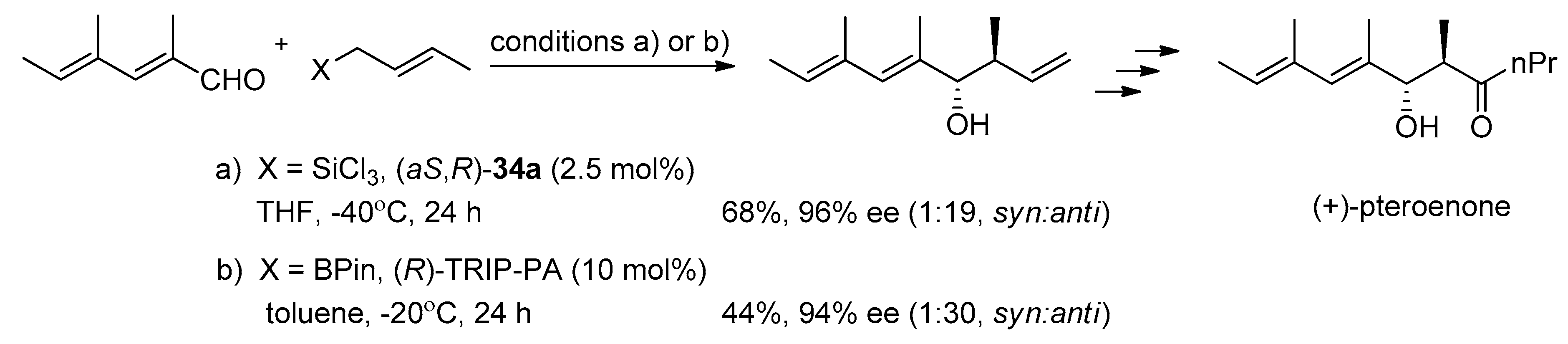
N-oxide catalysts were successfully applied to the synthesis of a few natural products [
12,
13,
14,
15,
16,
17]. For example, duloxetine, which is used in the treatment of major depression, was synthesized involving the allylation of 2-thiophenecarboxaldehyde with (
aR,
S)-
34a as catalyst (see
Scheme 7) [
12].
The advantage of the N,N’-dioxide catalysts is in their rather low loading, (only 1 mol%) being sufficient to bring about the desired allylation. In the case of phosphorous catalyst TRIP PA, the level of asymmetric induction depends on the catalyst loading, optimally it is 10 mol%, which is rather high (
Scheme 8) [
15].
Malkov and Kočovský [
48] have compared the effectiveness of catalyst
34a and METHOX in the reaction of different aldehydes with allyldisilane. Generally, obtained results were high and similar for both catalysts, however, reactions applying METHOX needed higher loading (15–20 mol% versus 5–10 mol%) and much longer time (7 days versus 12 h) to be completed. Optimal solvent for use of
34a as catalyst was THF, while CH
3CN for METHOX (temp. −35 °C for both). At the enantioselectivity of a similar order, the yields obtained were slightly higher for
34a (for both isomers 71–83% yield and 73–98% ee). In all cases, only anti isomers were obtained. For both catalyst types decrease of efficiency and enantioselectivity was observed in case benzaldehydes substituted by electron-donating group (methoxy group) in para-position. For (
aR,
R)-
34a two non-aromatic aldehydes were used–hexanal and
α,
β-unsaturated hex-2-enal. Great enantioselectivity 98% ee and good yield 83% has been received for the second one. For (
aS,
R)-
34a observed that decreasing the amount from 10 mol% to 1 mol% with simultaneous elongation of the reaction time from 12 h to 24 h caused a slight decrease of both the yield and enantioselectivity (from 82%, 96% ee to 70%, 91% ee).
Zhao et al. have designed symmetrically and unsymmetrically substituted, axially chiral 2,2’- bipyridine
N,
N’-dioxides combined with central chirality introduced with
α-amino acid residues:
C1-symmetric
36 and
C2-symmetric
35 (see
Figure 7) [
53]. Using 10 mol% of catalyst for allylation of
p-nitrobenzaldehyde in CH
2Cl
2 at −78 °C (with the addition of DIPEA) rather mediocre results were obtained 41–66% yield, 27–40% ee. It was observed that the levorotary catalyst gave comparable or even slightly better results in shorter reaction times. Also, levorotary catalyst gave (
R)-product, while dextrorotary catalyst gave (
S)-product. Nine different polar and nonpolar solvents were tested in allylation using (−)-
35 as catalyst and CH
2Cl
2 (48%, 33% ee)
, next to MeCN (35%, 41% ee), seemed to be better solvents for the allylation reactions. But in MeCN, nearly 3 times longer reaction time was needed to achieve shown results (11 h versus 4 h). Catalyst (–)-
36b showed similar enantioselectivity in the same test reaction, giving 66% yield and 35% ee,
Table 6, entry 1). Both,
C2-symmetric catalyst
35 and
C1-symmetric catalyst
36b have been evaluated in the allylation of other substituted benzaldehydes (see
Table 6). The best results were obtained with 4-methylbenzaldehyde and 1,4-benzodioxane-6-carboxaldehyde–61–65% ee and 53–63% ee, respectively (
Table 6, entries 6 and 8). By increasing the catalyst loading (for
35,
Table 6, entry 7), a bit higher yield was obtained, but a decrease in enantioselectivity was observed. The allylation of 4-methoxybenzaldehyde was also performed using (–)-
36b and, despite that methoxy group is electron-donating (similar to methyl group), in this case, the obtained product was racemic.
Kotora et al. synthesized a number of derivatives
34 (see
Figure 7), with variously modified aromatic substituents at the 3’ position [
54]. All these dioxide derivatives have been tested, as Lewis base catalysts, in allylation of aromatic aldehydes (0.5 mol%) in THF and CH
2Cl
2. Again, THF was a better reaction medium. As observed, the substitution of catalysts in the aryl group by EDG or EWG did not significantly change the catalytic activity of
34a. Slightly better results were observed for all (
S,
R) isomers, except the results of cinnamaldehyde allylation, which were similar for both epimers of
34b–
g. Asymmetric induction was not observed in the allylation of 2-thiophenecarboxaldehyde. Gradual increase of catalyst loading showed, that both the yield and enantioselectivity increased up to 74%, 80% ee for 5 mol% of (
aR,
R)-
34a and 90%, 88% ee for 5 mol% of (
aS,
R)-
34a. The allylations of benzaldehyde,
p-methoxybenzaldehyde and
p- trifluoromethylbenzaldehyde using
34, described in this report, were performed using 0.5 mol% of the catalyst [
54]. Comparing them with the same reactions applying 1mol% of the catalyst [
36] allows the conclusion that reducing the loading does not affect enantioselectivity. Only in case of
p-trifluorobenzaldehyde the yield slightly decreased using (
S,
R)-enantiomer, while with use (
R,
R)-enantiomer, the yield apparently decreased, when half amount of catalyst has been used.
The predictable utility of the expected product as a chiral building block in the synthesis of natural products [
54,
55] was the reason for the application of
34a–
g in allylation of
E-3-iodomethacrylaldehyde (
Table 7). For this reaction, the influence of modification of the phenyl group in catalyst turned out to be significant. The advantage of (
aR,
R)-catalysts
34b–
g over (
aR,
R)-
34a (with unsubstituted phenyl group in 3′ position) was especially noticeable in term of enantioselectivity (80–97% in comparison to 57%). When it comes to the yield, the highest result was observed for (
aR,
R)-
34e, having 4-trifluoromethylphenyl group (
Table 7, entry 5), while by using (
aR,
R)-
34b, having 4-methoxyphenyl group (
Table 7, entry 2), the lowest conversion was obtained. In all other cases, yields were comparable (approx. 70%). In the case of the (
aS,
R)-isomers, for all catalysts, including (
aS,
R)-
34a, the obtained enantioselectivity was excellent (98–99% ee). For compounds with EWG or EDG substituent present at the phenyl group, an increase of yield was observed–from 49% to 66–89%. The best and the worst results coincided with those obtained for (
aR,
R)-enantiomers (
Table 7, entries 5 and 2, respectively). The configuration of the obtained product was the same as the axial configuration in the catalyst.
Kotora and Lamaty presented bipyridine
N,
N’-dioxides
37, with
C2-symmetry (see
Figure 7) [
24,
25]. Their synthesis was based on catalytic [2+2+2] cyclotrimerization either hepta-1,6-diyne or propargyl ether as starting material with various nitriles. A detailed synthetic pathway, including two different approaches, was described. Obtained compounds
37a,
b were examined in the allylation of benzaldehyde. Although the use of acetonitrile as solvent notably improved the yields, (93% with (
aR,
S,
S)-
37b and 96% with (
aR,
S,
S)-
37a), compared to results in THF, the enantioselectivity remained moderate (45% ee and 33% ee, respectively). On the other hand, using DCM rise in enantioselectivity (72% ee using (
aR,
S,
S)-
37b and 46% ee using (
aR,
S,
S)-
37a) with noticeably decreased the yield. In both cases, slightly better enantioselectivity was observed for (
aS,
S,
S)-isomers of
37a,
b, but it was still rather moderate (51% ee and 40% ee respectively).
A recent report by Rubtsov and Malkov et al. [
56] presents the synthesis of atropoisomeric bipyridine
N,
N′-dioxides with terpene derived moieties
38 (see
Figure 7). The combination of axial chirality and terpene-derived structures seemed to be a promising direction. Especially compound
38e turned out to be an extremely efficient catalyst producing excellent enantio- and diastereoselectivities in the asymmetric crotylation over a whole range of aldehydes tested [
14]. Catalyst (−)-
38e proved particularly efficient with unsaturated aldehydes, though with aliphatic enantioselectivity dropped representing a common trend (see
Scheme 9).
Applying (−)-
38e in the same reaction conditions to a larger scale (5 mmol), provides excellent yield and enantioselectivity retained paving the way for the asymmetric synthesis of (−)-elisabethadione (see
Scheme 10) [
14].
Compounds
37 were also applied in aldol reaction of acetophenone with trichlorosilyl ketene acetal (
Scheme 11) [
24]. Good to excellent yields were achieved using
37a,
b catalysts, independently of their configuration (87–96%). For
37a, enantioselectivity was higher when (
aS,
S,
S)-isomer was used and for
37b,
c with (
aR,
S,
S)-isomer. The difference between the structure of catalysts is the presence of oxygen in the five-membered ring of compounds. The more selective were catalysts bearing the oxygen atom in the five-membered ring (
37b,
c). The best result received for (
aR,
S,
S)-
37b was 96% yield and 78% ee, which is better than obtained with the catalyst (
aR,
R)-
34a in the same conditions.



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
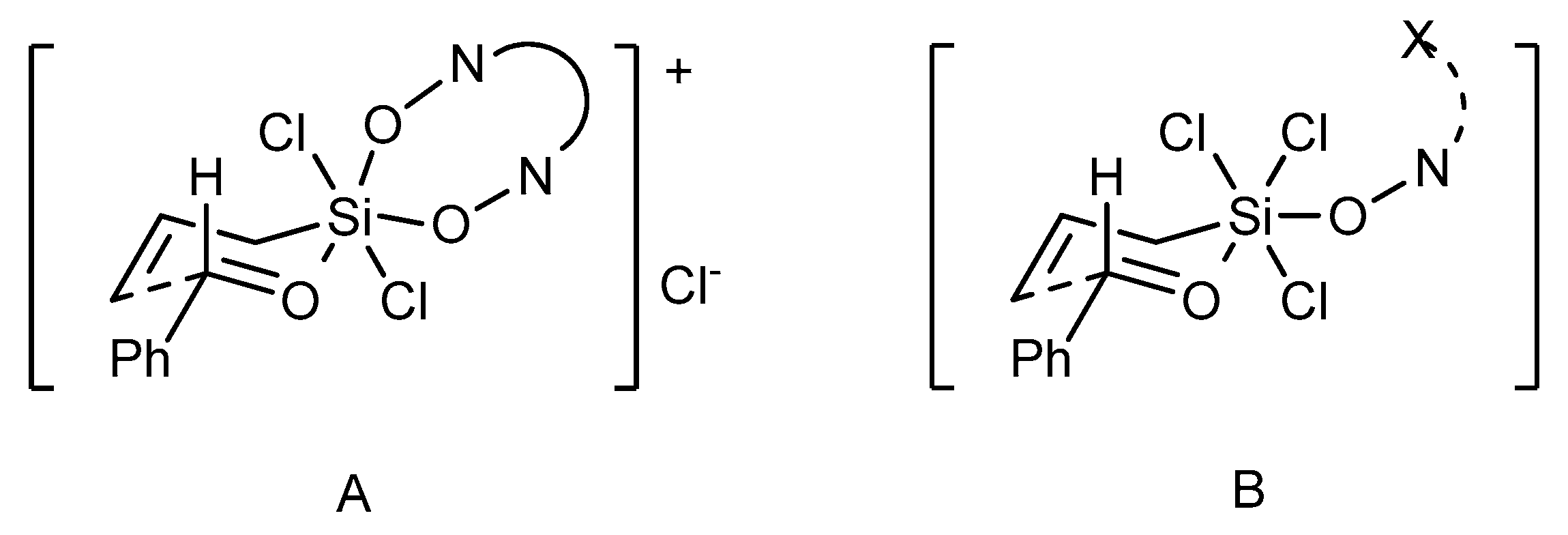{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







