
Bioadhesive Polymeric Films Based on Red Onion Skins Extract for Wound Treatment: An Innovative and Eco-Friendly Formulation

 ,
,  , ,
, ,  ,
,
Abstract
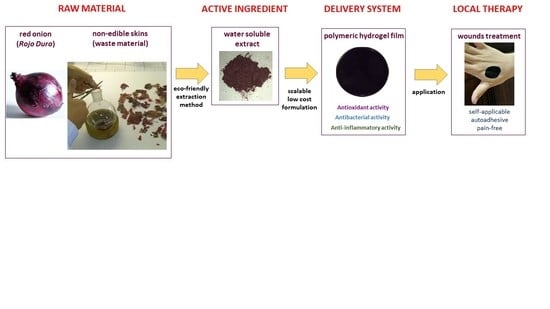
1. Introduction
2. Results
2.1. Optimization of the Extraction Procedure
2.2. Extract Characterization
2.2.1. Fingerprint Analysis of OLE Constituents
2.2.2. Antibacterial Activity Assay
2.2.3. Cytotoxicity Studies on RAW 264.7 and HaCaT Cell Lines
2.2.4. Anti-Inflammatory Activity
2.3. Hydrogel Film Preparation
2.4. Hydrogel Film Characterization
2.4.1. Hydrogel Film Antibacterial Activity
2.4.2. Hydrogel Film Thickness, Swelling Behavior and Matrix Erosion Capacity
2.4.3. Ex Vivo Adhesion Studies
2.4.4. OLE In Vitro Release and Correlation with the Anti-Inflammatory Activity
2.4.5. In Vitro Safety Studies of Hydrogel Film on HaCaT Cell Line
3. Materials and Methods
3.1. Materials
3.2. Extraction Procedure
3.3. Extract Characterization
3.3.1. Fingerprint Analysis of OLE Constituents
3.3.2. Antibacterial Activity Assay
3.3.3. Cell Lines and Cytotoxicity Studies
3.3.4. Anti-Inflammatory Activity
3.4. Hydrogel Film Preparation
3.5. Hydrogel Film Characterization
3.5.1. Hydrogel Film Antibacterial Activity
3.5.2. Hydrogel Film Thickness, Swelling Behavior and Matrix Erosion
3.5.3. Thermal Properties Measurement
3.5.4. Ex Vivo Adhesion Studies
3.5.5. In Vitro Release of Extract from The Hydrogel Film
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ninfali, P.; Mea, G.; Giorgini, S.; Rocchi, M.; Bacchiocca, M. Antioxidant capacity of vegetables, spices and dressings relevant to nutrition. Br. J. Nutr. 2005, 93, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Leong, L.; Williamkoh, J. Antioxidant activities of aqueous extracts of selected plants. Food Chem. 2006, 99, 775–783. [Google Scholar] [CrossRef]
- Slimestad, R.; Fossen, T.; Vågen, I.M. Onions: A Source of Unique Dietary Flavonoids. J. Agric. Food Chem. 2007, 55, 10067–10080. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cha, Y.-J.; Lee, K.-H.; Park, E. Effect of onion peel extract supplementation on the lipid profile and antioxidative status of healthy young women: A randomized, placebo-controlled, double-blind, crossover trial. Nutr. Res. Pract. 2013, 7, 373–379. [Google Scholar] [CrossRef]
- Lee, J.-S.; Cha, Y.-J.; Lee, K.-H.; Yim, J.-E. Onion peel extract reduces the percentage of body fat in overweight and obese subjects: A 12-week, randomized, double-blind, placebo-controlled study. Nutr. Res. Pract. 2016, 10, 175–181. [Google Scholar] [CrossRef]
- Kim, K.-A.; Yim, J.-E. Antioxidative Activity of Onion Peel Extract in Obese Women: A Randomized, Double-blind, Placebo Controlled Study. J. Cancer Prev. 2015, 20, 202–207. [Google Scholar] [CrossRef]
- Jung, J.Y.; Lim, Y.; Moon, M.S.; Kim, J.Y.; Kwon, O. Onion peel extracts ameliorate hyperglycemia and insulin resistance in high fat diet/streptozotocin-induced diabetic rats. Nutr. Metab. 2011, 8, 18. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jo, S.-H.; Kwon, Y.-I.; Hwang, J.-K. Effects of Onion (Allium cepa L.) Extract Administration on Intestinal α-Glucosidases Activities and Spikes in Postprandial Blood Glucose Levels in SD Rats Model. Int. J. Mol. Sci. 2011, 12, 3757–3769. [Google Scholar] [CrossRef]
- Albishi, T.; John, J.A.; Al-Khalifa, A.S.; Shahidi, F. Antioxidant, anti-inflammatory and DNA scission inhibitory activities of phenolic compounds in selected onion and potato varieties. J. Funct. Foods 2013, 5, 930–939. [Google Scholar] [CrossRef]
- Kim, O.Y.; Lee, S.-M.; Do, H.; Moon, J.; Lee, K.-H.; Cha, Y.-J.; Shin, M.-J. Influence of Quercetin-rich Onion Peel Extracts on Adipokine Expression in the Visceral Adipose Tissue of Rats. Phyther. Res. 2011, 26, 432–437. [Google Scholar] [CrossRef]
- Kim, K.-A.; Yim, J.-E. The Effect of Onion Peel Extract on Inflammatory Mediators in Korean Overweight and Obese Women. Clin. Nutr. Res. 2016, 5, 261–269. [Google Scholar] [CrossRef] [PubMed]
- De Dicastillo, C.; Navarro, R.; Guarda, A.; Galotto, M. Development of Biocomposites with Antioxidant Activity Based on Red Onion Extract and Acetate Cellulose. Antioxidants 2015, 4, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.L.; Dai, D.H.; Hu, W.L. Antimicrobial and antioxidant activities of the essential oil from onion (Allium cepa L.). Food Control 2013, 30, 48–53. [Google Scholar] [CrossRef]
- Shon, M.Y.; Choi, S.D.; Kahng, G.G.; Nam, S.H.; Sung, N.J. Antimutagenic, antioxidant and free radical scavenging activity of ethyl acetate extracts from white, yellow and red onions. Food Chem. Toxicol. 2004, 42, 659–666. [Google Scholar] [CrossRef]
- Petropoulos, S.A.; Fernandes, Â.; Barros, L.; Ferreira, I.C.F.R.; Ntatsi, G. Morphological, nutritional and chemical description of “Vatikiotiko”, an onion local landrace from Greece. Food Chem. 2015, 182, 156–163. [Google Scholar] [CrossRef]
- Benkeblia, N. Free-radical scavenging capacity and antioxidant properties of some selected onions (Allium cepa L.) and garlic (Allium sativum L.) extracts. Braz. Arch. Biol. Technol. 2005, 48, 753–759. [Google Scholar] [CrossRef]
- Ianni, F.; Marinozzi, M.; Scorzoni, S.; Sardella, R.; Natalini, B. Quantitative Evaluation of the Pyruvic Acid Content in Onion Samples with a Fully Validated High-Performance Liquid Chromatography Method. Int. J. Food Prop. 2016, 19, 752–759. [Google Scholar] [CrossRef]
- Lisanti, A.; Formica, V.; Ianni, F.; Albertini, B.; Marinozzi, M.; Sardella, R.; Natalini, B. Antioxidant activity of phenolic extracts from different cultivars of Italian onion (Allium cepa) and relative human immune cell proliferative induction. Pharm. Biol. 2016, 54, 799–806. [Google Scholar] [CrossRef]
- Rodrigues, A.S.; Almeida, D.P.F.; Simal-Gándara, J.; Pérez-Gregorio, M.R. Onions: A Source of Flavonoids. In Flavonoids—From Biosynthesis to Human Health; InTech: London, UK, 2017. [Google Scholar]
- Zuin, V.G.; Ramin, L.Z. Green and Sustainable Separation of Natural Products from Agro-Industrial Waste: Challenges, Potentialities, and Perspectives on Emerging Approaches. Top. Curr. Chem. 2018, 376, 3. [Google Scholar] [CrossRef]
- Ko, M.-J.; Cheigh, C.-I.; Cho, S.-W.; Chung, M.-S. Subcritical water extraction of flavonol quercetin from onion skin. J. Food Eng. 2011, 102, 327–333. [Google Scholar] [CrossRef]
- Brüll, V.; Burak, C.; Stoffel-Wagner, B.; Wolffram, S.; Nickenig, G.; Müller, C.; Langguth, P.; Alteheld, B.; Fimmers, R.; Naaf, S.; et al. Effects of a quercetin-rich onion skin extract on 24 h ambulatory blood pressure and endothelial function in overweight-to-obese patients with (pre-)hypertension: A randomised double-blinded placebo-controlled cross-over trial. Br. J. Nutr. 2015, 114, 1263–1277. [Google Scholar] [CrossRef]
- Ramos, F.A.; Takaishi, Y.; Shirotori, M.; Kawaguchi, Y.; Tsuchiya, K.; Shibata, H.; Higuti, T.; Tadokoro, T.; Takeuchi, M. Antibacterial and Antioxidant Activities of Quercetin Oxidation Products from Yellow Onion (Allium cepa) Skin. J. Agric. Food Chem. 2006, 54, 3551–3557. [Google Scholar] [CrossRef]
- Suh, H.; Lee, J.; Cho, J.; Kim, Y.; Chung, S. Radical scavenging compounds in onion skin. Food Res. Int. 1999, 32, 659–664. [Google Scholar] [CrossRef]
- Lee, K.A.; Kim, K.-T.; Kim, H.J.; Chung, M.-S.; Chang, P.-S.; Park, H.; Pai, H.-D. Antioxidant activities of onion (Allium cepa L.) peel extracts produced by ethanol, hot water, and subcritical water extraction. Food Sci. Biotechnol. 2014, 23, 615–621. [Google Scholar] [CrossRef]
- Frankel, E.N.; Meyer, A.S. The problems of using one-dimensional methods to evaluate multifunctional food and biological antioxidants. J. Sci. Food Agric. 2000, 80, 1925–1941. [Google Scholar] [CrossRef]
- Marinozzi, M.; Sardella, R.; Scorzoni, S.; Ianni, F.; Lisanti, A.; Natalini, B. Validated Pungency Assessment of Three Italian Onion (Allium cepa L.) Cultivars. Agric. Food 2014, 2, 532–541. [Google Scholar]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Kwon, H.K.; Song, M.J.; Lee, H.J.; Park, T.S.; Kim, M.; Park, H.J. Pediococcus pentosaceus-Fermented Cordyceps militaris Inhibits Inflammatory Reactions and Alleviates Contact Dermatitis. Int. J. Mol. Sci. 2018, 19, 3504. [Google Scholar] [CrossRef]
- Chen, B.-C.; Liao, C.-C.; Hsu, M.-J.; Liao, Y.-T.; Lin, C.-C.; Sheu, J.-R.; Lin, C.-H. Peptidoglycan-Induced IL-6 Production in RAW 264.7 Macrophages Is Mediated by Cyclooxygenase-2, PGE 2/PGE 4 Receptors, Protein Kinase A, IκB Kinase, and NF-κB. J. Immunol. 2006, 177, 681–693. [Google Scholar] [CrossRef]
- Jeong, D.; Dong, G.Z.; Lee, H.J.; Ryu, J.H. Anti-Inflammatory Compounds from Atractylodes macrocephala. Molecules 2019, 24, 1859. [Google Scholar] [CrossRef]
- An, Z.; Su, J. Acinetobacter baumannii outer membrane protein 34 elicits NLRP3 inflammasome activation via mitochondria-derived reactive oxygen species in RAW264.7 macrophages. Microbes Infect. 2018, 3, 143–153. [Google Scholar] [CrossRef]
- Escandell, J.; Recio, M.; Giner, R.; Máñez, S.; Ríos, J. Bcl-2 is a negative regulator of interleukin-1β secretion in murine macrophages in pharmacological-induced apoptosis. Br. J. Pharmacol. 2010, 160, 1844–1856. [Google Scholar] [CrossRef]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef]
- Latifa, K.; Sondess, S.; Hajer, G.; Manel, B.-H.-M.; Souhir, K.; Nadia, B.; Abir, J.; Salima, F.; Abdelhedi, M. Evaluation of physiological risk factors, oxidant-antioxidant imbalance, proteolytic and genetic variations of matrix metalloproteinase-9 in patients with pressure ulcer. Sci. Rep. 2016, 6, 29371. [Google Scholar] [CrossRef][Green Version]
- Pagano, C.; Ceccarini, M.R.; Calarco, P.; Scuota, S.; Conte, C.; Primavilla, S.; Ricci, M.; Perioli, L. Bioadhesive polymeric films based on usnic acid for burn wound treatment: Antibacterial and cytotoxicity studies. Colloids Surf. B Biointerfaces 2019, 178, 488–499. [Google Scholar] [CrossRef]
- Djekic, L.; Martinović, M.; Dobričić, V.; Čalija, B.; Medarević, Đ.; Primorac, M. Comparison of the Effect of Bioadhesive Polymers on Stability and Drug Release Kinetics of Biocompatible Hydrogels for Topical Application of Ibuprofen. J. Pharm. Sci. 2019, 108, 1326–1333. [Google Scholar] [CrossRef]
- Poonguzhali, R.; Basha, S.K.; Kumari, V.S. Nanostarch Reinforced with Chitosan/Poly (vinyl pyrrolidone) Blend for In Vitro Wound Healing Application. Polym. Plast. Technol. Eng. 2018, 57, 1400–1410. [Google Scholar] [CrossRef]
- Jose, T.; George, S.C.; Maya, M.G.; Maria, H.J.; Wilson, R.; Thomas, S. Effect of Bentonite Clay on the Mechanical, Thermal, and Pervaporation Performance of the Poly (vinyl alcohol) Nanocomposite Membranes. Ind. Eng. Chem. Res. 2014, 53, 16820–16831. [Google Scholar] [CrossRef]
- Abu-Jdayil, B. Rheology of sodium and calcium bentonite–water dispersions: Effect of electrolytes and aging time. Int. J. Miner. Process. 2011, 98, 208–213. [Google Scholar] [CrossRef]
- Silván, J.M.; Mingo, E.; Hidalgo, M.; de Pascual-Teresa, S.; Carrascosa, A.V.; Martinez-Rodriguez, A.J. Antibacterial activity of a grape seed extract and its fractions against Campylobacter spp. Food Control 2013, 29, 25–31. [Google Scholar] [CrossRef]
- Ceccarini, M.R.; Vannini, S.; Cataldi, S.; Moretti, M.; Villarini, M.; Fioretti, B.; Albi, E.; Beccari, T.; Codini, M. In Vitro Protective Effects of Lycium barbarum Berries Cultivated in Umbria (Italy) on Human Hepatocellular Carcinoma Cells. BioMed Res. Int. 2016, 2016, 7529521. [Google Scholar] [CrossRef]
- Pagano, C.; Perioli, L.; Latterini, L.; Nocchetti, M.; Ceccarini, M.R.; Marani, M.; Ramella, D.; Ricci, M. Folic acid-layered double hydroxides hybrids in skin formulations: Technological, photochemical and in vitro cytotoxicity on human keratinocytes and fibroblasts. Appl. Clay Sci. 2019, 168, 382–395. [Google Scholar] [CrossRef]
- Perioli, L.; Ambrogi, V.; Angelici, F.; Ricci, M.; Giovagnoli, S.; Capuccella, M.; Rossi, C. Development of mucoadhesive patches for buccal administration of ibuprofen. J. Control Release 2004, 99, 73–82. [Google Scholar] [CrossRef]
Sample Availability: Samples of OLE and films are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extraction Conditions: Solvent, Temperature | Recovery Yield (%) a |
|---|---|
| abs EtOH, RT b | 6.4 |
| 70% EtOH, RT b | 8.7 |
| abs EtOH, 60 °C | 7.9 |
| 70% EtOH, 60 °C | 9.7 |
| S. epidermidis | S. aureus | L. innocua | E. faecalis | |
|---|---|---|---|---|
| OLE MIC | 0.47 ± 0.00 | 0.94 ± 0.00 | 3.75 ± 0.00 | 3.75 ± 0.00 |
| OLE MBC | 0.94 ± 0.00 | 1.88 ± 0.00 | 7.50 ± 0.00 | 7.50 ± 0.00 |
| Ampicillin MIC | 0.13 ± 0.00 | 0.13 ± 0.00 | 0.50 ± 0.00 | 0.50 ± 0.00 |
| Ampicillin MBC | 0.50 ± 0.00 | 0.25 ± 0.00 | 1.00 ± 0.00 | 4.00 ± 0.00 |
| Hydrogel Film (OLE mg) | S. epidermidis | S. aureus | L. innocua | E. faecalis |
|---|---|---|---|---|
| B1 (3.64) | 23.00 ± 0.00 | 16.33 ± 0.58 | n.i. | n.i. |
| B2 (10.92) | 28.67 ± 0.58 | 20.33 ± 0.58 | 21.67 ± 0.58 | 21.00 ± 0.00 |
| B3 (18.21) | 25.67 ± 0.58 | 21.00 ± 0.00 | 21.67 ± 0.58 | 21.33 ± 0.58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, C.; Marinozzi, M.; Baiocchi, C.; Beccari, T.; Calarco, P.; Ceccarini, M.R.; Chielli, M.; Orabona, C.; Orecchini, E.; Ortenzi, R.; et al. Bioadhesive Polymeric Films Based on Red Onion Skins Extract for Wound Treatment: An Innovative and Eco-Friendly Formulation. Molecules 2020, 25, 318. https://doi.org/10.3390/molecules25020318
Pagano C, Marinozzi M, Baiocchi C, Beccari T, Calarco P, Ceccarini MR, Chielli M, Orabona C, Orecchini E, Ortenzi R, et al. Bioadhesive Polymeric Films Based on Red Onion Skins Extract for Wound Treatment: An Innovative and Eco-Friendly Formulation. Molecules. 2020; 25(2):318. https://doi.org/10.3390/molecules25020318
Chicago/Turabian StylePagano, Cinzia, Maura Marinozzi, Claudio Baiocchi, Tommaso Beccari, Paola Calarco, Maria Rachele Ceccarini, Michela Chielli, Ciriana Orabona, Elena Orecchini, Roberta Ortenzi, and et al. 2020. "Bioadhesive Polymeric Films Based on Red Onion Skins Extract for Wound Treatment: An Innovative and Eco-Friendly Formulation" Molecules 25, no. 2: 318. https://doi.org/10.3390/molecules25020318
APA StylePagano, C., Marinozzi, M., Baiocchi, C., Beccari, T., Calarco, P., Ceccarini, M. R., Chielli, M., Orabona, C., Orecchini, E., Ortenzi, R., Ricci, M., Scuota, S., Tiralti, M. C., & Perioli, L. (2020). Bioadhesive Polymeric Films Based on Red Onion Skins Extract for Wound Treatment: An Innovative and Eco-Friendly Formulation. Molecules, 25(2), 318. https://doi.org/10.3390/molecules25020318