
Evaluation of Self-Assembly Pathways to Control Crystallization-Driven Self-Assembly of a Semicrystalline P(VDF-co-HFP)-b-PEG-b-P(VDF-co-HFP) Triblock Copolymer



Abstract

{kind=link}
{kind=link}
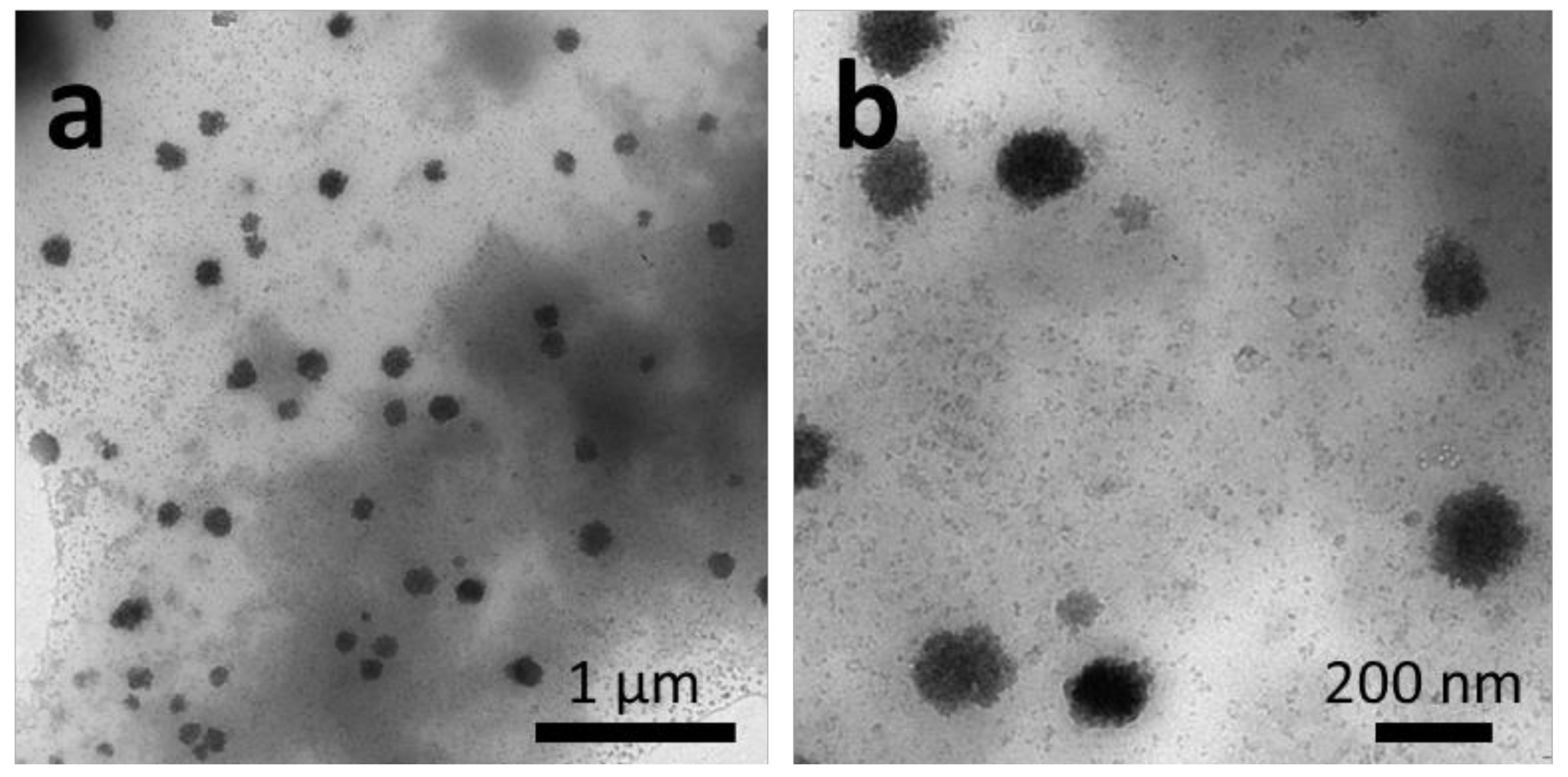{kind=link}
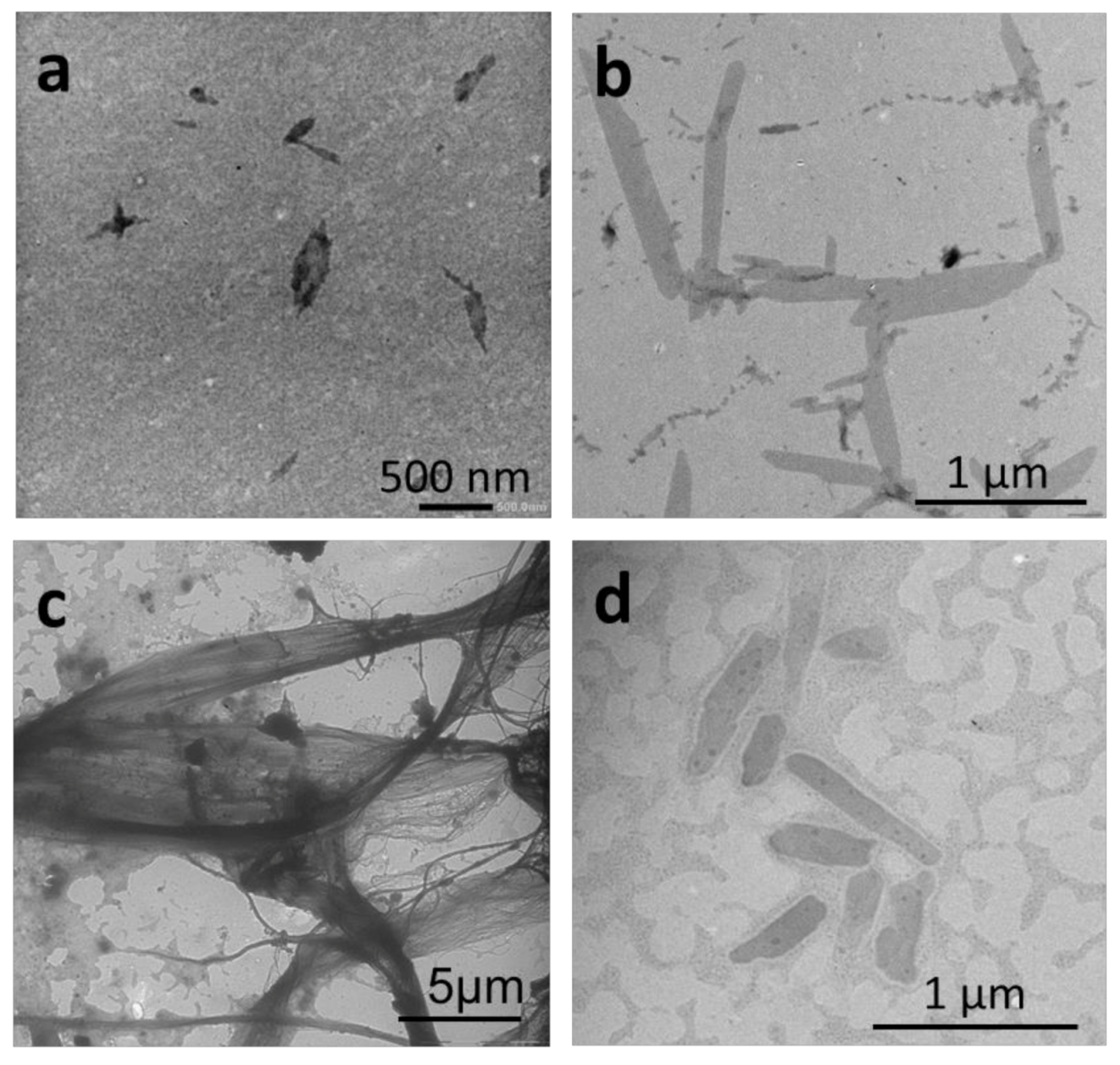{kind=link}
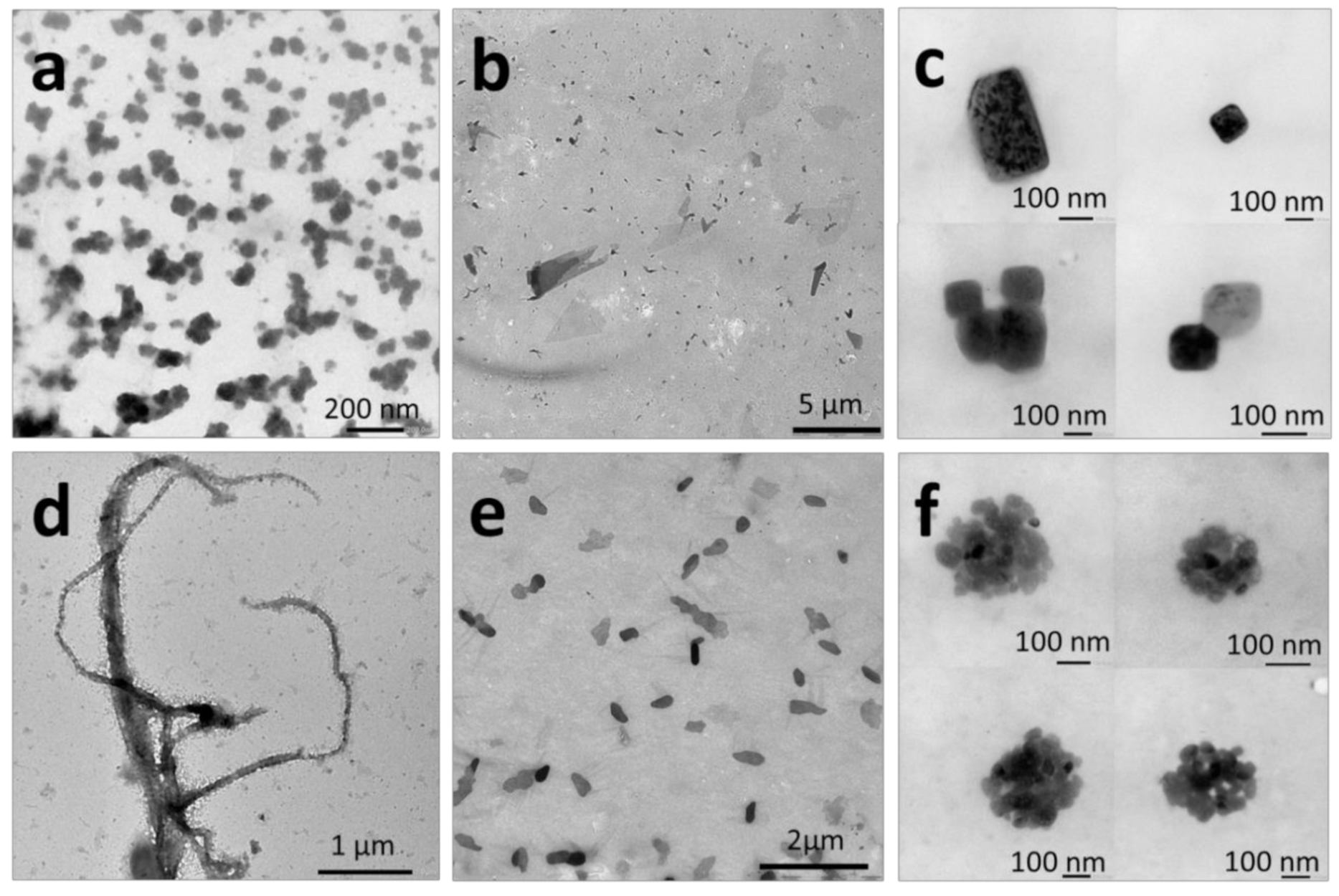{kind=link}
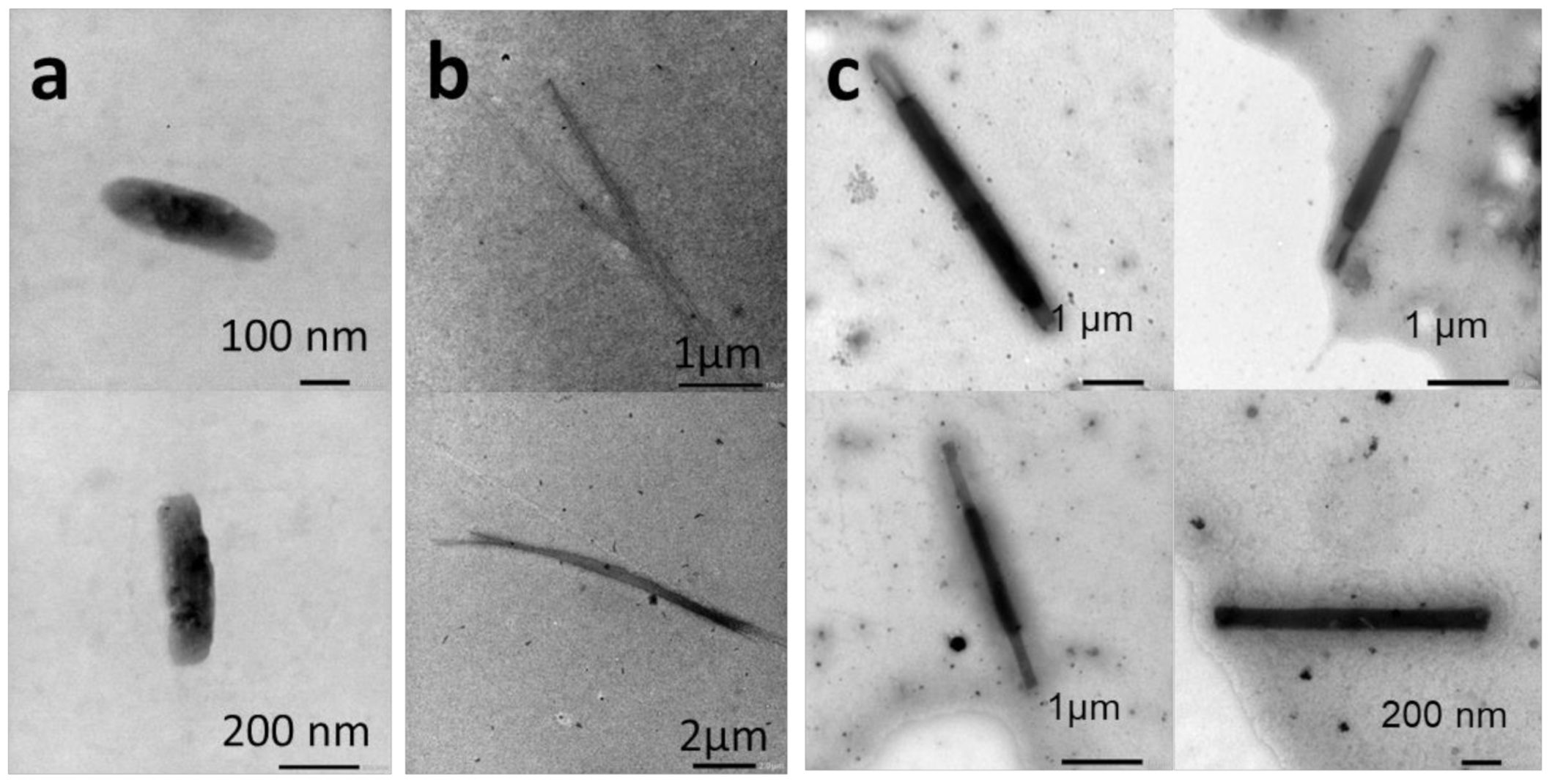{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
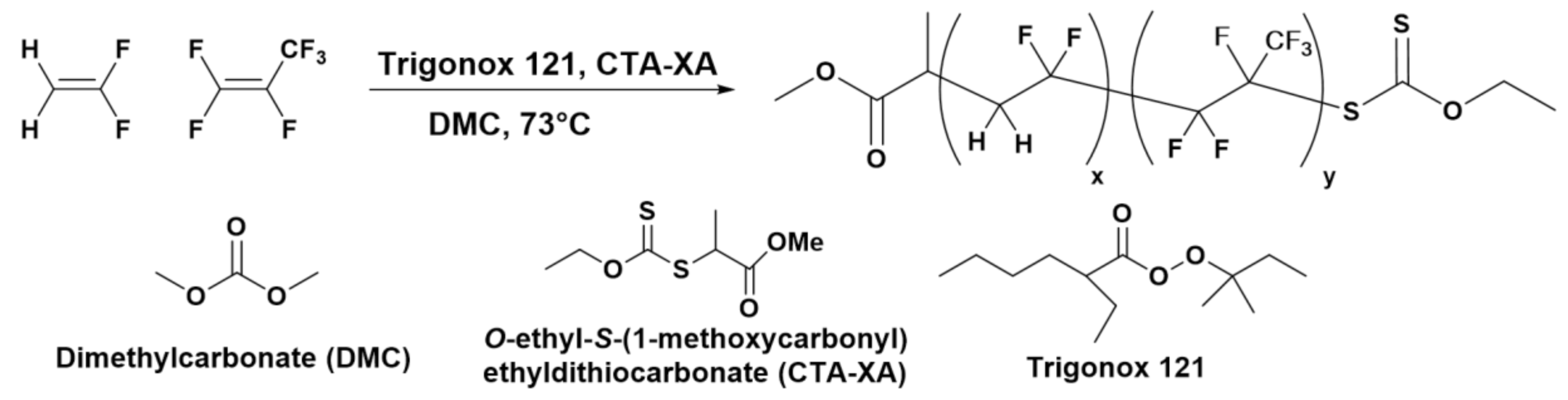
2.1. Polymers Syntheses and Characterizations
2.2. Self-Assembly
2.2.1. Thin-Film Rehydration
2.2.2. Micellization
2.2.3. Nanoprecipitation
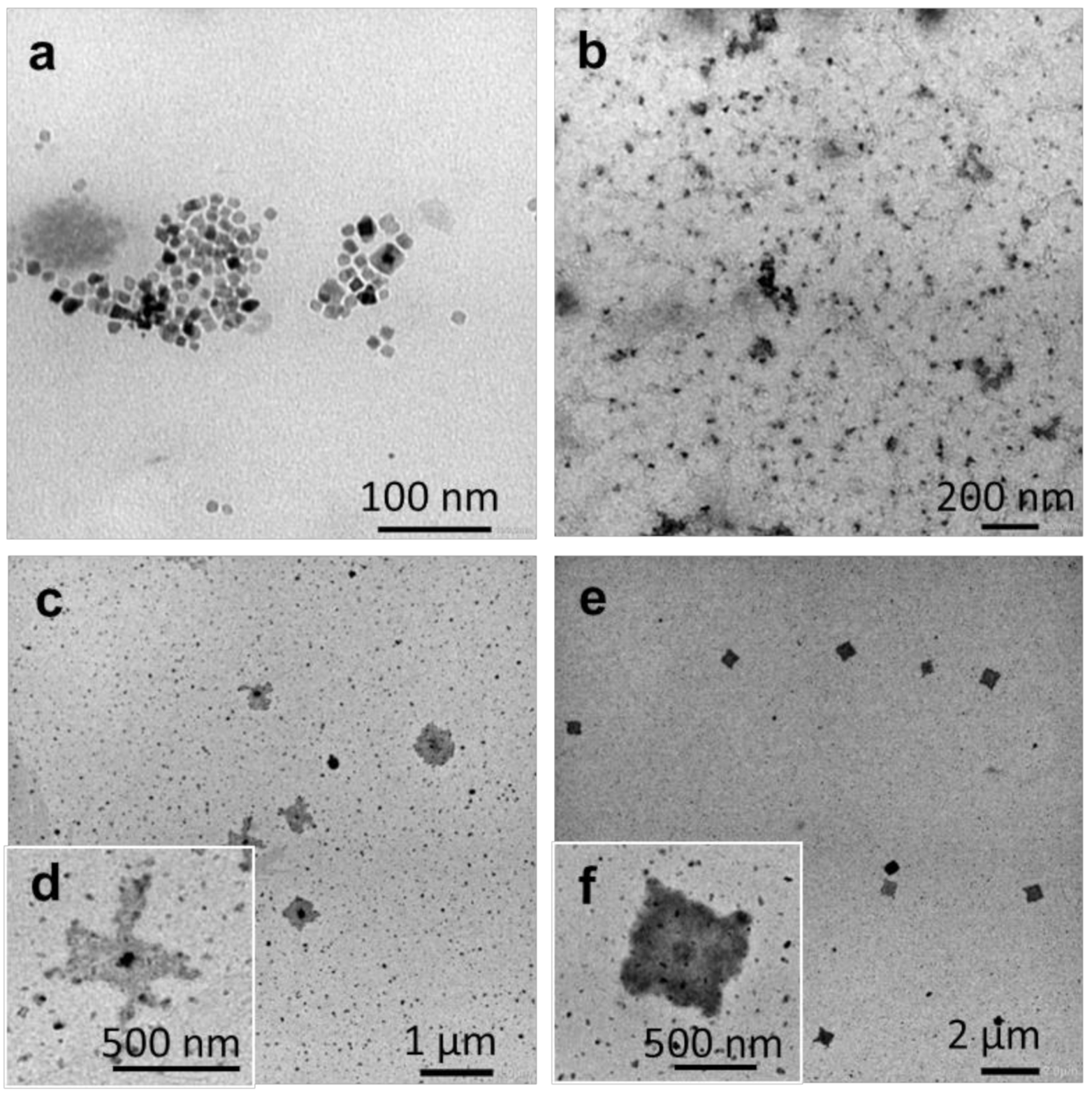
2.2.4. Temperature-Induced Crystallization-Driven Self-Assembly (TI-CDSA)
3. Materials and Methods
3.1. Materials
3.2. Nuclear Magnetic Resonance (NMR)
3.3. Size-Exclusion Chromatography (SEC)
3.4. Differential Scanning Calorimetry (DSC)
3.5. Thermogravimetric Analysis (TGA)
3.6. Transmission Electron Microscopy (TEM)
3.7. X-Ray Diffraction (XRD)
3.8. Dynamic Light Scattering (DLS)
3.9. Synthesis
3.9.1. P(VDF51-co-HFP4)-XA Synthesis in Autoclave
3.9.2. PEG136-DA Synthesis
3.9.3. P(VDF-co-HFP)-b-PEG-b-P(VDF-co-HFP) Synthesis
3.10. Self-Assembly
3.10.1. Preparation of the Block Copolymer Solutions
3.10.2. Nanoprecipitation
3.10.3. Micellization
3.10.4. Thin Film Hydration
3.10.5. Temperature-Induced Crystallization-Driven Self-Assembly (TI-CDSA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, J.K.; Yang, S.Y.; Lee, Y.; Kim, Y. Functional nanomaterials based on block copolymer self-assembly. Prog. Polym. Sci. 2010, 35, 1325–1349. [Google Scholar] [CrossRef]
- Smart, T.; Lomas, H.; Massignani, M.; Flores-merino, M.V.; Perez, L.R.; Battaglia, G. Block copolymer nanostructures One of the most important classes of synthetic systems for creating. Nanotoday 2008, 3, 38–46. [Google Scholar] [CrossRef]
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Crassous, J.J.; Schurtenberger, P.; Ballauff, M.; Mihut, A.M. Design of block copolymer micelles via crystallization. Polymer 2015, 62, A1–A13. [Google Scholar] [CrossRef]
- Lotz, B.; Kovacs, A.J.; Bassett, G.A.; Keller, A. Properties of copolymers composed of one poly-ethylene-oxide and one polystyrene block. Kolloid-Z. Z. Polym. 1966, 209, 115–128. [Google Scholar] [CrossRef]
- Arno, M.C.; Inam, M.; Coe, Z.; Cambridge, G.; Macdougall, L.J.; Keogh, R.; Dove, A.P.; O’Reilly, R.K. Precision Epitaxy for Aqueous 1D and 2D Poly(ε-caprolactone) Assemblies. J. Am. Chem. Soc. 2017, 139, 16980–16985. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Feng, C.; Cui, Y.; Yang, X.; Manners, I.; Winnik, M.A.; Huang, X. Monodisperse Fiber-like Micelles of Controlled Length and Composition with an Oligo(p-phenylenevinylene) Core via “Living” Crystallization-Driven Self-Assembly. J. Am. Chem. Soc. 2017, 139, 7136–7139. [Google Scholar] [CrossRef]
- Yu, W.; Inam, M.; Jones, J.R.; Dove, A.P.; O’Reilly, R.K. Understanding the CDSA of poly(lactide) containing triblock copolymers. Polym. Chem. 2017, 8, 5504–5512. [Google Scholar] [CrossRef]
- Finnegan, J.R.; He, X.; Street, S.T.G.; Garcia-Hernandez, J.D.; Hayward, D.W.; Harniman, R.L.; Richardson, R.M.; Whittell, G.R.; Manners, I. Extending the Scope of “living” Crystallization-Driven Self-Assembly: Well-Defined 1D Micelles and Block Comicelles from Crystallizable Polycarbonate Block Copolymers. J. Am. Chem. Soc. 2018, 140, 17127–17140. [Google Scholar] [CrossRef]
- Shi, Z.; Wei, Y.; Zhu, C.; Sun, J.; Li, Z. Crystallization-Driven Two-Dimensional Nanosheet from Hierarchical Self-Assembly of Polypeptoid-Based Diblock Copolymers. Macromolecules 2018, 51, 6344–6351. [Google Scholar] [CrossRef]
- Shin, S.; Menk, F.; Kim, Y.; Lim, J.; Char, K.; Zentel, R.; Choi, T.L. Living Light-Induced Crystallization-Driven Self-Assembly for Rapid Preparation of Semiconducting Nanofibers. J. Am. Chem. Soc. 2018, 140, 6088–6094. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Jarrett-Wilkins, C.; Rahman, M.A.; Zhu, T.; Sha, Y.; Manners, I.; Tang, C. Crystallization-Driven Self-Assembly of Metallo-Polyelectrolyte Block Copolymers with a Polycaprolactone Core-Forming Segment. ACS Macro Lett. 2019, 835–840. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Wu, L.; Yu, W.; Wilks, T.R.; Dove, A.P.; Ding, H.M.; O’Reilly, R.K.; Chen, G.; Jiang, M. Glyco-Platelets with Controlled Morphologies via Crystallization-Driven Self-Assembly and Their Shape-Dependent Interplay with Macrophages. ACS Macro Lett. 2019, 8, 596–602. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Oliver, A.M.; Pearce, S.; Harniman, R.L.; Whittell, G.R.; Liu, Y.; Du, S.; Leng, J.; Manners, I. Low length dispersity fiber-like micelles from an A–B–A triblock copolymer with terminal crystallizable poly(ferrocenyldimethylsilane) segments via living crystallization-driven self-assembly. Polym. Chem. 2019. [Google Scholar] [CrossRef]
- Guerin, G.; Rupar, P.; Molev, G.; Manners, I.; Jinnai, H.; Winnik, M.A. Lateral Growth of 1D Core-Crystalline Micelles upon Annealing in Solution. Macromolecules 2016, 49, 7004–7014. [Google Scholar] [CrossRef]
- Inam, M.; Cambridge, G.; Pitto-Barry, A.; Laker, Z.P.L.; Wilson, N.R.; Mathers, R.T.; Dove, A.P.; O’Reilly, R.K. 1D: Vs. 2D shape selectivity in the crystallization-driven self-assembly of polylactide block copolymers. Chem. Sci. 2017, 8, 4223–4230. [Google Scholar] [CrossRef]
- Qi, H.; Zhou, T.; Mei, S.; Chen, X.; Li, C.Y. Responsive Shape Change of Sub-5 nm Thin, Janus Polymer Nanoplates. ACS Macro Lett. 2016, 5, 651–655. [Google Scholar] [CrossRef]
- He, X.; He, Y.; Hsiao, M.S.; Harniman, R.L.; Pearce, S.; Winnik, M.A.; Manners, I. Complex and Hierarchical 2D Assemblies via Crystallization-Driven Self-Assembly of Poly(l-lactide) Homopolymers with Charged Termini. J. Am. Chem. Soc. 2017, 139, 9221–9228. [Google Scholar] [CrossRef]
- Qiu, H.; Gao, Y.; Boott, C.E.; Gould, O.E.C.; Harniman, R.L.; Miles, M.J.; Webb, S.E.D.; Winnik, M.A.; Manners, I. Uniform patchy and hollow rectangular platelet micelles from crystallizable polymer blends. Science 2016, 352, 697–702. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Y.; Chen, Y. Fabrication of 2D surface-functional polymer platelets via crystallization-driven self-assembly of poly(ε-caprolactone)-contained block copolymers. Polymer 2019, 160, 196–203. [Google Scholar] [CrossRef]
- Ganda, S.; Dulle, M.; Drechsler, M.; Förster, B.; Förster, S.; Stenzel, M.H. Two-Dimensional Self-Assembled Structures of Highly Ordered Bioactive Crystalline-Based Block Copolymers. Macromolecules 2017, 50, 8544–8553. [Google Scholar] [CrossRef]
- Hsiao, M.S.; Zheng, J.X.; Leng, S.; Van Horn, R.M.; Quirk, R.P.; Thomas, E.L.; Chen, H.L.; Hsiao, B.S.; Rong, L.; Lotz, B.; et al. Crystal orientation change and its origin in one-dimensional nanoconfinement constructed by polystyrene-block-poly(ethylene oxide) single crystal mats. Macromolecules 2008, 41, 8114–8123. [Google Scholar] [CrossRef]
- Yin, L.; Hillmyer, M.A. Disklike micelles in water from polyethylene-containing diblock copolymers. Macromolecules 2011, 44, 3021–3028. [Google Scholar] [CrossRef]
- Yusoff, S.F.M.; Hsiao, M.S.; Schacher, F.H.; Winnik, M.A.; Manners, I. Formation of lenticular platelet micelles via the interplay of crystallization and chain stretching: Solution self-assembly of poly(ferrocenyldimethylsilane)-Block-poly(2-vinylpyridine) with a crystallizable core-forming metalloblock. Macromolecules 2012, 45, 3883–3891. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, Y.; Yu, Q.; Manners, I.; Winnik, M.A. Monitoring Collapse of Uniform Cylindrical Brushes with a Thermoresponsive Corona in Water. ACS Macro Lett. 2018, 7, 166–171. [Google Scholar] [CrossRef]
- Barbosa, J.C.; Dias, J.P.; Lanceros-Méndez, S.; Costa, C.M. Recent advances in poly(vinylidene fluoride) and its copolymers for lithium-ion battery separators. Membranes 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Costa, C.M.; Correia, D.M.; Nunes-Pereira, J.; Oliveira, J.; Martins, P.; Gonçalves, R.; Cardoso, V.F.; Lanceros-Méndez, S. Electroactive poly(vinylidene fluoride)-based structures for advanced applications. Nat. Protoc. 2018, 13, 681–704. [Google Scholar] [CrossRef]
- Yu, Y.; Sun, H.; Orbay, H.; Chen, F.; England, C.G.; Cai, W.; Wang, X. Biocompatibility and in vivo operation of implantable mesoporous PVDF-based nanogenerators. Nano Energy 2016, 27, 275–281. [Google Scholar] [CrossRef]
- Guerre, M.; Schmidt, J.; Talmon, Y.; Améduri, B.; Ladmiral, V. An amphiphilic poly(vinylidene fluoride)-b-poly(vinyl alcohol) block copolymer: synthesis and self-assembly in water. Polym. Chem. 2017, 8, 1125–1128. [Google Scholar] [CrossRef]
- Guerre, M.; Semsarilar, M.; Godiard, F.; Améduri, B.; Ladmiral, V. Polymerization-induced self-assembly of PVAc-b-PVDF block copolymers via RAFT dispersion polymerization of vinylidene fluoride in dimethyl carbonate. Polym. Chem. 2017, 8, 1477–1487. [Google Scholar] [CrossRef]
- Guerre, M.; Semsarilar, M.; Totée, C.; Silly, G.; Améduri, B.; Ladmiral, V. Self-assembly of poly(vinylidene fluoride)-block-poly(2-(dimethylamino)ethylmethacrylate) block copolymers prepared by CuAAC click coupling. Polym. Chem. 2017, 5203–5211. [Google Scholar] [CrossRef]
- Lopez, G.; Guerre, M.; Schmidt, J.; Talmon, Y.; Ladmiral, V.; Habas, J.-P.; Améduri, B. An amphiphilic PEG-b-PFPE-b-PEG triblock copolymer: Synthesis by CuAAC click chemistry and self-assembly in water. Polym. Chem. 2016, 7, 402–409. [Google Scholar] [CrossRef]
- Guerre, M.; Semsarilar, M.; Godiard, F.; Ameduri, B.; Ladmiral, V. An amphiphilic poly(vinylidene fluoride)-b-poly (vinyl alcohol) block copolymer: Synthesis and self-assembly in water. Polym. Chem. 2017, 9, 1477–1487. [Google Scholar] [CrossRef]
- Guerre, M.; Campagne, B.; Gimello, O.; Parra, K.; Ameduri, B.; Ladmiral, V. Deeper Insight into the MADIX Polymerization of Vinylidene Fluoride. Macromolecules 2015, 48, 7810–7822. [Google Scholar] [CrossRef]
- Guerre, M.; Lopez, G.; Soulestin, T.; Totée, C.; Améduri, B.; Silly, G.; Ladmiral, V. A Journey into the Microstructure of PVDF Made by RAFT. Macromol. Chem. Phys. 2016, 217, 2275–2285. [Google Scholar] [CrossRef]
- Guerre, M.; Rahaman, S.M.W.; Améduri, B.; Poli, R.; Ladmiral, V. Limits of Vinylidene Fluoride RAFT Polymerization. Macromolecules 2016, 49, 5386–5396. [Google Scholar] [CrossRef]
- Folgado, E.; Guerre, M.; Da Costa, A.; Ferri, A.; Addad, A.; Ladmiral, V.; Semsarilar, M. “One-Pot” Aminolysis/Thia-Michael Addition preparation of well-defined amphiphilic PVDF-b-PEG-b-PVDF triblock copolymers: Self-assembly behaviour in mixed solvents. Polym. Chem. 2020, 11, 401–410. [Google Scholar] [CrossRef]
- Apostolides, D.E.; Patrickios, C.S.; Sakai, T.; Guerre, M.; Lopez, G.; Améduri, B.; Ladmiral, V.; Simon, M.; Gradzielski, M.; Clemens, D.; et al. Near-Model Amphiphilic Polymer Conetworks Based on Four-Arm Stars of Poly(vinylidene fluoride) and Poly(ethylene glycol): Synthesis and Characterization. Macromolecules 2018, 51, 2476–2488. [Google Scholar] [CrossRef]
- Guerre, M.; Uchiyama, M.; Folgado, E.; Semsarilar, M.; Améduri, B.; Satoh, K.; Kamigaito, M.; Ladmiral, V. Combination of Cationic and Radical RAFT Polymerizations: A Versatile Route to Well-Defined Poly(ethyl vinyl ether)-block-poly(vinylidene fluoride) Block Copolymers. ACS Macro Lett. 2017, 6, 393–398. [Google Scholar] [CrossRef]
- Guerre, M.; Ameduri, B.; Ladmiral, V. RAFT synthesis of well-defined PVDF-b-PVAc block copolymers. Polym. Chem. 2016, 7, 6918–6933. [Google Scholar] [CrossRef]
- Twum, E.B.; McCord, E.F.; Fox, P.A.; Lyons, D.F.; Rinaldi, P.L. Characterization of backbone structures in poly(vinylidene fluoride-co-hexafluoropropylene) copolymers by multidimensional 19F NMR spectroscopy. Macromolecules 2013, 46, 4892–4908. [Google Scholar] [CrossRef]
- Ameduri, B. From vinylidene fluoride (VDF) to the applications of VDF-Containing polymers and copolymers: Recent developments and future trends. Chem. Rev. 2009, 109, 6632–6686. [Google Scholar] [CrossRef] [PubMed]
- Folgado, E.; Ladmiral, V.; Semsarilar, M. Towards permanent hydrophilic PVDF membranes. Amphiphilic PVDF-b-PEG-b-PVDF triblock copolymer as membrane additive. Eur. Polym. J. 2020, 131, 109708. [Google Scholar] [CrossRef]
- Guerre, M.; Ameduri, B.; Ladmiral, V. One-pot synthesis of poly(vinylidene fluoride) methacrylate macromonomers via thia-Michael addition. Polym. Chem. 2016, 7, 441–450. [Google Scholar] [CrossRef]
- Mckee, J.R.; Ladmiral, V.; Niskanen, J.; Tenhu, H.; Armes, S.P. Synthesis of Sterically-Stabilized Polystyrene Latexes Using Well-Defined Thermoresponsive Poly(N-isopropylacrylamide) Macromonomers. Macromolecules 2011, 44, 7692–7703. [Google Scholar] [CrossRef]
- Lopez, G.; Guerre, M.; Améduri, B.; Habas, J.P.; Ladmiral, V. Photocrosslinked PVDF-based star polymer coatings: An all-in-one alternative to PVDF/PMMA blends for outdoor applications. Polym. Chem. 2017, 8, 3045–3049. [Google Scholar] [CrossRef]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. In Liposomes. Methods in Molecular Biology; D’Souza, G., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1522. [Google Scholar]
- Owen, S.C.; Chan, D.P.Y.; Shoichet, M.S. Polymeric micelle stability. Nano Today 2012, 7, 53–65. [Google Scholar] [CrossRef]
- Pustulka, K.M.; Wohl, A.R.; Lee, H.S.; Michel, A.R.; Han, J.; Hoye, T.R.; McCormick, A.V.; Panyam, J.; Macosko, C.W. Flash Nanoprecipitation: Particle Structure and Stability. Mol. Pharm. 2013, 10, 4367–4377. [Google Scholar] [CrossRef]
- Zinn, T.; Willner, L.; Pipich, V.; Richter, D.; Lund, R. Molecular Exchange Kinetics of Micelles: Corona Chain Length Dependence. ACS Macro Lett. 2016, 5, 884–888. [Google Scholar] [CrossRef]
- Foster, J.C.; Varlas, S.; Couturaud, B.; Coe, Z.; O’Reilly, R.K. Getting into Shape: Reflections on a New Generation of Cylindrical Nanostructures’ Self-Assembly Using Polymer Building Blocks. J. Am. Chem. Soc. 2019, 141, 2742–2753. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, J.; Tang, B.Z. Kinetic trapping—A strategy for directing the self-assembly of unique functional nanostructures. Chem. Commun. 2016, 52, 11870–11884. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Gao, Y.; Du, V.A.; Harniman, R.; Winnik, M.A.; Manners, I. Branched Micelles by Living Crystallization-Driven Block Copolymer Self-Assembly under Kinetic Control. J. Am. Chem. Soc. 2015, 137, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Chow, S.F.; Zheng, Y. Application of flash nanoprecipitation to fabricate poorly water-soluble drug nanoparticles. Acta Pharm. Sin. B 2019, 9, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Grundy, L.S.; Lee, V.E.; Li, N.; Sosa, C.; Mulhearn, W.D.; Liu, R.; Register, R.A.; Nikoubashman, A.; Prud’homme, R.K.; Panagiotopoulos, A.Z.; et al. Rapid Production of Internally Structured Colloids by Flash Nanoprecipitation of Block Copolymer Blends. ACS Nano 2018, 12, 4660–4668. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, M.; Hatton, F.L.; Slater, R.A.; Chambon, P.; North, J.; Peacock, A.K.; He, T.; McDonald, T.O.; Owen, A.; Rannard, S.P. Stable, polymer-directed and SPION-nucleated magnetic amphiphilic block copolymer nanoprecipitates with readily reversible assembly in magnetic fields. Nanoscale 2016, 8, 7224–7231. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Zou, D.; Hui, Y.; Nigam, K.; Middelberg, A.P.J.; Zhao, C.-X. Formulation of Nanoparticles Using Mixing-Induced Nanoprecipitation for Drug Delivery. Ind. Eng. Chem. Res. 2020, 59, 4134–4149. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, H.; He, H.; Ribbe, A.E.; Thayumanavan, S. Blended Assemblies of Amphiphilic Random and Block Copolymers for Tunable Encapsulation and Release of Hydrophobic Guest Molecules. Macromolecules 2020, 53, 2713–2723. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Baby, T.; Chen, D.; Weitz, D.A.; Zhao, C.-X. Stable Polymer Nanoparticles with Exceptionally High Drug Loading by Sequential Nanoprecipitation. Angew. Chem. Int. Ed. 2020, 59, 4720–4728. [Google Scholar] [CrossRef]
- Macedo, A.S.; Carvalho, E.O.; Cardoso, V.F.; Correia, D.M.; Tubio, C.R.; Fidalgo-Marijuan, A.; Botelho, G.; Lanceros-Méndez, S. Tailoring electroactive poly(vinylidene fluoride-co-trifluoroethylene) microspheres by a nanoprecipitation method. Mater. Lett. 2020, 261, 127018. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Tran, T.T.D.; Zhang, J.; Kong, L. Manufacturing techniques and surface engineering of polymer based nanoparticles for targeted drug delivery to cancer. Nanomaterials 2016, 6. [Google Scholar] [CrossRef]
- Han, L.; Wang, M.; Jia, X.; Chen, W.; Qian, H.; He, F. Uniform two-dimensional square assemblies from conjugated block copolymers driven by π-π Interactions with controllable sizes. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Coutelier, O.; Harrisson, S.; Tassaing, T.; Marty, J.; Destarac, M. Enhanced Solubility of Polyvinyl Esters in scCO 2 by Means of Vinyl Tri fl uorobutyrate Monomer. ACS Macro Lett. 2015, 4, 89–93. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folgado, E.; Mayor, M.; Ladmiral, V.; Semsarilar, M. Evaluation of Self-Assembly Pathways to Control Crystallization-Driven Self-Assembly of a Semicrystalline P(VDF-co-HFP)-b-PEG-b-P(VDF-co-HFP) Triblock Copolymer. Molecules 2020, 25, 4033. https://doi.org/10.3390/molecules25174033
Folgado E, Mayor M, Ladmiral V, Semsarilar M. Evaluation of Self-Assembly Pathways to Control Crystallization-Driven Self-Assembly of a Semicrystalline P(VDF-co-HFP)-b-PEG-b-P(VDF-co-HFP) Triblock Copolymer. Molecules. 2020; 25(17):4033. https://doi.org/10.3390/molecules25174033
Chicago/Turabian StyleFolgado, Enrique, Matthias Mayor, Vincent Ladmiral, and Mona Semsarilar. 2020. "Evaluation of Self-Assembly Pathways to Control Crystallization-Driven Self-Assembly of a Semicrystalline P(VDF-co-HFP)-b-PEG-b-P(VDF-co-HFP) Triblock Copolymer" Molecules 25, no. 17: 4033. https://doi.org/10.3390/molecules25174033
APA StyleFolgado, E., Mayor, M., Ladmiral, V., & Semsarilar, M. (2020). Evaluation of Self-Assembly Pathways to Control Crystallization-Driven Self-Assembly of a Semicrystalline P(VDF-co-HFP)-b-PEG-b-P(VDF-co-HFP) Triblock Copolymer. Molecules, 25(17), 4033. https://doi.org/10.3390/molecules25174033