
New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer’s Disease Treatment

 , , , , , ,
, , , , , ,  and
and 
Abstract

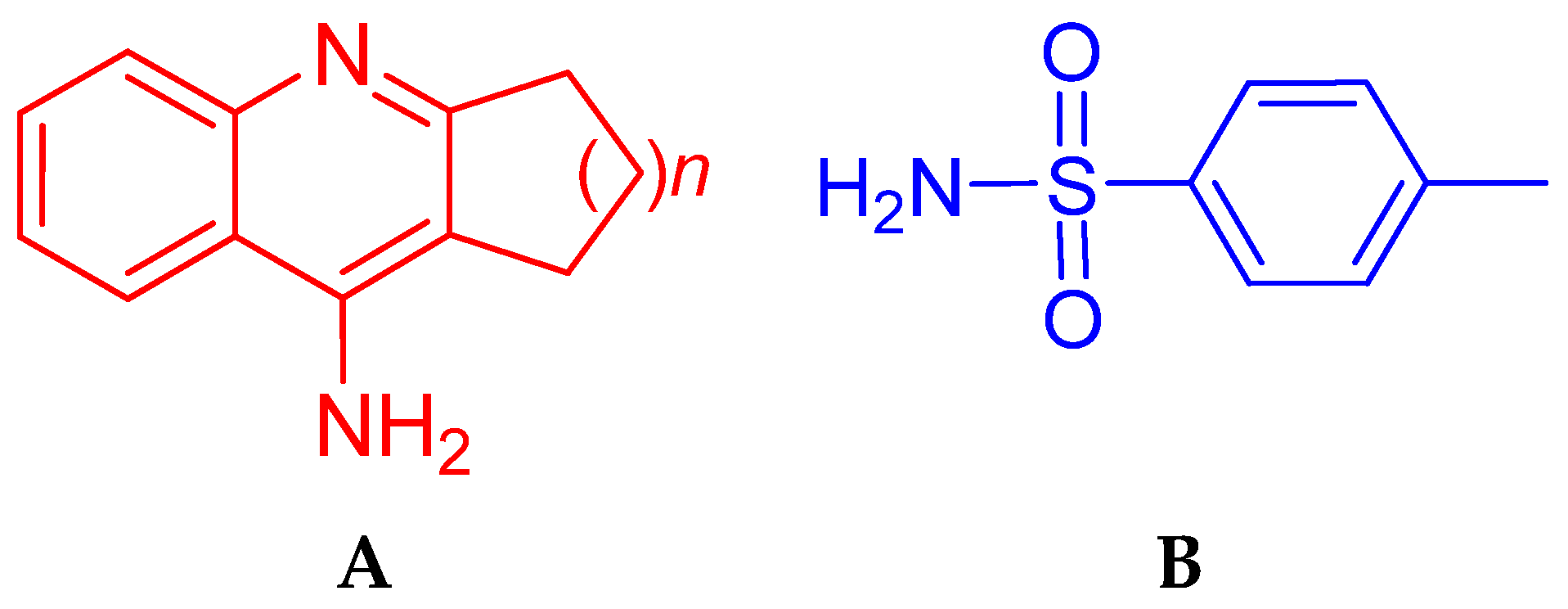
1. Introduction
2. Results and Discussion
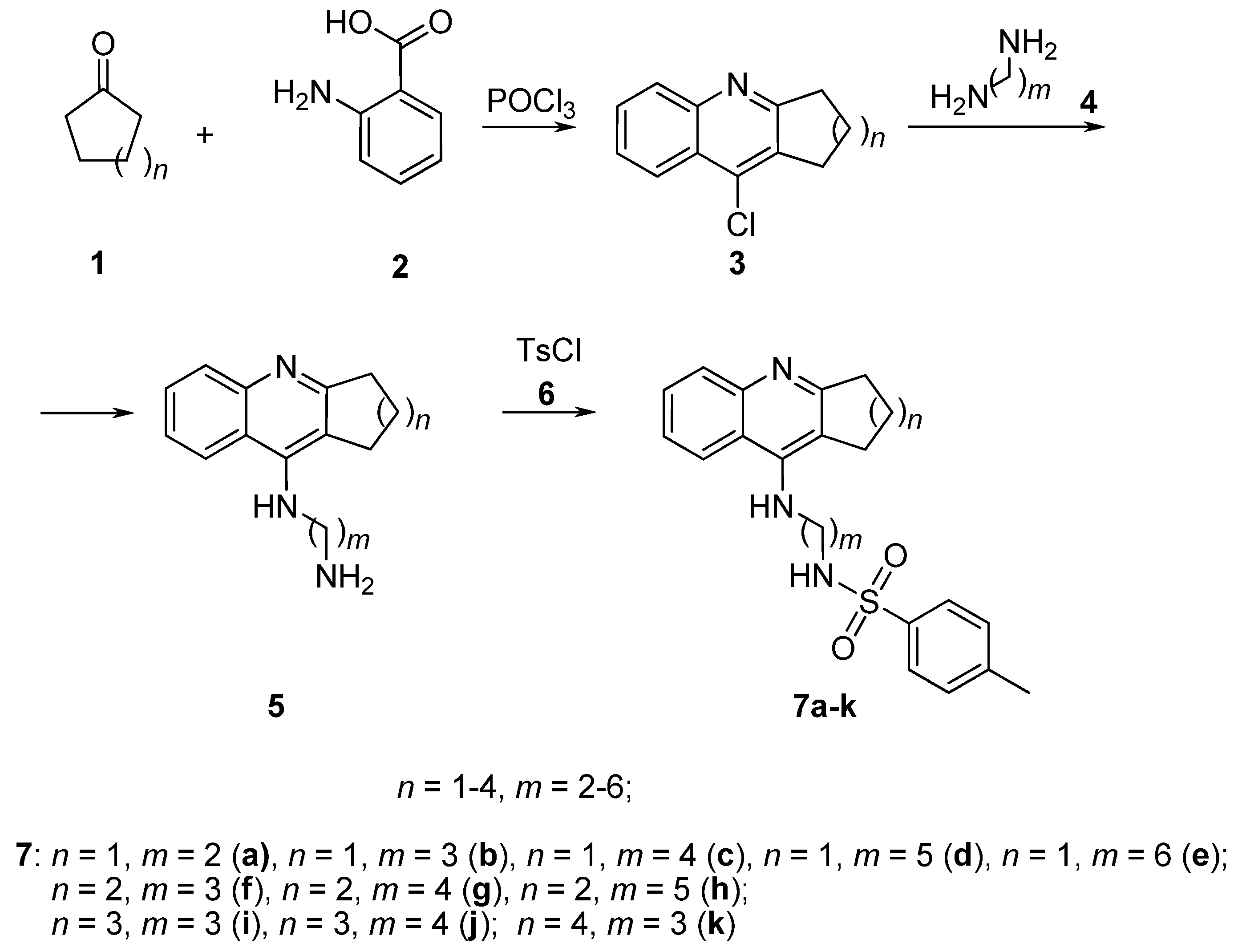
2.1. Chemistry
2.2. Inhibition Studies of AChE, BChE and CES. Structure-Activity Relationships
2.3. Kinetic Studies of AChE and BChE Inhibition
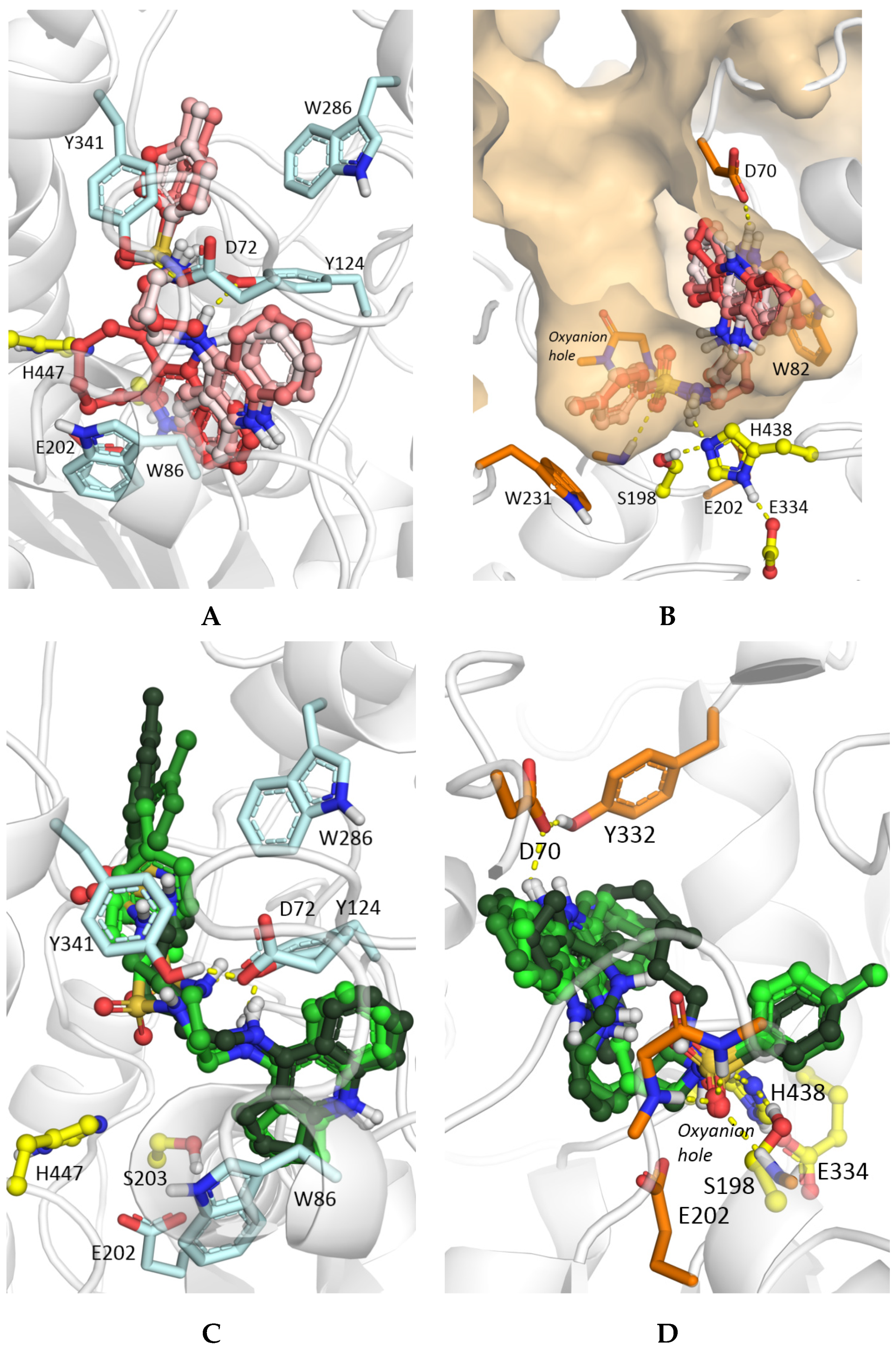
2.4. Molecular Docking Studies
2.5. Displacement of Propidium Iodide from the PAS of EeAChE
2.6. Antioxidant Activity
2.7. Predicted ADMET Profiles and PAINS Analysis
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of Compounds
General Procedure for the Preparation of Derivatives 7a–k
3.3. Biological Assays
3.3.1. Enzymatic Assays
- In vitro AChE, BChE and CES Inhibition
- Kinetic Study of AChE and BChE Inhibition. Determination of Steady-State Inhibition Constants
3.3.2. Propidium Displacement Studies
3.3.3. Antioxidant Activity: ABTS Radical Cation Scavenging Activity Assay
3.4. Molecular Modeling Studies
3.5. Prediction of ADMET Profiles and PAINS Analysis
3.6. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimer Dement. 2015, 11, 332–384. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, E.; Schelterns, P.; Feldman, H. Treatment of Alzheimer’s disease; current status and new perspectives. Lancet Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Perry, E.K.; Tomlinson, B.E.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J. 1978, 2, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Allgaier, M.; Allgaier, C. An update on drug treatment options of Alzheimer’s disease. Front. Biosci. 2014, 19, 1345–1354. [Google Scholar] [CrossRef]
- Reid, G.A.; Chilukuri, N.; Darvesh, S. Butyrylcholinesterase and the cholinergic system. Neuroscience 2013, 234, 53–68. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterases: New roles in brain function and in Alzheimer’s disease. Neurochem. Res. 2003, 28, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Castro, A. Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2006, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M.; Geula, C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann. Neurol. 1994, 36, 722–727. [Google Scholar] [CrossRef]
- Ballard, C.; Greig, N.; Guillozet-Bongaarts, A.; Enz, A.; Darvesh, S. Cholinesterases: Roles in the Brain during Health and Disease. Curr. Alzheimer. Res. 2005, 2, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [PubMed]
- Furukawa-Hibi, Y.; Alkam, T.; Nitta, A.; Matsuyama, A.; Mizoguchi, H.; Suzuki, K.; Moussaoui, S.; Yu, Q.S.; Greig, N.H.; Nagai, T.; et al. Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-beta peptide in mice. Behav. Brain Res. 2011, 225, 222–229. [Google Scholar] [CrossRef]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Kosak, U.; Brus, B.; Knez, D.; Sink, R.; Zakelj, S.; Trontelj, J.; Pislar, A.; Slenc, J.; Gobec, M.; Zivin, M.; et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [PubMed]
- Bartorelli, L.; Giraldi, C.; Saccardo, M.; Cammarata, S.; Bottini, G.; Fasanaro, A.M.; Trequattrini, A. Effects of switching from an AChE inhibitor to a dual AChE-BuChE inhibitor in patients with Alzheimer’s disease. Curr. Med. Res. Opin. 2005, 21, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.A.; Mufson, E.J.; Gomez-Ramos, P. Cholinesterases colocalize with sites of neurofibrillary degeneration in aged and Alzheimer’s brains. Acta Neuropathol. 1994, 87, 284–292. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Cash, M.K.; Reid, G.A.; Martin, E.; Mitnitski, A.; Geula, C. Butyrylcholinesterase is associated with beta-amyloid plaques in the transgenic APPSWE/PSEN1dE9 mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 2–14. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Arce, M.P.; Rodriguez-Franco, M.I.; Gonzalez-Munoz, G.C.; Perez, C.; Lopez, B.; Villarroya, M.; Lopez, M.G.; Garcia, A.G.; Conde, S. Neuroprotective and cholinergic properties of multifunctional glutamic acid derivatives for the treatment of Alzheimer’s disease. J. Med. Chem. 2009, 52, 7249–7257. [Google Scholar] [CrossRef]
- Lushchekina, S.V.; Kots, E.D.; Novichkova, D.A.; Petrov, K.A.; Masson, P. Role of Acetylcholinesterase in β-Amyloid Aggregation Studied by Accelerated Molecular Dynamics. BioNanoScience 2017, 7, 396–402. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase Accelerates Assembly of Amyloid-β-Peptides into Alzheimer’s Fibrils: Possible Role of the Peripheral Site of the Enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Muñoz-Ruiz, P.; Rubio, L.; García-Palomero, E.; Dorronsoro, I.; del Monte-Millán, M.; Valenzuela, R.; Usán, P.; de Austria, C.; Bartolini, M.; Andrisano, V.; et al. Design, Synthesis, and Biological Evaluation of Dual Binding Site Acetylcholinesterase Inhibitors: New Disease-Modifying Agents for Alzheimer’s Disease. J. Med. Chem. 2005, 48, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; Formosa, X.; Galdeano, C.; Gomez, T.; Munoz-Torrero, D.; Ramirez, L.; Viayna, E.; Gomez, E.; Isambert, N.; Lavilla, R.; et al. Tacrine-based dual binding site acetylcholinesterase inhibitors as potential disease-modifying anti-Alzheimer drug candidates. Chem. Biol. Interact. 2010, 187, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer’s disease. Curr. Alzheimer. Res. 2016, 13, 1–5. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Risacher, S.L.; Nho, K.; Kim, S.; Swaminathan, S.; Shen, L.; Foroud, T.M.; Hakonarson, H.; Huentelman, M.J.; Aisen, P.S.; et al. APOE and BCHE as modulators of cerebral amyloid deposition: A florbetapir PET genome-wide association study. Mol. Psychiatry 2014, 19, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med. Chem. 2011, 18, 4949–4975. [Google Scholar] [CrossRef]
- Oset-Gasque, M.J.; Marco-Contelles, J. Alzheimer’s Disease, the “One-Molecule, One-Target” Paradigm, and the Multitarget Directed Ligand Approach. ACS Chem. Neurosci. 2018, 9, 401–403. [Google Scholar] [CrossRef]
- De Freitas Silva, M.; Dias, K.S.T.; Gontijo, V.S.; Ortiz, C.J.C.; Viegas, C., Jr. Multi-Target Directed Drugs as a Modern Approach for Drug Design Towards Alzheimer’s Disease: An Update. Curr. Med. Chem. 2018, 25, 3491–3525. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Kumar, A.; Panda, G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef] [PubMed]
- Mesiti, F.; Chavarria, D.; Gaspar, A.; Alcaro, S.; Borges, F. The chemistry toolbox of multitarget-directed ligands for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 181, 111572. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Soriano, E.; Marco-Contelles, J.; Andrisano, V.; Monti, B.; Windisch, M.; Hutter-Paier, B.; McClymont, D.W.; et al. The bivalent ligand approach as a tool for improving the in vitro anti-Alzheimer multitarget profile of dimebon. ChemMedChem 2013, 8, 1276–1281. [Google Scholar] [CrossRef]
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Shafiee, A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 128, 332–345. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Perez, C.; Monjas, L.; Rademann, J.; Rodriguez-Franco, M.I. New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with cholinergic, antioxidant, and beta-amyloid-reducing properties. J. Med. Chem. 2012, 55, 1303–1317. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Rudakova, E.V.; Kovaleva, N.V.; Lushchekina, S.V.; Boltneva, N.P.; Proshin, A.N.; Shchegolkov, E.V.; Burgart, Y.V.; Saloutin, V.I. Cholinesterase and carboxylesterase inhibitors as pharmacological agents. Russ. Chem. Bull. 2019, 68, 967–984. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Radchenko, E.V.; et al. Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer’s disease treatment: Synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg. Chem. 2020, 94, 103387. [Google Scholar] [CrossRef]
- Minarini, A.; Milelli, A.; Simoni, E.; Rosini, M.; Bolognesi, M.L.; Marchetti, C.; Tumiatti, V. Multifunctional tacrine derivatives in Alzheimer’s disease. Curr. Top. Med. Chem. 2013, 13, 1771–1786. [Google Scholar] [CrossRef] [PubMed]
- Girek, M.; Szymański, P. Tacrine hybrids as multi-target-directed ligands in Alzheimer’s disease: Influence of chemical structures on biological activities. Chem. Pap. Chem. Zvesti 2018, 73, 269–289. [Google Scholar] [CrossRef]
- Milelli, A.; De Simone, A.; Ticchi, N.; Chen, H.H.; Betari, N.; Andrisano, V.; Tumiatti, V. Tacrine-based multifunctional agents in Alzheimer’s disease: An old story in continuous development section sign. Curr. Med. Chem. 2017, 24, 3522–3546. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Nepovimova, E.; Dolezal, R.; Mezeiova, E.; Soukup, O.; Kuca, K. Multitarget tacrine hybrids with neuroprotective properties to confront Alzheimer’s disease. Curr. Top. Med. Chem. 2017, 17, 1006–1026. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Dai, Y.C.; Li, N.G.; Dong, Z.X.; Gu, T.; Shi, Z.H.; Xue, X.; Tang, Y.P.; Duan, J.A. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2017, 32, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Przybylowska, M.; Kowalski, S.; Dzierzbicka, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multifunctional Tacrine Analogues. Curr. Neuropharmacol. 2019, 17, 472–490. [Google Scholar] [CrossRef]
- AlFadly, E.D.; Elzahhar, P.A.; Tramarin, A.; Elkazaz, S.; Shaltout, H.; Abu-Serie, M.M.; Janockova, J.; Soukup, O.; Ghareeb, D.A.; El-Yazbi, A.F.; et al. Tackling neuroinflammation and cholinergic deficit in Alzheimer’s disease: Multi-target inhibitors of cholinesterases, cyclooxygenase-2 and 15-lipoxygenase. Eur. J. Med. Chem. 2019, 167, 161–186. [Google Scholar] [CrossRef]
- Supuran, C.T. Special Issue: Sulfonamides. Molecules 2017, 22, 1642. [Google Scholar] [CrossRef]
- Apaydin, S.; Torok, M. Sulfonamide derivatives as multi-target agents for complex diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [Google Scholar] [CrossRef]
- Bonardi, A.; Nocentini, A.; Bua, S.; Combs, J.; Lomelino, C.; Andring, J.; Lucarini, L.; Sgambellone, S.; Masini, E.; McKenna, R.; et al. Sulfonamide Inhibitors of Human Carbonic Anhydrases Designed through a Three-Tails Approach: Improving Ligand/Isoform Matching and Selectivity of Action. J. Med. Chem. 2020, 63, 7422–7444. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 2019, 14, 1175–1197. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 28, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Bag, S.; Tulsan, R.; Sood, A.; Cho, H.; Redjeb, H.; Zhou, W.; LeVine, H., III; Torok, B.; Torok, M. Sulfonamides as multifunctional agents for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015, 25, 626–630. [Google Scholar] [CrossRef]
- Soyer, Z.; Uysal, S.; Parlar, S.; Tarikogullari Dogan, A.H.; Alptuzun, V. Synthesis and molecular docking studies of some 4-phthalimidobenzenesulfonamide derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Akocak, S.; Boga, M.; Lolak, N.; Tuneg, M.; Sanku, R.K.K. Design, synthesis and biological evaluation of 1,3-diaryltriazene-substituted sulfonamides as antioxidant, acetylcholinesterase and butyrylcholinesterase inhibitors. J. Turk. Chem. Soc. Sect. Chem. 2019. [Google Scholar] [CrossRef]
- Taslimi, P.; Sujayev, A.; Mamedova, S.; Kalin, P.; Gulcin, I.; Sadeghian, N.; Beydemir, S.; Kufrevioglu, O.I.; Alwasel, S.H.; Farzaliyev, V.; et al. Synthesis and bioactivity of several new hetaryl sulfonamides. J. Enzym. Inhib. Med. Chem. 2017, 32, 137–145. [Google Scholar] [CrossRef]
- Kosak, U.; Knez, D.; Coquelle, N.; Brus, B.; Pislar, A.; Nachon, F.; Brazzolotto, X.; Kos, J.; Colletier, J.P.; Gobec, S. N-Propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: Potential multifunctional anti-Alzheimer’s agents. Bioorg. Med. Chem. 2017, 25, 633–645. [Google Scholar] [CrossRef]
- Gouda, M.A.; Hussein, B.H.M. Synthesis and Anti-Oxidant Evaluation of Some Novel Sulfa Drugs. Lett. Drug Des. Discov. 2017, 14, 1425–1432. [Google Scholar] [CrossRef]
- Ulus, R.; Zengin Kurt, B.; Gazioglu, I.; Kaya, M. Microwave assisted synthesis of novel hybrid tacrine-sulfonamide derivatives and investigation of their antioxidant and anticholinesterase activities. Bioorg. Chem. 2017, 70, 245–255. [Google Scholar] [CrossRef]
- Swetha, R.; Kumar, D.; Gupta, S.K.; Ganeshpurkar, A.; Singh, R.; Gutti, G.; Kumar, D.; Jana, S.; Krishnamurthy, S.; Singh, S.K. Multifunctional hybrid sulfonamides as novel therapeutic agents for Alzheimer’s disease. Future Med. Chem. 2019, 11, 3161–3178. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Lushchekina, S.V.; Rudakova, E.V.; Boltneva, N.P.; Proshin, A.N.; Lednev, B.V.; Serkov, I.V.; Bachurin, S.O. Conjugates of Tacrine and Its Cyclic Homologues with p-Toluenesulfonamide as Novel Acetylcholinesterase and Butyrylcholinesterase Inhibitors. Dokl. Biochem. Biophys. 2018, 483, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, L.Z.; Zhao, H.T.; Huang, S.L.; Zhong, S.M.; Qin, J.K.; Chen, Z.F.; Huang, Z.S.; Liang, H. Hybrids of oxoisoaporphine-tacrine congeners: Novel acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation inhibitors. Eur. J. Med. Chem. 2011, 46, 4970–4979. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Sokolov, V.B.; Grigoriev, V.V.; Serebryakova, O.G.; Vikhareva, E.A.; Aksinenko, A.Y.; Barreto, G.E.; Aliev, G.; et al. Conjugates of g-carbolines and phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer disease. Sci. Rep. 2015, 5, 13164. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Rudakova, E.V.; Serebryakova, O.G.; Aksinenko, A.Y.; Lushchekina, S.V.; Bachurin, S.O.; Richardson, R.J. Esterase profiles of organophosphorus compounds in vitro predict their behavior in vivo. Chem. Biol. Interact. 2016, 259, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Boltneva, N.P.; Lushchekina, S.V.; Serebryakova, O.G.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Bachurin, S.O.; Richardson, R.J. Synthesis, molecular docking and biological evaluation of N,N-disubstituted 2-aminothiazolines as a new class of butyrylcholinesterase and carboxylesterase inhibitors. Bioorg. Med. Chem. 2016, 24, 1050–1062. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Radchenko, E.V.; Palyulin, V.A.; Rudakova, E.V.; Aksinenko, A.Y.; Sokolov, V.B.; Zefirov, N.S.; Richardson, R.J. Organophosphorus compound esterase profiles as predictors of therapeutic and toxic effects. Chem. Biol. Interact. 2013, 203, 231–237. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Radchenko, E.V.; Baskin, I.I.; Palyulin, V.A.; Richardson, R.J.; Zefirov, N.S. Combined QSAR studies of inhibitor properties of O-phosphorylated oximes toward serine esterases involved in neurotoxicity, drug metabolism and Alzheimer’s disease. SAR QSAR Environ. Res. 2012, 23, 627–647. [Google Scholar] [CrossRef]
- Pang, Y.P.; Quiram, P.; Jelacic, T.; Hong, F.; Brimijoin, S. Highly potent, selective, and low cost bis-tetrahydroaminacrine inhibitors of acetylcholinesterase. Steps toward novel drugs for treating Alzheimer’s disease. J. Biol. Chem. 1996, 271, 23646–23649. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Peng, S.; Liao, H.; Zhang, Y.; Lehmann, J. Tacrine-flurbiprofen hybrids as multifunctional drug candidates for the treatment of Alzheimer’s disease. Arch. Pharm. (Weinh.) 2013, 346, 865–871. [Google Scholar] [CrossRef]
- Czarnecka, K.; Szymanski, P.; Girek, M.; Mikiciuk-Olasik, E.; Skibinski, R.; Kabzinski, J.; Majsterek, I.; Malawska, B.; Jonczyk, J.; Bajda, M. Tetrahydroacridine derivatives with fluorobenzoic acid moiety as multifunctional agents for Alzheimer’s disease treatment. Bioorg. Chem. 2017, 72, 315–322. [Google Scholar] [CrossRef]
- Korabecny, J.; Andrs, M.; Nepovimova, E.; Dolezal, R.; Babkova, K.; Horova, A.; Malinak, D.; Mezeiova, E.; Gorecki, L.; Sepsova, V.; et al. 7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment. Molecules 2015, 20, 22084–22101. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Horova, A.; Musilek, K.; Nepovimova, E.; Drtinova, L.; Gazova, Z.; Siposova, K.; Dolezal, R.; Jun, D.; et al. Design, synthesis and in vitro testing of 7-methoxytacrine-amantadine analogues: A novel cholinesterase inhibitors for the treatment of Alzheimer’s disease. Med. Chem. Res. 2015, 24, 2645–2655. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Shevtsova, E.F.; Boltneva, N.P.; Lushchekina, S.V.; Kovaleva, N.V.; Rudakova, E.V.; Bachurin, S.O.; Richardson, R.J. Overview of novel multifunctional agents based on conjugates of gamma-carbolines, carbazoles, tetrahydrocarbazoles, phenothiazines, and aminoadamantanes for treatment of Alzheimer’s disease. Chem. Biol. Interact. 2019, 308, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Shevtsova, E.F.; Aksinenko, A.Y.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Pushkareva, E.A.; Serkova, T.P.; Dubova, L.G.; et al. Bis-γ-carbolines as new potential multitarget agents for Alzheimer’s disease. Pure Appl. Chem. 2020. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Saxena, A.; Redman, A.M.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in active site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Biochemistry 1997, 36, 14642–14651. [Google Scholar] [CrossRef]
- Wlodek, S.T.; Clark, T.W.; Scott, L.R.; McCammon, J.A. Molecular Dynamics of Acetylcholinesterase Dimer Complexed with Tacrine. J. Am. Chem. Soc. 1997, 119, 9513–9522. [Google Scholar] [CrossRef]
- Masson, P.; Lushchekina, S.V. Slow-binding inhibition of cholinesterases, pharmacological and toxicological relevance. Arch. Biochem. Biophys. 2016, 593, 60–68. [Google Scholar] [CrossRef]
- Masson, P.; Froment, M.-T.; Fortier, P.-L.; Visicchio, J.-E.; Bartels, C.F.; Lockridge, O. Butyrylcholinesterase-catalysed hydrolysis of aspirin, a negatively charged ester, and aspirin-related neutral esters. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1998, 1387, 41–52. [Google Scholar] [CrossRef]
- Kharlamova, A.D.; Lushchekina, S.V.; Petrov, K.A.; Kots, E.D.; Nachon, F.; Villard-Wandhammer, M.; Zueva, I.V.; Krejci, E.; Reznik, V.S.; Zobov, V.V.; et al. Slow-binding inhibition of acetylcholinesterase by an alkylammonium derivative of 6-methyluracil: Mechanism and possible advantages for myasthenia gravis treatment. Biochem. J. 2016, 473, 1225–1236. [Google Scholar] [CrossRef]
- Sawatzky, E.; Wehle, S.; Kling, B.; Wendrich, J.; Bringmann, G.; Sotriffer, C.A.; Heilmann, J.; Decker, M. Discovery of Highly Selective and Nanomolar Carbamate-Based Butyrylcholinesterase Inhibitors by Rational Investigation into Their Inhibition Mode. J. Med. Chem. 2016, 59, 2067–2082. [Google Scholar] [CrossRef] [PubMed]
- Lushchekina, S.V.; Makhaeva, G.F.; Novichkova, D.A.; Zueva, I.V.; Kovaleva, N.V.; Richardson, R.J. Supercomputer modeling of dual-site acetylcholinesterase (AChE) inhibition. Supercomput. Front. Innov. 2018, 5, 89–97. [Google Scholar] [CrossRef]
- Taylor, P.; Lappi, S. Interaction of fluorescence probes with acetylcholinesterase. Site and specificity of propidium binding. Biochemistry 1975, 14, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Sterri, S.H.; Johnsen, B.A.; Fonnum, F. A radiochemical assay method for carboxylesterase, and comparison of enzyme activity towards the substrates methyl [1-14C] butyrate and 4-nitrophenyl butyrate. Biochem. Pharmacol. 1985, 34, 2779–2785. [Google Scholar] [CrossRef]
- Taylor, P.; Lwebuga-Mukasa, J.; Lappi, S.; Rademacher, J. Propidium—A fluorescence probe for a peripheral anionic site on acetylcholinesterase. Mol. Pharmacol. 1974, 10, 703–708. [Google Scholar]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef]
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: Kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Löwdin, P.-O. On the nonorthogonality problem. In Advances in Quantum Chemistry; Per-Olov, L., Ed.; Academic Press: New York, NY, USA, 1970; Volume 5, pp. 185–199. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A.; Zefirov, N.S. Prediction of human intestinal absorption of drug compounds. Russ. Chem. Bull. 2016, 65, 576–580. [Google Scholar] [CrossRef]
- Dyabina, A.S.; Radchenko, E.V.; Palyulin, V.A.; Zefirov, N.S. Prediction of blood-brain barrier permeability of organic compounds. Dokl. Biochem. Biophys. 2016, 470, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, E.V.; Rulev, Y.A.; Safanyaev, A.Y.; Palyulin, V.A.; Zefirov, N.S. Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components. Dokl. Biochem. Biophys. 2017, 473, 128–131. [Google Scholar] [CrossRef] [PubMed]
- ADMET Prediction Service. Available online: http://qsar.chem.msu.ru/admet/ (accessed on 1 April 2020).
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided. Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics Software. Available online: http://www.rdkit.org (accessed on 1 December 2019).
- Voevodin, V.; Antonov, A.; Nikitenko, D.; Shvets, P.; Sobolev, S.; Sidorov, I.; Stefanov, K.; Voevodin, V.; Zhumatiy, S. Supercomputer Lomonosov-2: Large scale, deep monitoring and fine analytics for the user community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibitory Activity Against AChE, BChE and CES and Inhibitor Selectivity | Displacement of Propidium from the EeAChE PAS, (%) 1 | |||||
|---|---|---|---|---|---|---|---|
| N | n | m | Human Erythrocyte AChE, IC50 (µM) | Equine Serum BChE, IC50 (µM) | Porcine Liver CES, (%)1 | Selectivity, S = IC50 AChE/IC50 BChE | |
| 7a | 1 | 2 | 9.03 ± 0.64 | 0.924 ± 0.031 | 11.9 ± 1.5 | 9.8 | 9.2 ± 1.0 |
| 7b | 1 | 3 | 7.76 ± 0.61 | 0.327 ± 0.004 | 8.1 ± 0.6 | 23.7 | 12.3 ± 1.1 |
| 7c | 1 | 4 | 2.08 ± 0.08 | 0.578 ± 0.025 | 9.0 ± 0.1 | 3.6 | 14.9 ± 1.2 |
| 7d | 1 | 5 | 1.97 ± 0.05 | 0.459 ± 0.044 | 29.6 ± 1.2 | 4.3 | 15.1 ± 1.4 |
| 7e | 1 | 6 | 4.00 ± 0.09 | 0.209 ± 0.008 | 18.6 ± 0.2 | 1.9 | 15.9 ± 1.3 |
| 7f | 2 | 3 | 1.88 ± 0.03 | 0.110 ± 0.005 | 2.0 ± 0.8 | 17.1 | 9.8 ± 0.8 |
| 7g | 2 | 4 | 0.668 ± 0.17 | 0.0617 ± 0.0003 | 17.1 ± 1.4 | 10.8 | 14.8 ± 1.3 |
| 7h | 2 | 5 | 0.131 ± 0.01 | 0.0680 ± 0.0014 | 6.7 ± 0.8 | 1.9 | 17.5 ± 1.5 |
| 7i | 3 | 3 | 2.76 ± 0.04 | 0.0431 ± 0.0011 | 1.9 ± 0.9 | 64.0 | 13.9 ± 1.3 |
| 7j | 3 | 4 | 1.16 ± 0.03 | 0.0788 ± 0.006 | 10.3 ± 0.8 | 14.7 | 16.1 ± 1.4 |
| 7k | 4 | 3 | 11.1 ± 0.2 | 0.461 ± 0.007 | 5.2 ± 1.5 | 24.1 | 15.9 ± 1.7 |
| Tacrine | 0.601 ± 0.047 | 0.0295 ± 0.0020 | n.a. | 20.4 | 4.4 ± 0.6 | ||
| Donepezil | 0.040 ± 0.004 | 19.2 ± 3.0 | n.a. | 0.002 | 10.1 ± 0.6 | ||
| BNPP | n.a. | n.a. | 1.80 ± 0.11 | n.d. | n.d. | ||
| Compound | LogBB | HIA% | hERG, pKi | hERG, pIC50 | LogPow | pS | QED | ||
|---|---|---|---|---|---|---|---|---|---|
| N | n | m | |||||||
| 7a | 1 | 2 | −0.94 | 97 | 4.67 | 5.54 | 2.89 | 3.60 | 0.64 |
| 7b | 1 | 3 | −0.86 | 100 | 4.88 | 5.76 | 3.21 | 3.73 | 0.60 |
| 7c | 1 | 4 | −0.78 | 100 | 4.96 | 6.14 | 3.62 | 4.02 | 0.55 |
| 7d | 1 | 5 | −0.70 | 100 | 5.25 | 6.18 | 4.04 | 4.34 | 0.49 |
| 7e | 1 | 6 | −0.63 | 100 | 5.03 | 6.35 | 4.47 | 4.68 | 0.44 |
| 7f | 2 | 3 | −0.78 | 100 | 4.92 | 5.64 | 3.71 | 4.09 | 0.57 |
| 7g | 2 | 4 | −0.70 | 100 | 5.25 | 6.02 | 4.09 | 4.38 | 0.52 |
| 7h | 2 | 5 | −0.63 | 100 | 5.30 | 6.37 | 4.49 | 4.70 | 0.46 |
| 7i | 3 | 3 | −0.70 | 100 | 4.93 | 5.75 | 4.11 | 4.40 | 0.43 |
| 7j | 3 | 4 | −0.63 | 100 | 5.01 | 6.46 | 4.48 | 4.69 | 0.38 |
| 7k | 4 | 3 | −0.63 | 100 | 5.04 | 5.91 | 4.55 | 4.75 | 0.51 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Rudakova, E.V.; Proshin, A.N.; Serkov, I.V.; Radchenko, E.V.; Palyulin, V.A.; et al. New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer’s Disease Treatment. Molecules 2020, 25, 3915. https://doi.org/10.3390/molecules25173915
Makhaeva GF, Kovaleva NV, Boltneva NP, Lushchekina SV, Astakhova TY, Rudakova EV, Proshin AN, Serkov IV, Radchenko EV, Palyulin VA, et al. New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer’s Disease Treatment. Molecules. 2020; 25(17):3915. https://doi.org/10.3390/molecules25173915
Chicago/Turabian StyleMakhaeva, Galina F., Nadezhda V. Kovaleva, Natalia P. Boltneva, Sofya V. Lushchekina, Tatiana Yu. Astakhova, Elena V. Rudakova, Alexey N. Proshin, Igor V. Serkov, Eugene V. Radchenko, Vladimir A. Palyulin, and et al. 2020. "New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer’s Disease Treatment" Molecules 25, no. 17: 3915. https://doi.org/10.3390/molecules25173915
APA StyleMakhaeva, G. F., Kovaleva, N. V., Boltneva, N. P., Lushchekina, S. V., Astakhova, T. Y., Rudakova, E. V., Proshin, A. N., Serkov, I. V., Radchenko, E. V., Palyulin, V. A., Bachurin, S. O., & Richardson, R. J. (2020). New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer’s Disease Treatment. Molecules, 25(17), 3915. https://doi.org/10.3390/molecules25173915