
Fatty Acid and Stable Carbon Isotope Composition of Slovenian Milk: Year, Season, and Regional Variability


 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Milk Fatty Acids Composition
2.1.1. Influence of the Year and Season of Production and Impact of Feed
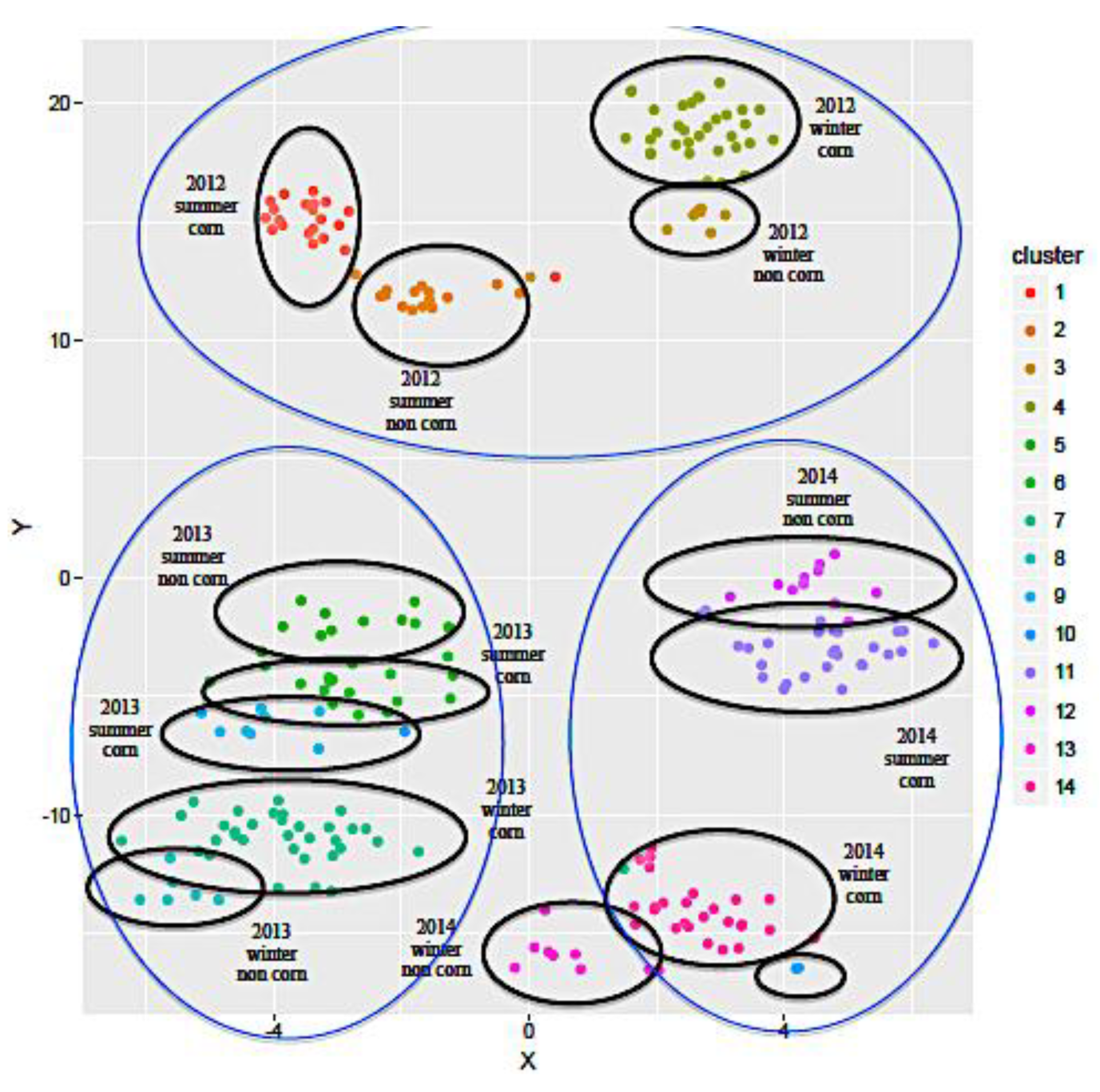
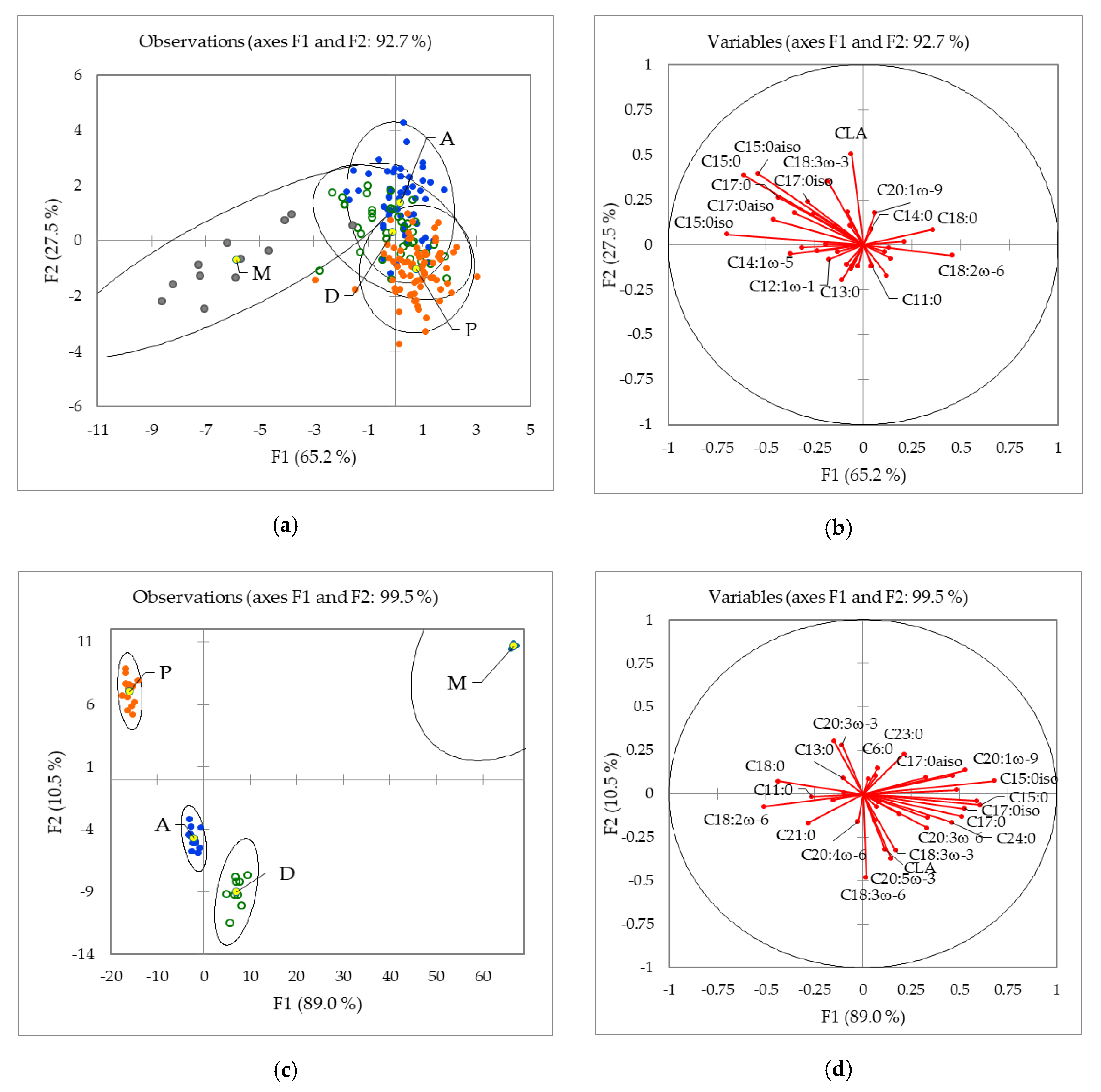
2.1.2. Influence of the Geographical Origin on FA Composition
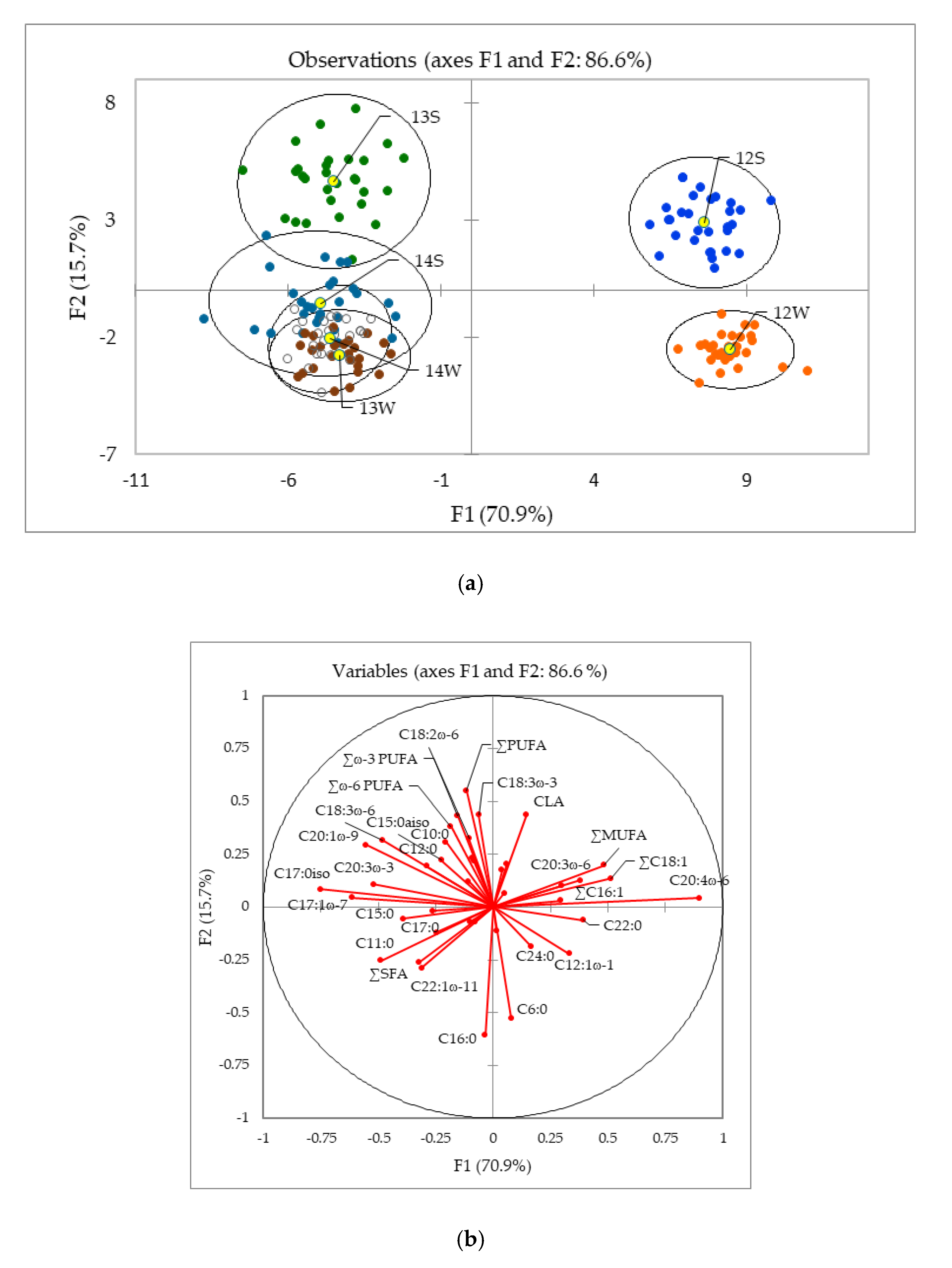
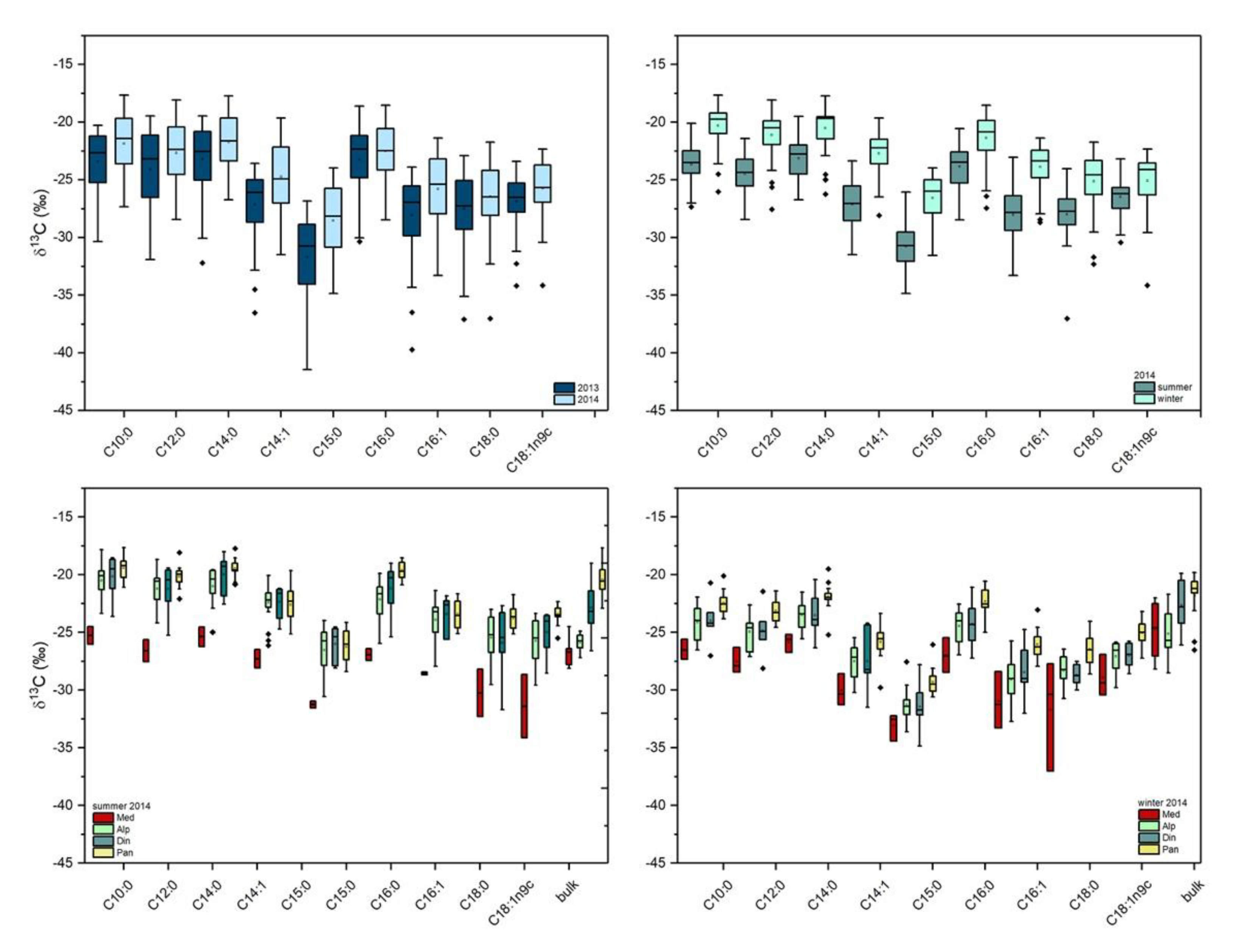
2.2. Stable Carbon Isotope Composition of FA in Milk
2.3. Quality of Milk
3. Materials and Methods
3.1. Milk Collection and Storage
3.2. Chemicals and Standards
3.3. Analytical Procedures
3.3.1. Extraction and Esterification of Milk FAs
3.3.2. Gas Chromatography of Fatty Acids
3.3.3. Elemental Analysis-Isotope Ratio Mass Spectrometry (EA-IRMS)
3.3.4. Compound Specific Isotope Analysis of δ13C in Fatty Acids
3.4. Statistical Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ehtesham, E.; Baisden, W.T.; Keller, E.D.; Hayman, A.R.; Van Hale, R.; Frew, R.D. Correlation between precipitation and geographical location of the δ2H values of the fatty acids in milk and bulk milk powder. Geochim. Cosmochim Acta 2013, 111, 105–116. [Google Scholar] [CrossRef]
- Haug, A.; Høstmark, A.T.; Harstad, O.M. Bovine milk in human nutrition—A review. Lipids Health Dis. 2007, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.; Ross, R.P.; Hill, C.; Fitzgerald, G.F.; Stanton, C. Milk intelligence: Mining milk for bioactive substances associated with human health. Int. Dairy J. 2011, 21, 377–401. [Google Scholar] [CrossRef]
- Dairy Production and Products: Products. Available online: http://www.fao.org/dairy-production-products/products/en/ (accessed on 18 April 2020).
- EU Agricultural Outlook for Markets and Income, 2019–2030; European Commission, DG Agriculture and Rural Development: Brussels, Belgium, 2019.
- Milk and Milk Products, Detailed Data, Slovenia. 2018. Available online: https://www.stat.si/StatWeb/en/News/Index/8208 (accessed on 18 April 2020).
- Yayota, M.; Tsukamoto, M.; Yamada, Y.; Ohtani, S. Milk composition and flavor under different feeding systems: A survey of dairy farms. J. Dairy Sci. 2013, 96, 5174–5183. [Google Scholar] [CrossRef]
- Ellis, K.A.; Innocent, G.; Grove-White, D.; Cripps, P.; McLean, W.G.; Howard, C.V.; Mihm, M. Comparing the fatty acid composition of organic and conventional milk. J. Dairy Sci. 2006, 89, 1938–1950. [Google Scholar] [CrossRef]
- Hanus, O.; Samkova, E.; Křížova, L.; Hasoňova, L.; Kala, R. Role of fatty acids in milk fat and the influence of selected factors on their variability—A review. Molecules 2018, 23, 1636. [Google Scholar] [CrossRef]
- Benjamin, S.; Spener, F. Conjugated linoleic acids as functional food: An insight into their health benefits. Nutr. Metab. (Lond.) 2009, 6, 36. [Google Scholar] [CrossRef]
- Mooney, D.; McCarthy, C.; Belton, O. Effects of conjugated linoleic acid isomers on monocyte, macrophage and foam cell phenotype in atherosclerosis. Prostaglandins Other Lipid Mediat. 2012, 98, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Chilliard, Y.; Glasser, F.; Ferlay, A.; Bernard, L.; Rouel, J.; Doreau, M. Diet, rumen biohydrogenation and nutritional quality of cow and goat milk fat. Eur. J. Lipid Sci. Technol. 2007, 109, 828–855. [Google Scholar] [CrossRef]
- Kliem, K.E.; Shingfield, K.J.; Livingstone, K.M.; Givens, D.I. Seasonal variation in the fatty acid composition of milk available at retail in the United Kingdom and implications for dietary intake. Food Chem. 2013, 141, 274–281. [Google Scholar] [CrossRef]
- Collomb, M.; Bisig, W.; Bütikofer, U.; Sieber, R.; Bregy, M.; Etter, L. Seasonal variation in the fatty acid composition of milk supplied to dairies in the mountain regions of Switzerland. Dairy Sci. Technol. 2008, 88, 631–647. [Google Scholar] [CrossRef]
- Cossignani, L.; Blasi, F.; Bosi, A.; D’Arco, G.; Maurelli, S.; Simonetti, M.S.; Damiani, P. Detection of cow milk in donkey milk by chemometric procedures on triacylglycerol stereospecific analysis results. J. Dairy Res. 2011, 78, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Camin, F.; Perini, M.; Colombari, G.; Bontempo, L.; Versini, G. Influence of dietary composition on the carbon, nitrogen, oxygen and hydrogen stable isotope ratios of milk. Rapid Commun. Mass Spectrom. 2008, 22, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.; Giesemann, A. Differentiation of organically and conventionally produced milk by stable isotope and fatty acid analysis. Anal. Bioanal. Chem. 2007, 388, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.K.; Spangenberg, J.E.; Klevenhusen, F.; Soliva, C.R.; Kreuzer, M.; Leiber, F. Stable carbon isotope composition of c9,t11-conjugated linoleic acid in cow’s milk as related to dietary fatty acids. Lipids 2012, 47, 161–169. [Google Scholar] [CrossRef]
- Luo, D.; Dong, H.; Luo, H.; Xian, Y.; Guo, X.; Wu, Y. Multi-element (C, N, H, O) stable isotope ratio analysis for determining the geographical origin of pure milk from different regions. Food Anal. Methods 2016, 9, 437–442. [Google Scholar] [CrossRef]
- Bontempo, L.; Lombardi, G.; Paoletti, R.; Ziller, L.; Camin, F. H, C, N and O stable isotope characteristics of alpine forage, milk and cheese. Int. Dairy J. 2012, 23, 99–104. [Google Scholar] [CrossRef]
- Crittenden, R.G.; Andrew, A.S.; LeFournour, M.; Young, M.D.; Middleton, H.; Stockmann, R. Determining the geographic origin of milk in Australasia using multi-element stable isotope ratio analysis. Int. Dairy J. 2007, 17, 421–428. [Google Scholar] [CrossRef]
- Potočnik, D.; Nečemer, M.; Perišić, I.; Jagodic, M.; Mazej, D.; Camin, F.; Eftimov, T.; Strojnik, L.; Ogrinc, N. Geographical verification of Slovenian milk using stable isotope ratio, multi-element and multivariate modelling approaches. Food Chem. 2020, 326, 126958. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J. Authentication of organic milk using δ13C and the α-linolenic acid content of milk fat. J. Agric. Food Chem. 2009, 57, 785–790. [Google Scholar] [CrossRef]
- Ehtesham, E.; Hayman, A.R.; McComb, K.A.; Van Hale, R.; Frew, R.D. Correlation of Geographical Location with Stable Isotope Values of Hydrogen and Carbon of Fatty Acids from New Zealand Milk and Bulk Milk Powder. J. Agric. Food Chem. 2013, 61, 8914–8923. [Google Scholar] [CrossRef]
- Ehtesham, E.; Hayman, A.; Van Hale, R.; Frew, R. Influence of feed and water on the stable isotopic composition of dairy milk. Int. Dairy J. 2015, 47, 37–45. [Google Scholar] [CrossRef]
- Gaspardo, B.; Lavrenčič, A.; Levart, A.; Del Zotto, S.; Stefanon, B. Use of milk fatty acids composition to discriminate area of origin of bulk milk. J. Dairy Sci. 2010, 93, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Malacarne, M.; Martuzzi, F.; Summer, A.; Mariani, P. Protein and fat composition of mare’s milk: Some nutritional remarks with reference to human and cow’s milk. Int. Dairy J. 2002, 12, 869–877. [Google Scholar] [CrossRef]
- Coppa, M.; Ferlay, A.; Chassaing, C.; Agabriel, C.; Glasser, F.; Chilliard, Y.; Borreani, G.; Barcarolo, R.; Baars, T.; Kusche, D.; et al. Prediction of bulk milk fatty acid composition based on farming practices collected through on-farm surveys. J. Dairy Sci. 2013, 96, 4197–4211. [Google Scholar] [CrossRef]
- Kaufman, L.; Rousseeuw, P.J. Partitioning around Medoids (Program PAM). In Finding Groups in Data an Introduction to Cluster Analysis; Kaufman, L., Rousseeuw, P.J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; pp. 68–125. [Google Scholar]
- Cossignani, L.; Blasi, F.; Simonetti, M.S.; Montesano, D. Fatty Acids and Phytosterols to Discriminate Geographic Origin of Lycium barbarum Berry. Food Anal. Methods 2018, 11, 1180–1188. [Google Scholar] [CrossRef]
- Potočnik, T.; Ogrinc, N.; Potočnik, D.; Košir, I.J. Fatty acid composition and δ13C isotopic ratio characterisation of pumpkin seed oil. J. Food Comp. Anal. 2016, 53, 85–90. [Google Scholar] [CrossRef]
- Hrastar, R.; Gams Petrišič, M.; Ogrinc, N.; Košir, I.J. Fatty acid and stable carbon isotope charcaterization of Camelina sativa oil: Implications for authentification. J. Agric. Food Chem. 2009, 57, 579–585. [Google Scholar] [CrossRef]
- Logan, A.; Müller, W.; Versteeg, C. Australian milk fat-seasonal and regional composition of fatty acids. J. Dairy Res. Technol. 2019, 2, 011. [Google Scholar] [CrossRef]
- Rutkowska, J.; Bialek, M.; Adamska, A.; Zbikowska, A. Differentiation of geographical origin of cream products in poland according to their fatty acid profile. Food Chem. 2015, 178, 26–31. [Google Scholar] [CrossRef]
- Bauman, D.E.; Griinari, J.M. Nutritional regulation of milk fat synthesis. Annu. Rev. Nutr. 2003, 23, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Dudd, S.N.; Evershed, R.P. Direct demonstration of milk as an element of archaeological economies. Science 1998, 282, 1478–1481. [Google Scholar] [CrossRef]
- Stergiadis, S.; Leifert, C.; Seal, C.J.; Eyre, M.D.; Steinshamn, H.; Butler, G. Improving the fatty acid profile of winter milk from housed cows with contrasting feeding regimes by oilseed supplementation. Food Chem. 2014, 164, 293–300. [Google Scholar] [CrossRef] [PubMed]
- MacGibbon, A.K.H.; Taylor, M.W. Composition and structure of bovine milk lipids. In Advanced Dairy Chemistry, 3rd ed.; Fox, P.F., McSweeney, P.L.H., Eds.; Springer: New York, NY, USA, 2009; Volume 2, pp. 1–42. [Google Scholar]
- Schwendel, B.H.; Wester, T.J.; Morel, P.C.H.; Tavendale, M.H.; Deadman, C.; Shadbolt, N.M.; Otter, D.E. Invited review: Organic and conventionally produced milk-An evaluation of factors influencing milk composition. J. Dairy Sci. 2015, 98, 721–746. [Google Scholar] [CrossRef] [PubMed]
- Engel, E.; Ferlay, A.; Cornu, A.; Chilliard, Y.; Agabriel, C.; Bielicki, G.; Martin, B. Relevance of Isotopic and Molecular Biomarkers for the Authentication of Milk According to Production Zone and Type of Feeding of the Cow. J. Agric. Food Chem. 2007, 55, 9099–9108. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, A.; Martin, B.; Pradel, P.; Coulon, J.B.; Chilliard, Y. Influence of grass-based diets on milk fatty acid composition and milk lipolytic system in Tarentaise and Montbéliarde cow breeds. J. Dairy Sci. 2006, 89, 4026–4041. [Google Scholar] [CrossRef]
- Nantapo, C.T.W.; Muchenje, V.; Hugo, A. Atherogenicity index and health-related fatty acids in different stages of lactation from Friesian, Jersey and Friesian x Jersey cross cow milk under a pasture-based dairy system. Food Chem. 2014, 146, 127–133. [Google Scholar] [CrossRef]
- Bauman, D.E.; Lock, A.L. Conjugated Linoleic Acid: Biosynthesis and Nutritional Significance. In Advanced Dairy Chemistry, 3rd ed.; Fox, P.F., McSweeney, P.L.H., Eds.; Springer: New York, NY, USA, 2009; Volume 2, pp. 93–136. [Google Scholar]
- Khan, N.A.; Yu, P.; Ali, M.; Cone, J.W.; Hendriks, W.H. Nutritive value of maize silage in relation to dairy cow performance and milk quality. J. Sci. Food Agric. 2015, 95, 238–252. [Google Scholar] [CrossRef]
- Larqué, E.; Garaulet, M.; Pérez-Llamas, F.; Zamora, S.; Tebar, F.J. Fatty Acid Composition and Nutritional Relevance of Most Widely Consumed Margarines in Spain. Grasas Aceites 2003, 54, 65–70. [Google Scholar]
- Christie, W.W.; Han, X. Lipid Analysis: Isolation, Separation, Identification and Lipidomic Analysis, 4th ed.; The Oily Press: Bridgwater, UK, 2010. [Google Scholar]
- Brand, W.A.; Coplen, T.B.; Vogl, J.; Rosner, M.; Prohaska, T. Assessment of international reference materials for isotope-ratio analysis (IUPAC technical report). Pure Appl. Chem. 2014, 86, 425–467. [Google Scholar] [CrossRef]
- Paul, D.; Skrzypek, G.; Fórizs, I. Normalization of measured stable isotopic compositions to isotope reference scales—A review. Rapid Commun. Mass Spectrom. 2007, 21, 3006–3014. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2012 | 2013 | 2014 | Winter | Summer | Corn | Non-Corn | p-Value 1 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| FA (% of Total FA) | Feed Type | Mean ± SD | Year | Season | ||||||
| SFA | ||||||||||
| C6:0 | Corn | 1.25 ± 0.26 | 1.20 ± 0.46 | 1.30 ± 0.44 | 1.44 ± 0.32 | 1.05 ± 0.37 | 1.30 ± 0.39 | 1.12 ± 0.40 | * | *** |
| C8:0 | Corn, concentrates | 1.22 ± 0.08 | 1.36 ± 0.41 | 1.22 ± 0.34 | 1.28 ± 0.26 | 1.25 ± 0.37 | 1.29 ± 0.33 | 1.20 ± 0.28 | *** | * |
| C10:0 | Corn, concentrates | 3.03 ± 0.25 | 3.57 ± 1.02 | 3.18 ± 0.82 | 3.28 ± 0.62 | 3.23 ± 0.95 | 3.33 ± 0.81 | 3.07 ± 0.74 | ** | * |
| C11:0 | Corn, concentrates | 0.04 ± 0.03 | 0.06 ± 0.03 | 0.08 ± 0.02 | 0.06 ± 0.02 | 0.05 ± 0.04 | 0.06 ± 0.03 | 0.05 ± 0.03 | *** | *** |
| C12:0 | Corn, concentrates | 3.57 ± 0.33 | 4.10 ± 0.80 | 3.90 ± 0.82 | 3.97 ± 0.61 | 3.74 ± 0.80 | 3.94 ± 0.67 | 3.65 ± 0.79 | NS | *** |
| C13:0 | 0.11 ± 0.01 | 0.11 ± 0.03 | 0.11 ± 0.03 | 0.12 ± 0.02 | 0.10 ± 0.03 | 0.11 ± 0.03 | 0.10 ± 0.02 | NS | *** | |
| C14:0 | Corn | 12.0 ± 0.65 | 12.8 ± 1.86 | 12.9 ± 1.26 | 12.9 ± 0.94 | 12.3 ± 1.70 | 12.6 ± 1.41 | 12.5 ± 1.39 | NS | *** |
| C15:0iso | Hay, grass silage | 0.32 ± 0.05 | 0.34 ± 0.13 | 0.34 ± 0.05 | 0.32 ± 0.05 | 0.35 ± 0.12 | 0.32 ± 0.09 | 0.37 ± 0.07 | *** | *** |
| C15:0aiso | Hay, grass silage | 0.51 ± 0.08 | 0.53 ± 0.09 | 0.56 ± 0.09 | 0.50 ± 0.07 | 0.56 ± 0.10 | 0.50 ± 0.07 | 0.61 ± 0.11 | NS | *** |
| C15:0 | Hay, grass silage | 1.17 ± 0.11 | 1.24 ± 0.13 | 1.33 ± 0.15 | 1.24 ± 0.14 | 1.25 ± 0.16 | 1.21 ± 0.12 | 1.34 ± 0.16 | NS | NS |
| C16:0 | Calcium salts of palm oil, canola and soy, corn | 32.60 ± 2.13 | 32.7 ± 2.48 | 33.2 ± 2.11 | 34.1 ± 1.73 | 31.6 ± 2.01 | 32.9 ± 1.86 | 32.4 ± 3.04 | *** | *** |
| C17:0iso | Hay, grass silage | 0.37 ± 0.07 | 0.50 ± 0.06 | 0.53 ± 0.06 | 0.44 ± 0.10 | 0.49 ± 0.09 | 0.45 ± 0.09 | 0.51 ± 0.10 | *** | *** |
| C17:0aiso | Hay, grass silage | 0.47 ± 0.05 | 0.44 ± 0.06 | 0.48 ± 0.07 | 0.44 ± 0.05 | 0.48 ± 0.07 | 0.45 ± 0.05 | 0.51 ± 0.08 | *** | *** |
| C17:0 | Hay, grass silage | 0.61 ± 0.08 | 0.65 ± 0.11 | 0.69 ± 0.15 | 0.63 ± 0.10 | 0.67 ± 0.14 | 0.61 ± 0.09 | 0.73 ± 0.15 | NS | *** |
| C18:0 | Fresh herbage, grass silage, concentrates | 10.2 ± 1.01 | 9.87 ± 1.17 | 9.99 ± 1.66 | 9.41 ± 0.90 | 10.6 ± 1.36 | 9.97 ± 1.06 | 10.2 ± 1.81 | *** | *** |
| C20:0 | 0.18 ± 0.02 | 0.19 ± 0.15 | 0.14 ± 0.08 | 0.16 ± 0.05 | 0.18 ± 0.13 | 0.17 ± 0.11 | 0.17 ± 0.07 | NS | *** | |
| C21:0 | 0.03 ± 0.01 | 0.05 ± 0.07 | 0.02 ± 0.02 | 0.04 ± 0.03 | 0.03 ± 0.05 | 0.03 ± 0.05 | 0.03 ± 0.03 | *** | NS | |
| C22:0 | 0.07 ± 0.02 | 0.04 ± 0.04 | 0.03 ± 0.03 | 0.05 ± 0.03 | 0.05 ± 0.04 | 0.05 ± 0.03 | 0.05 ± 0.03 | *** | NS | |
| C23:0 | 0.03 ± 0.01 | 0.04 ± 0.05 | 0.03 ± 0.03 | 0.04 ± 0.04 | 0.03 ± 0.02 | 0.03 ± 0.04 | 0.04 ± 0.03 | *** | *** | |
| C24:0 | 0.05 ± 0.01 | 0.04 ± 0.04 | 0.04 ± 0.03 | 0.05 ± 0.02 | 0.04 ± 0.03 | 0.04 ± 0.03 | 0.05 ± 0.03 | *** | NS | |
| ∑SFA | Conserved forage and concentrate rich diet | 67.76 ± 2.49 | 69.90 ± 2.99 | 70.10 ± 2.74 | 70.40 ± 2.06 | 68.04 ± 3.20 | 69.43 ± 2.61 | 68.71 ± 3.60 | *** | *** |
| MUFA | Rapeseed, seeds oils, fresh grass, non-conserved forage | |||||||||
| C12:1 | 0.12 ± 0.03 | 0.10 ± 0.03 | 0.09 ± 0.03 | 0.12 ± 0.03 | 0.09 ± 0.03 | 0.10 ± 0.03 | 0.10 ± 0.04 | *** | *** | |
| C14:1 | 1.04 ± 0.11 | 1.29 ± 1.41 | 1.17 ± 0.25 | 1.18 ± 0.18 | 1.15 ± 1.17 | 1.19 ± 0.95 | 1.12 ± 0.29 | NS | *** | |
| C16:1 | 2.03 ± 0.11 | 1.85 ± 0.20 | 1.94 ± 0.17 | 1.92 ± 0.14 | 1.96 ± 0.21 | 1.95 ± 0.18 | 1.92 ± 0.18 | *** | NS | |
| C17:1 | 0.28 ± 0.03 | 0.34 ± 0.05 | 0.37 ± 0.06 | 0.31 ± 0.06 | 0.34 ± 0.07 | 0.32 ± 0.05 | 0.36 ± 0.07 | *** | *** | |
| C18:1ω-9c/9t | Grass, rapeseeds | 23.7 ± 2.17 | 20.12 ± 2.8 | 20.7 ± 2.56 | 20.5 ± 2.01 | 22.6 ± 3.38 | 21.4 ± 2.76 | 21.9 ± 3.42 | *** | *** |
| C20:1ω-9 | 0.04 ± 0.01 | 0.25 ± 0.17 | 0.14 ± 0.08 | 0.12 ± 0.09 | 0.17 ± 0.17 | 0.14 ± 0.13 | 0.16 ± 0.16 | *** | NS | |
| C22:1ω-9 | 0.01 ± 0.01 | 0.02 ± 0.02 | 0.04 ± 0.03 | 0.02 ± 0.02 | 0.02 ± 0.02 | 0.02 ± 0.02 | 0.02 ± 0.02 | NS | *** | |
| ∑MUFA | 27.26 ± 2.10 | 23.97 ± 2.83 | 24.42 ± 2.38 | 24.20 ± 1.87 | 26.30 ± 3.23 | 25.10 ± 2.71 | 25.54 ± 3.17 | *** | *** | |
| PUFA | ||||||||||
| CLA (c-9, t-11 C18:2) | Grass silage, fish oil, rapeseed, linseed | 0.68 ± 0.28 | 0.61 ± 0.26 | 0.55 ± 0.20 | 0.51 ± 0.12 | 0.72 ± 0.31 | 0.55 ± 0.19 | 0.78 ± 0.31 | *** | *** |
| ω-6 PUFA | Corn, hay, straw, | |||||||||
| C18:2ω-6c | Grassing, red clover silage, corn | 2.00 ± 0.22 | 2.09 ± 0.32 | 1.98 ± 0.28 | 1.94 ± 0.23 | 2.11 ± 0.30 | 2.05 ± 0.24 | 1.95 ± 0.35 | NS | *** |
| C18:3ω-6 | 0.11 ± 0.02 | 0.43 ± 0.29 | 0.23 ± 0.12 | 0.20 ± 0.12 | 0.32 ± 0.30 | 0.24 ± 0.21 | 0.31 ± 0.28 | *** | *** | |
| C20:3ω-6 | Fish oil | 0.08 ± 0.01 | 0.06 ± 0.04 | 0.06 ± 0.04 | 0.06 ± 0.04 | 0.06 ± 0.04 | 0.07 ± 0.04 | 0.06 ± 0.03 | *** | NS |
| C20:4ω-6 | Fish oil | 0.12 ± 0.02 | 0.03 ± 0.02 | 0.03 ± 0.03 | 0.06 ± 0.05 | 0.06 ± 0.05 | 0.06 ± 0.05 | 0.06 ± 0.05 | *** | NS |
| ∑ω-6 PUFA | 2.31 ± 0.24 | 2.61 ± 0.58 | 2.30 ± 0.33 | 2.27 ± 0.26 | 2.55 ± 0.53 | 2.41 ± 0.39 | 2.38 ± 0.54 | *** | *** | |
| ω-3 PUFA | Grassing, | |||||||||
| C18:3ω-3 | Grassing, red clover silage, legume silage, cereals, oil seeds | 0.62 ± 0.20 | 0.69 ± 0.33 | 0.58 ± 0.20 | 0.54 ± 0.14 | 0.72 ± 0.30 | 0.57 ± 0.23 | 0.78 ± 0.25 | NS | *** |
| C20:3ω-3 | Fish oil | 0.02 ± 0.01 | 0.10 ± 0.09 | 0.09 ± 0.03 | 0.06 ± 0.05 | 0.07 ± 0.08 | 0.07 ± 0.07 | 0.05 ± 0.03 | *** | NS |
| C20:5ω-3 | Fish oil, linseed | 0.06 ± 0.02 | 0.05 ± 0.05 | 0.04 ± 0.03 | 0.05 ± 0.02 | 0.05 ± 0.06 | 0.05 ± 0.04 | 0.07 ± 0.06 | *** | NS |
| ∑ω-3 PUFA | 0.70 ± 0.22 | 0.84 ± 0.34 | 0.71 ± 0.21 | 0.65 ± 0.15 | 0.85 ± 0.32 | 0.69 ± 0.25 | 0.90 ± 0.25 | *** | *** | |
| ∑PUFA | 3.69 ± 0.50 | 4.05 ± 0.98 | 3.55 ± 0.56 | 3.43 ± 0.33 | 4.11 ± 0.87 | 3.65 ± 0.65 | 4.06 ± 0.86 | *** | *** | |
| ω-6:ω-3 | 3.64 ± 1.14 | 3.36 ± 0.79 | 3.80 ± 3.18 | 3.71 ± 1.19 | 3.48 ± 2.58 | 3.87 ± 2.14 | 2.87 ± 1.35 | NS | *** | |
| δ13C (‰) Mean ± SD | ||||||||
|---|---|---|---|---|---|---|---|---|
| FA | Source of FA | 2013 | 2014 | Winter | Summer | Corn | Non-Corn | Average |
| C10:0 | metabolism | −23.9 ± 2.7 | −22.1 ± 2.5 | −21.3 ± 2.0 | −24.7 ± 2.3 | −22.2 ± 2.4 | −26.2 ± 1.8 | −22.9 ± 2.8 |
| C12:0 | metabolism | −24.6 ± 3.2 | −22.8 ± 2.6 | −21.8 ± 2.2 | −25.7 ± 2.5 | −22.8 ± 2.6 | −27.3 ± 2.0 | −23.6 ± 3.0 |
| C14:1 | biosynthesis | −27.4 ± 3.2 | −24.9 ± 3.0 | −24.3 ± 2.5 | −28.0 ± 3.0 | −25.2 ± 2.9 | −29.9 ± 2.2 | −26.0 ± 3.3 |
| C14:0 | metabolism | −23.5 ± 3.0 | −21.9 ± 2.6 | −21.2 ± 2.0 | −24.3 ± 2.8 | −21.9 ± 2.4 | −26.2 ± 2.2 | −22.6 ± 2.9 |
| C15:0 | microbial biosynthesis | −32.1 ± 3.7 | −28.8 ± 2.9 | −28.2 ± 2.6 | −32.6 ± 3.4 | −29.5 ± 3.4 | −33.9 ± 2.7 | −30.3 ± 3.7 |
| C16:1 | biosynthesis | −28.5 ± 3.5 | −26.0 ± 3.0 | −25.2 ± 2.3 | −29.4 ± 3.3 | −26.3 ± 3.0 | −31.3 ± 2.6 | −27.2 ± 3.5 |
| C16:0 | diet and metabolism | −23.6 ± 2.9 | −22.6 ± 2.5 | −21.8 ± 2.0 | −24.5 ± 2.7 | −22.2 ± 2.2 | −26.7 ± 1.7 | −23.0 ± 2.7 |
| C18:1ω-9c | biosynthesis | −27.2 ± 2.5 | −25.9 ± 2.2 | −25.6 ± 2.1 | −27.5 ± 2.5 | −25.8 ± 1.9 | −29.8 ± 2.1 | −26.5 ± 2.5 |
| C18:0 | biosynthesis and diet | −27.8 ± 3.2 | −26.6 ± 2.8 | −25.6 ± 2.3 | −29.0 ± 2.7 | −26.4 ± 2.5 | −31.0 ± 2.6 | −27.2 ± 3.0 |
| Bulk | diet | −21.4 ± 1.9 | −21.5 ± 2.2 | −20.6 ± 1.7 | −22.5 ± 1.9 | −20.7 ± 1.4 | −24.8 ± 0.9 | −21.4 ± 2.0 |
| Fatty Acids | Year | Season | Alpine | Mediterranean | Pannonian | Dinaric |
|---|---|---|---|---|---|---|
| (% of Total FA) | Mean ± SD | |||||
| Saturated FA (SFA) | 2012 | summer | 64.15 ± 1.62 | 69.57 ± 3.30 | 66.33 ± 0.74 | 66.74 ± 2.14 |
| winter | 69.57 ± 0.82 | 71.64 ± 3.10 | 68.89 ± 0.66 | 69.52 ± 0.53 | ||
| average | 66.81 ± 3.09 | 70.60 ± 3.08 | 67.69 ± 1.47 | 68.12 ± 2.05 | ||
| 2013 | summer | 68.78 ± 1.60 | 68.19 ± 7.58 | 70.20 ± 5.04 | 70.63 ± 0.71 | |
| winter | 70.50 ± 0.75 | 70.39 ± 3.68 | 69.63 ± 1.99 | 70.08 ± 2.46 | ||
| average | 69.64 ± 1.51 | 69.29 ± 5.46 | 69.91 ± 3.77 | 70.34 ± 1.82 | ||
| 2014 | summer | 68.08 ± 1.33 | 69.55 ± 4.65 | 68.42 ± 1.22 | 68.62 ± 3.48 | |
| winter | 72.45 ± 1.02 | 73.23 ± 4.25 | 70.56 ± 2.76 | 72.16 ± 0.79 | ||
| average | 70.26 ± 2.51 | 71.39 ± 4.47 | 69.42 ± 2.31 | 70.49 ± 2.99 | ||
| Monounsaturated FA (MUFA) | 2012 | summer | 29.93 ± 1.28 | 24.95 ± 3.35 | 28.73 ± 0.62 | 27.96 ± 2.00 |
| winter | 25.79 ± 0.86 | 23.59 ± 3.26 | 26.56 ± 0.52 | 25.94 ± 0.51 | ||
| average | 27.86 ± 2.37 | 24.27 ± 3.05 | 27.57 ± 1.23 | 26.89 ± 1.72 | ||
| 2013 | summer | 24.98 ± 1.38 | 26.40 ± 7.03 | 24.36 ± 2.00 | 23.93 ± 0.86 | |
| winter | 23.73 ± 0.84 | 24.16 ± 3.09 | 24.16 ± 0.53 | 23.26 ± 1.13 | ||
| average | 24.36 ± 1.29 | 25.28 ± 5.01 | 24.25 ± 1.42 | 23.57 ± 1.04 | ||
| 2014 | summer | 26.31 ± 0.92 | 25.53 ± 3.66 | 26.74 ± 1.14 | 25.13 ± 2.27 | |
| winter | 22.20 ± 1.13 | 21.59 ± 3.56 | 23.15 ± 1.37 | 22.64 ± 0.57 | ||
| average | 24.25 ± 2.33 | 23.56 ± 3.89 | 25.07 ± 2.20 | 23.81 ± 2.01 | ||
| Conjugated linoleic acid (C18:2c9,t11) | 2012 | summer | 1.20 ± 0.30 | 0.67 ± 0.12 | 0.62 ± 0.12 | 0.72 ± 0.20 |
| winter | 0.61 ± 0.09 | 0.59 ± 0.10 | 0.48 ± 0.06 | 0.55 ± 0.10 | ||
| average | 0.90 ± 0.37 | 0.63 ± 0.11 | 0.54 ± 0.11 | 0.63 ± 0.17 | ||
| 2013 | summer | 0.89 ± 0.24 | 0.67 ± 0.61 | 0.63 ± 0.34 | 0.54 ± 0.12 | |
| winter | 0.62 ± 0.15 | 0.52 ± 0.23 | 0.44 ± 0.07 | 0.54 ± 0.15 | ||
| average | 0.75 ± 0.24 | 0.60 ± 0.42 | 0.54 ± 0.26 | 0.54 ± 0.13 | ||
| 2014 | summer | 0.86 ± 0.18 | 0.51 ± 0.31 | 0.50 ± 0.14 | 0.58 ± 0.21 | |
| winter | 0.47 ± 0.09 | 0.53 ± 0.20 | 0.44 ± 0.08 | 0.43 ± 0.06 | ||
| average | 0.67 ± 0.24 | 0.52 ± 0.23 | 0.47 ± 0.12 | 0.51 ± 0.16 | ||
| α-linolenic acid (C18:3ω-3) | 2012 | summer | 0.84 ± 0.14 | 0.98 ± 0.34 | 0.52 ± 0.07 | 0.70 ± 0.16 |
| winter | 0.58 ± 0.09 | 0.76 ± 0.33 | 0.45 ± 0.06 | 0.57 ± 0.16 | ||
| average | 0.71 ± 0.18 | 0.87 ± 0.32 | 0.48 ± 0.07 | 0.63 ± 0.17 | ||
| 2013 | summer | 1.08 ± 0.54 | 0.46 ± 0.07 | 0.78 ± 0.30 | 0.71 ± 0.21 | |
| winter | 0.61 ± 0.12 | 0.56 ± 0.14 | 0.50 ± 0.05 | 0.56 ± 0.11 | ||
| average | 0.84 ± 0.45 | 0.51 ± 0.11 | 0.64 ± 0.26 | 0.63 ± 0.18 | ||
| 2014 | summer | 0.69 ± 0.26 | 0.89 ± 0.24 | 0.54 ± 0.13 | 0.66 ± 0.21 | |
| winter | 0.60 ± 0.13 | 0.81 ± 0.08 | 0.45 ± 0.09 | 0.45 ± 0.17 | ||
| average | 0.64 ± 0.21 | 0.85 ± 0.17 | 0.50 ± 0.12 | 0.55 ± 0.22 | ||
| Eicosapentaenoic acid (C20:5ω-3) | 2012 | summer | 0.08 ± 0.01 | 0.11 ± 0.02 | 0.06 ± 0.01 | 0.07 ± 0.02 |
| winter | 0.06 ± 0.01 | 0.08 ± 0.02 | 0.05 ± 0.02 | 0.06 ± 0.02 | ||
| average | 0.07 ± 0.01 | 0.09 ± 0.03 | 0.05 ± 0.01 | 0.07 ± 0.02 | ||
| 2013 | summer | 0.02 ± 0.03 | 0.16 ± 0.26 | 0.06 ± 0.10 | 0.02 ± 0.02 | |
| winter | 0.05 ± 0.04 | 0.04 ± 0.01 | 0.03 ± 0.02 | 0.05 ± 0.02 | ||
| average | 0.04 ± 0.04 | 0.10 ± 0.17 | 0.05 ± 0.07 | 0.04 ± 0.02 | ||
| 2014 | summer | 0.04 ± 0.02 | 0.03 ± 0.03 | 0.03 ± 0.04 | 0.03 ± 0.02 | |
| winter | 0.04 ± 0.02 | 0.07 ± 0.01 | 0.04 ± 0.01 | 0.07 ± 0.02 | ||
| average | 0.04 ± 0.02 | 0.05 ± 0.03 | 0.03 ± 0.03 | 0.05 ± 0.03 | ||
| Polyunsaturated FA (PUFA) | 2012 | summer | 4.59 ± 0.46 | 3.71 ± 0.18 | 3.68 ± 0.19 | 3.90 ± 0.27 |
| winter | 3.40 ± 0.17 | 3.43 ± 0.25 | 3.39 ± 0.21 | 3.32 ± 0.26 | ||
| average | 4.00 ± 0.70 | 3.57 ± 0.25 | 3.52 ± 0.25 | 3.59 ± 0.39 | ||
| 2013 | summer | 4.98 ± 1.04 | 3.76 ± 1.38 | 4.58 ± 1.36 | 4.06 ± 0.61 | |
| winter | 3.81 ± 0.38 | 3.24 ± 0.44 | 3.44 ± 0.19 | 3.66 ± 0.29 | ||
| average | 4.39 ± 0.97 | 3.50 ± 0.96 | 4.01 ± 1.12 | 3.85 ± 0.50 | ||
| 2014 | summer | 4.23 ± 0.66 | 3.64 ± 0.73 | 3.53 ± 0.39 | 3.69 ± 0.67 | |
| winter | 3.41 ± 0.31 | 3.21 ± 0.72 | 3.32 ± 0.31 | 3.20 ± 0.37 | ||
| average | 3.82 ± 0.66 | 3.42 ± 0.69 | 3.43 ± 0.37 | 3.43 ± 0.57 | ||
| ω-3 PUFA | 2012 | summer | 0.94 ± 0.16 | 1.12 ± 0.37 | 0.59 ± 0.08 | 0.79 ± 0.19 |
| winter | 0.66 ± 0.10 | 0.86 ± 0.35 | 0.51 ± 0.06 | 0.64 ± 0.18 | ||
| average | 0.80 ± 0.19 | 0.99 ± 0.35 | 0.55 ± 0.08 | 0.71 ± 0.20 | ||
| 2013 | summer | 1.17 ± 0.54 | 0.65 ± 0.28 | 1.00 ± 0.37 | 0.83 ± 0.16 | |
| winter | 0.75 ± 0.14 | 0.70 ± 0.14 | 0.63 ± 0.06 | 0.68 ± 0.12 | ||
| average | 0.96 ± 0.44 | 0.67 ± 0.20 | 0.82 ± 0.32 | 0.75 ± 0.16 | ||
| 2014 | summer | 0.81 ± 0.28 | 1.01 ± 0.28 | 0.67 ± 0.16 | 0.75 ± 0.22 | |
| winter | 0.73 ± 0.16 | 0.95 ± 0.07 | 0.58 ± 0.10 | 0.61 ± 0.17 | ||
| average | 0.77 ± 0.22 | 0.98 ± 0.18 | 0.63 ± 0.14 | 0.68 ± 0.20 | ||
| ω-6 PUFA | 2012 | summer | 2.45 ± 0.13 | 1.92 ± 0.16 | 2.48 ± 0.15 | 2.38 ± 0.17 |
| winter | 2.14 ± 0.15 | 1.98 ± 0.08 | 2.39 ± 0.23 | 2.14 ± 0.25 | ||
| average | 2.29 ± 0.21 | 1.95 ± 0.12 | 2.43 ± 0.20 | 2.25 ± 0.24 | ||
| 2013 | summer | 2.92 ± 0.70 | 2.43 ± 1.23 | 2.94 ± 0.85 | 2.70 ± 0.51 | |
| winter | 2.44 ± 0.18 | 2.03 ± 0.22 | 2.36 ± 0.14 | 2.43 ± 0.21 | ||
| average | 2.68 ± 0.55 | 2.23 ± 0.82 | 2.65 ± 0.67 | 2.56 ± 0.39 | ||
| 2014 | summer | 2.56 ± 0.34 | 2.12 ± 0.18 | 2.36 ± 0.30 | 2.35 ± 0.33 | |
| winter | 2.20 ± 0.16 | 1.72 ± 0.55 | 2.29 ± 0.22 | 2.16 ± 0.34 | ||
| average | 2.38 ± 0.32 | 1.92 ± 0.43 | 2.33 ± 0.26 | 2.25 ± 0.34 | ||
| ω-6:ω-3 PUFA | 2012 | summer | 2.69 ± 0.55 | 1.90 ± 0.85 | 4.26 ± 0.72 | 3.19 ± 0.94 |
| winter | 3.32 ± 0.61 | 2.58 ± 1.03 | 4.79 ± 0.94 | 3.60 ± 1.08 | ||
| average | 3.01 ± 0.66 | 2.24 ± 0.92 | 4.54 ± 0.87 | 3.41 ± 1.01 | ||
| 2013 | summer | 2.86 ± 1.05 | 3.64 ± 0.42 | 3.15 ± 0.86 | 3.34 ± 0.73 | |
| winter | 3.35 ± 0.52 | 3.01 ± 0.78 | 3.79 ± 0.39 | 3.69 ± 0.91 | ||
| average | 3.11 ± 0.84 | 3.32 ± 0.66 | 3.47 ± 0.73 | 3.52 ± 0.82 | ||
| 2014 | summer | 3.01 ± 0.58 | 2.19 ± 0.48 | 3.69 ± 0.89 | 3.30 ± 0.74 | |
| winter | 3.13 ± 0.61 | 1.83 ± 0.67 | 4.06 ± 0.72 | 3.32 ± 0.86 | ||
| average | 3.07 ± 0.59 | 2.01 ± 0.56 | 3.86 ± 0.82 | 3.31 ± 0.69 | ||
| AI | 2012 | summer | 2.31 ± 0.20 | 3.25 ± 0.53 | 2.58 ± 0.10 | 2.63 ± 0.33 |
| winter | 3.06 ± 0.15 | 3.51 ± 0.60 | 2.92 ± 0.10 | 3.06 ± 0.11 | ||
| average | 2.69 ± 0.42 | 3.38 ± 0.52 | 2.76 ± 0.20 | 2.86 ± 0.32 | ||
| 2013 | summer | 3.03 ± 0.28 | 2.89 ± 1.82 | 3.15 ± 0.46 | 3.33 ± 0.20 | |
| winter | 3.30 ± 0.19 | 3.34 ± 0.62 | 3.79 ± 0.39 | 3.71 ± 0.21 | ||
| average | 3.16 ± 0.27 | 3.13 ± 1.24 | 3.47 ± 0.73 | 3.35 ± 0.18 | ||
| 2014 | summer | 2.81 ± 0.27 | 2.92 ± 0.75 | 2.84 ± 0.20 | 3.07 ± 0.43 | |
| winter | 3.79 ± 0.25 | 4.13 ± 1.05 | 3.50 ± 0.31 | 3.75 ± 0.19 | ||
| average | 3.30 ± 0.59 | 3.53 ± 1.05 | 3.15 ± 0.42 | 3.43 ± 0.47 | ||
| TI | 2012 | summer | 2.69 ± 0.23 | 3.19 ± 0.23 | 3.05 ± 0.11 | 3.03 ± 0.28 |
| winter | 3.47 ± 0.11 | 3.62 ± 0.28 | 3.43 ± 0.11 | 3.46 ± 0.11 | ||
| average | 3.08 ± 0.43 | 3.41 ± 0.33 | 3.25 ± 0.22 | 3.26 ± 0.30 | ||
| 2013 | summer | 3.09 ± 0.34 | 3.18 ± 1.13 | 3.19 ± 0.42 | 3.41 ± 0.20 | |
| winter | 3.67 ± 0.14 | 3.68 ± 0.58 | 3.64 ± 0.12 | 3.71 ± 0.21 | ||
| average | 3.38 ± 0.40 | 3.43 ± 0.85 | 3.42 ± 0.38 | 3.57 ± 0.25 | ||
| 2014 | summer | 3.25 ± 0.34 | 3.35 ± 0.71 | 3.33 ± 0.20 | 3.46 ± 0.44 | |
| winter | 3.82 ± 0.14 | 3.88 ± 0.79 | 3.80 ± 0.16 | 3.95 ± 0.25 | ||
| average | 3.54 ± 0.39 | 3.61 ± 0.73 | 3.55 ± 0.42 | 3.72 ± 0.42 | ||
| Alpine | Dinaric | Mediterranean | Pannonian | Grand Total | |
|---|---|---|---|---|---|
| 2012 | 24 | 17 | 6 | 30 | 77 |
| summer | 12 | 8 | 3 | 14 | 37 |
| winter | 12 | 9 | 3 | 16 | 40 |
| 2013 | 24 | 17 | 6 | 30 | 77 |
| summer | 12 | 8 | 3 | 15 | 38 |
| winter | 12 | 9 | 3 | 15 | 39 |
| 2014 | 24 | 17 | 6 | 30 | 77 |
| summer | 12 | 8 | 3 | 16 | 39 |
| winter | 12 | 9 | 3 | 14 | 38 |
| Grand Total | 72 | 51 | 18 | 90 | 231 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potočnik, D.; Strojnik, L.; Eftimov, T.; Levart, A.; Ogrinc, N. Fatty Acid and Stable Carbon Isotope Composition of Slovenian Milk: Year, Season, and Regional Variability. Molecules 2020, 25, 2892. https://doi.org/10.3390/molecules25122892
Potočnik D, Strojnik L, Eftimov T, Levart A, Ogrinc N. Fatty Acid and Stable Carbon Isotope Composition of Slovenian Milk: Year, Season, and Regional Variability. Molecules. 2020; 25(12):2892. https://doi.org/10.3390/molecules25122892
Chicago/Turabian StylePotočnik, Doris, Lidija Strojnik, Tome Eftimov, Alenka Levart, and Nives Ogrinc. 2020. "Fatty Acid and Stable Carbon Isotope Composition of Slovenian Milk: Year, Season, and Regional Variability" Molecules 25, no. 12: 2892. https://doi.org/10.3390/molecules25122892
APA StylePotočnik, D., Strojnik, L., Eftimov, T., Levart, A., & Ogrinc, N. (2020). Fatty Acid and Stable Carbon Isotope Composition of Slovenian Milk: Year, Season, and Regional Variability. Molecules, 25(12), 2892. https://doi.org/10.3390/molecules25122892