
Optimization of QuEChERS Method for Simultaneous Determination of Neonicotinoid Residues in Pollinator Forage


Abstract
1. Introduction
2. Results
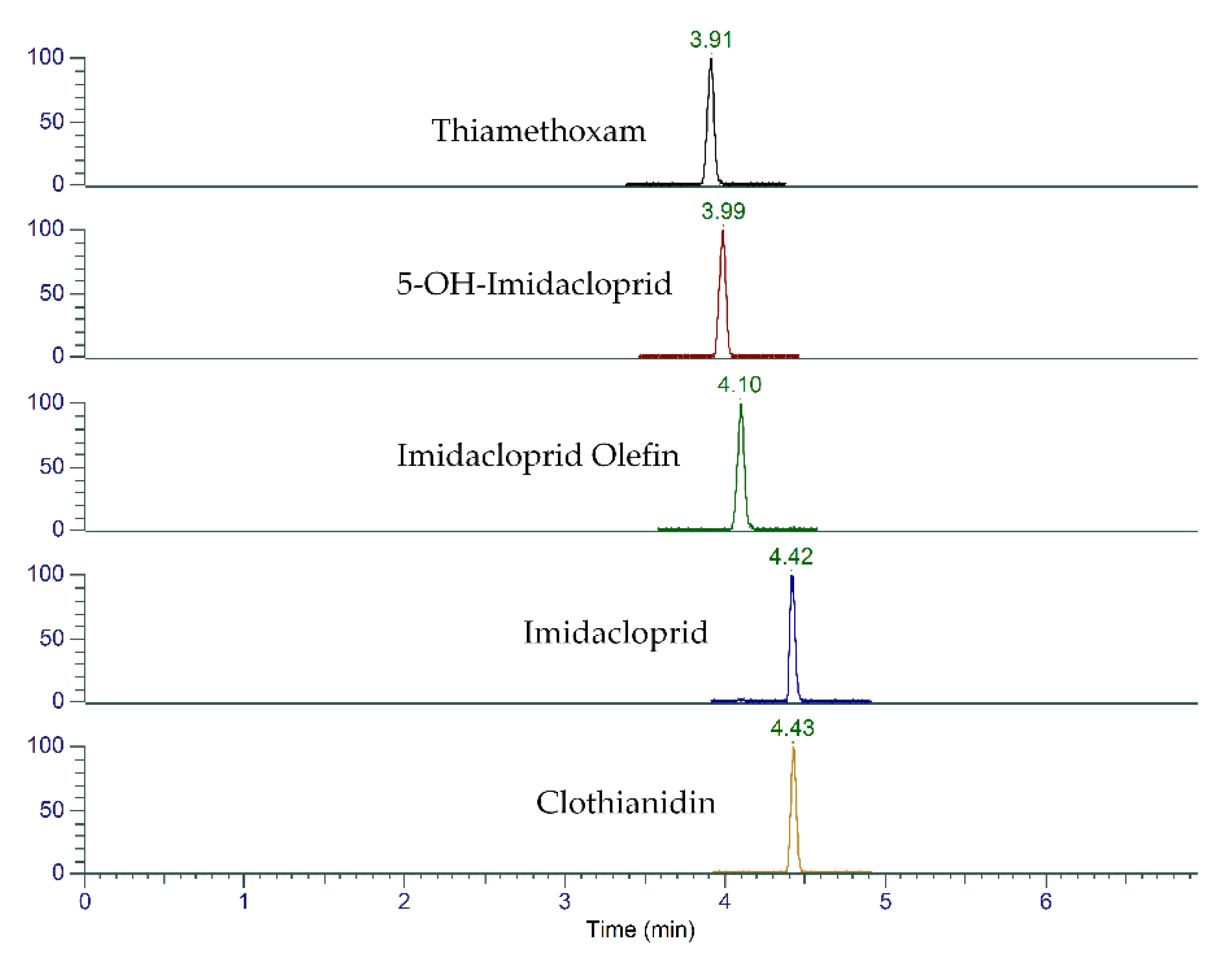
2.1. UHPLC-MS/MS Method Optimization
2.2. Optimization of Sample Preparation
2.2.1. Plant Tissue Matrix
2.2.2. Pollen Matrix
2.3. Method Validation
2.3.1. Evaluation of Linearity
2.3.2. Evaluation of the Method Detection Limit
2.3.3. Evaluation of Recovery
2.3.4. Evaluation of Trueness and Precision
3. Discussion
4. Materials and Methods
4.1. Standards, Reagents, and Solvents
4.2. Spiking Solution Preparation
4.3. Leaf Sample Preparation
4.4. Pollen Sample Preparation
4.5. LC-MS Conditions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Douglas, M.R.; Tooker, J.F. Large-Scale Deployment of Seed Treatments Has Driven Rapid Increase in Use of Neonicotinoid Insecticides and Preemptive Pest Management in U.S. Field Crops. Environ. Sci. Technol. 2015, 49, 5088–5097. [Google Scholar] [CrossRef] [PubMed]
- Sparks, T.C. Insecticide discovery: An evaluation and analysis. Pestic. Biochem. Physiol. 2013, 107, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Bass, C.; Denholm, I.; Williamson, M.S.; Nauen, R. The global status of insect resistance to neonicotinoid insecticides. Pestic. Biochem. Physiol. 2015, 121, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P.; Nauen, R.; Schindler, M.; Elbert, A. Overview of the Status and Global Strategy for Neonicotinoids. J. Agric. Food Chem. 2011, 59, 2897–2908. [Google Scholar] [CrossRef]
- Goulson, D. REVIEW: An overview of the environmental risks posed by neonicotinoid insecticides. J. Appl. Ecol. 2013, 50, 977–987. [Google Scholar] [CrossRef]
- Hladik, M.L.; Main, A.R.; Goulson, D. Environmental Risks and Challenges Associated with Neonicotinoid Insecticides. Environ. Sci. Technol. 2018, 52, 3329–3335. [Google Scholar] [CrossRef]
- Agriculture and Environment Research Unit (AERU). Pesticide Properties Database; Science and Technology Research Institute, University of Hertfordshire: U.K, September 2018. Available online: http://sitem.herts.ac.uk/aeru/footprint/en/index.htm (accessed on 12 April 2020).
- US Environmental Protection Agency. Imidacloprid Proposed Interim Registration Review Decision Case Number 7605. Washington, DC. 2020. Available online: https://www.regulations.gov/document?D=EPA-HQ-OPP-2008-0844-1619 (accessed on 28 March 2020).
- US Environmental Protection Agency. Clothianidin and Thiamethoxam Proposed Interim Registration Review Decision Case Numbers 7620 and 7614. Washington, DC. 2020. Available online: https://www.epa.gov/sites/production/files/202001/documents/clothianidin _and_thiamethoxam_pid_final_1.pdf (accessed on 12 April 2020).
- Gibbons, D.; Morrissey, C.A.; Mineau, P. A review of the direct and indirect effects of neonicotinoids and fipronil on vertebrate wildlife. Environ. Sci. Pollut. Res. 2014, 22, 103–118. [Google Scholar] [CrossRef]
- Bonmatin, J.-M.; Giorio, C.; Girolami, V.; Goulson, D.; Kreutzweiser, D.P.; Krupke, C.; Liess, M.; Long, E.; Marzaro, M.; Mitchell, E.A.D.; et al. Environmental fate and exposure; neonicotinoids and fipronil. Environ. Sci. Pollut. Res. 2014, 22, 35–67. [Google Scholar] [CrossRef]
- Botías, C.; David, A.; Horwood, J.; Abdul-Sada, A.; Nicholls, E.; Hill, E.; Goulson, D. Neonicotinoid Residues in Wildflowers, a Potential Route of Chronic Exposure for Bees. Environ. Sci. Technol. 2015, 49, 12731–12740. [Google Scholar] [CrossRef]
- Chen, M.; Tao, L.; McLean, J.; Lu, C. Quantitative Analysis of Neonicotinoid Insecticide Residues in Foods: Implication for Dietary Exposures. J. Agric. Food Chem. 2014, 62, 6082–6090. [Google Scholar] [CrossRef]
- David, A.; Botías, C.; Abdul-Sada, A.; Goulson, D.; Hill, E.M. Sensitive determination of mixtures of neonicotinoid and fungicide residues in pollen and single bumblebees using a scaled down QuEChERS method for exposure assessment. Anal. Bioanal. Chem. 2015, 407, 8151–8162. [Google Scholar] [CrossRef] [PubMed]
- Gbylik-Sikorska, M.; Śniegocki, T.; Posyniak, A. Determination of neonicotinoid insecticides and their metabolites in honey bee and honey by liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2015, 990, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Pecenka, J.R.; Lundgren, J.G. Non-target effects of clothianidin on monarch butterflies. Naturwissenschaften 2015, 102, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Collins, E.M.; Tao, L.; Lu, C. Simultaneous determination of residues in pollen and high-fructose corn syrup from eight neonicotinoid insecticides by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 9251–9264. [Google Scholar] [CrossRef]
- Bargar, T.A.; Hladik, M.L.; Daniels, J.C. Uptake and toxicity of clothianidin to monarch butterflies from milkweed consumption. PeerJ 2020, 8, e8669. [Google Scholar] [CrossRef]
- Olaya-Arenas, P.; Kaplan, I. Quantifying Pesticide Exposure Risk for Monarch Caterpillars on Milkweeds Bordering Agricultural Land. Front. Ecol. Evol. 2019, 7, 223. [Google Scholar] [CrossRef]
- Stenoien, C.; Nail, K.R.; Zalucki, J.M.; Parry, H.; Oberhauser, K.S.; Zalucki, M.P. Monarchs in decline: A collateral landscape-level effect of modern agriculture. Insect Sci. 2016, 25, 528–541. [Google Scholar] [CrossRef]
- Thogmartin, W.E.; Wiederholt, R.; Oberhauser, K.; Drum, R.G.; Diffendorfer, J.E.; Altizer, S.; Taylor, O.R.; Pleasants, J.; Semmens, D.; Semmens, B.; et al. Monarch butterfly population decline in North America: Identifying the threatening processes. R. Soc. Open Sci. 2017, 4, 170760. [Google Scholar] [CrossRef]
- Sánchez-Hernández, L.; Hernández-Domínguez, D.; Martín, M.T.; Del Nozal, J.B.; Higes, M.; Yagüe, J.L.B. Residues of neonicotinoids and their metabolites in honey and pollen from sunflower and maize seed dressing crops. J. Chromatogr. A 2016, 1428, 220–227. [Google Scholar] [CrossRef]
- Bonmatin, J.-M.; Moineau, I.; Charvet, R.; Fleche, C.; Colin, M.E.; Bengsch, E.R. A LC/APCI-MS/MS Method for Analysis of Imidacloprid in Soils, in Plants, and in Pollens. Anal. Chem. 2003, 75, 2027–2033. [Google Scholar] [CrossRef]
- Commission Decision (2002/657/EC) of 12 August 2002 Implementing Council Directive96/23/EC concerning the performance of analytical methods and the interpretation of results. 2002: Official Journal of the European Communities L221:8–36. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF (accessed on 28 March 2020).
- Menditto, A.; Patriarca, M.; Magnusson, B. Understanding the meaning of accuracy, trueness and precision. Accreditation Qual. Assur. 2006, 12, 45–47. [Google Scholar] [CrossRef]
- Krischik, V.; Rogers, M.A.; Gupta, G.; Varshney, A. Soil-Applied Imidacloprid Translocates to Ornamental Flowers and Reduces Survival of Adult Coleomegilla maculata, Harmonia axyridis, and Hippodamia convergens Lady Beetles, and Larval Danaus plexippus and Vanessa cardui Butterflies. PLoS ONE 2015, 10, e0119133. [Google Scholar] [CrossRef]
- Krupke, C.H.; Hunt, G.J.; Eitzer, B.D.; Andino, G.; Given, K. Multiple Routes of Pesticide Exposure for Honey Bees Living Near Agricultural Fields. PLoS ONE 2012, 7, e29268. [Google Scholar] [CrossRef] [PubMed]
- Long, E.Y.; Krupke, C.H. Non-cultivated plants present a season-long route of pesticide exposure for honey bees. Nat. Commun. 2016, 7, 11629. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Zhang, Y.; Bidne, K.G.; Hellmich, R.L.; Coats, J.R.; Bradbury, S.P. Assessing Field-Scale Risks of Foliar Insecticide Applications to Monarch Butterfly ( Danaus plexippus ) Larvae. Environ. Toxicol. Chem. 2020, 39, 923–941. [Google Scholar] [CrossRef] [PubMed]
- Mach, B.M.; Bondarenko, S.; Potter, D.A. Uptake and dissipation of neonicotinoid residues in nectar and foliage of systemically treated woody landscape plants. Environ. Toxicol. Chem. 2017, 37, 860–870. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |


{kind=link}
{kind=link}
| Compound | Retention Time (min) | Precursor (m/z) | Product (m/z) | Collision Energy (V) | RF Lens (V) |
|---|---|---|---|---|---|
| Imidacloprid olefin | 4.08 | 254 | 171 | 17.2 | 46 |
| Imidacloprid olefin | 4.08 | 254 | 205 | 13.8 | 46 |
| Imidacloprid | 4.41 | 256 | 175 | 19.5 | 41 |
| Imidacloprid | 4.41 | 256 | 209 | 15.4 | 41 |
| Imidacloprid | 4.41 | 256 | 212 | 10.2 | 41 |
| Clothianidin | 4.42 | 250 | 113 | 26.2 | 34 |
| Clothianidin | 4.42 | 250 | 169 | 14.3 | 34 |
| Thiamethoxam | 3.88 | 292 | 132 | 21.9 | 34 |
| Thiamethoxam | 3.88 | 292 | 181 | 22.3 | 34 |
| Thiamethoxam | 3.88 | 292 | 211 | 10.2 | 34 |
| 5-OH-imidacloprid | 3.96 | 272 | 134 | 41.2 | 53 |
| 5-OH-imidacloprid | 3.96 | 272 | 191 | 19.1 | 53 |
| 5-OH-imidacloprid | 3.96 | 272 | 225 | 15.0 | 53 |
| Imidacloprid olefin-13C3,15N | 4.08 | 259 | 176 | 17.2 | 43 |
| Imidacloprid olefin-13C3,15N | 4.08 | 259 | 211 | 10.2 | 43 |
| Imidacloprid olefin-13C3,15N | 4.08 | 259 | 241 | 10.2 | 43 |
| Imidacloprid-pyr-d4-CH3-d2,13C | 4.39 | 260 | 179 | 18.6 | 47 |
| Imidacloprid-pyr-d4-CH3-d2,13C | 4.39 | 260 | 213 | 16.1 | 47 |
| Imidacloprid-pyr-d4-CH3-d2,13C | 4.39 | 260 | 214 | 10.5 | 47 |
| Clothianidin-d3 | 4.41 | 253 | 132 | 16.6 | 44 |
| Clothianidin-d3 | 4.41 | 253 | 172 | 12.7 | 44 |
| Thiamethoxam-d3 | 3.88 | 295 | 184 | 22.4 | 45 |
| Thiamethoxam-d3 | 3.88 | 295 | 214 | 11.7 | 45 |
| 5-OH-imidacloprid-d4 | 3.95 | 276 | 195 | 19.7 | 51 |
| 5-OH-imidacloprid-d4 | 3.95 | 276 | 229 | 13.3 | 51 |
| Trueness | ||||
|---|---|---|---|---|
| Compound ID | MDL (ng/g) | Recovery (%) | Low QC | High QC |
| Thiamethoxam | 0.04 | 93.6 | 90.1 | 100 |
| 5-OH-imidacloprid | 0.1 | 78.4 | 96.6 | 78.4 |
| Imidacloprid olefin | 0.1 | 90.2 | 109 | 98.1 |
| Imidacloprid | 0.2 | 86.7 | 90.0 | 103 |
| Clothianidin | 0.3 | 85.4 | 90.0 | 100 |
| Trueness | ||||
|---|---|---|---|---|
| Compound ID | MDL (ng/g) | Recovery (%) | Low QC | High QC |
| Thiamethoxam | 0.04 | 101 | 99.7 | 95.5 |
| 5-OH-imidacloprid | 0.3 | 89.4 | 95.8 | 100 |
| Imidacloprid olefin | 1.0 | 94.5 | 111 | 108 |
| Imidacloprid | 0.06 | 99.7 | 93.6 | 95.0 |
| Clothianidin | 0.06 | 95.8 | 96.0 | 92.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, M.J.; Dang, V.; Bradbury, S.P.; Coats, J.R. Optimization of QuEChERS Method for Simultaneous Determination of Neonicotinoid Residues in Pollinator Forage. Molecules 2020, 25, 2732. https://doi.org/10.3390/molecules25122732
Hall MJ, Dang V, Bradbury SP, Coats JR. Optimization of QuEChERS Method for Simultaneous Determination of Neonicotinoid Residues in Pollinator Forage. Molecules. 2020; 25(12):2732. https://doi.org/10.3390/molecules25122732
Chicago/Turabian StyleHall, Maura J., Viet Dang, Steven P. Bradbury, and Joel R. Coats. 2020. "Optimization of QuEChERS Method for Simultaneous Determination of Neonicotinoid Residues in Pollinator Forage" Molecules 25, no. 12: 2732. https://doi.org/10.3390/molecules25122732
APA StyleHall, M. J., Dang, V., Bradbury, S. P., & Coats, J. R. (2020). Optimization of QuEChERS Method for Simultaneous Determination of Neonicotinoid Residues in Pollinator Forage. Molecules, 25(12), 2732. https://doi.org/10.3390/molecules25122732