
Method Validation and Investigation of the Levels of Pharmaceuticals and Personal Care Products in Sludge of Wastewater Treatment Plants and Soils of Irrigated Golf Course


Abstract

1. Introduction
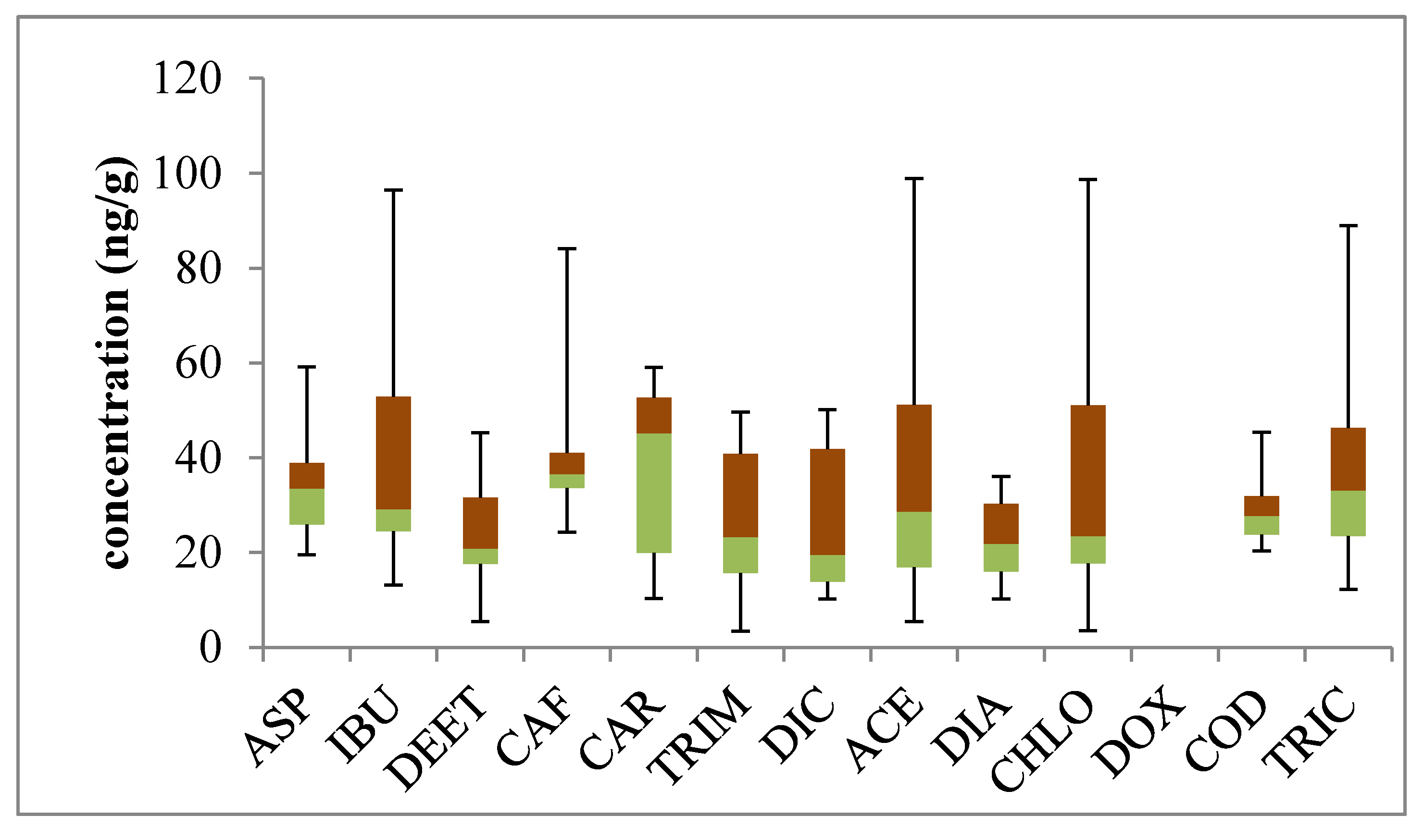
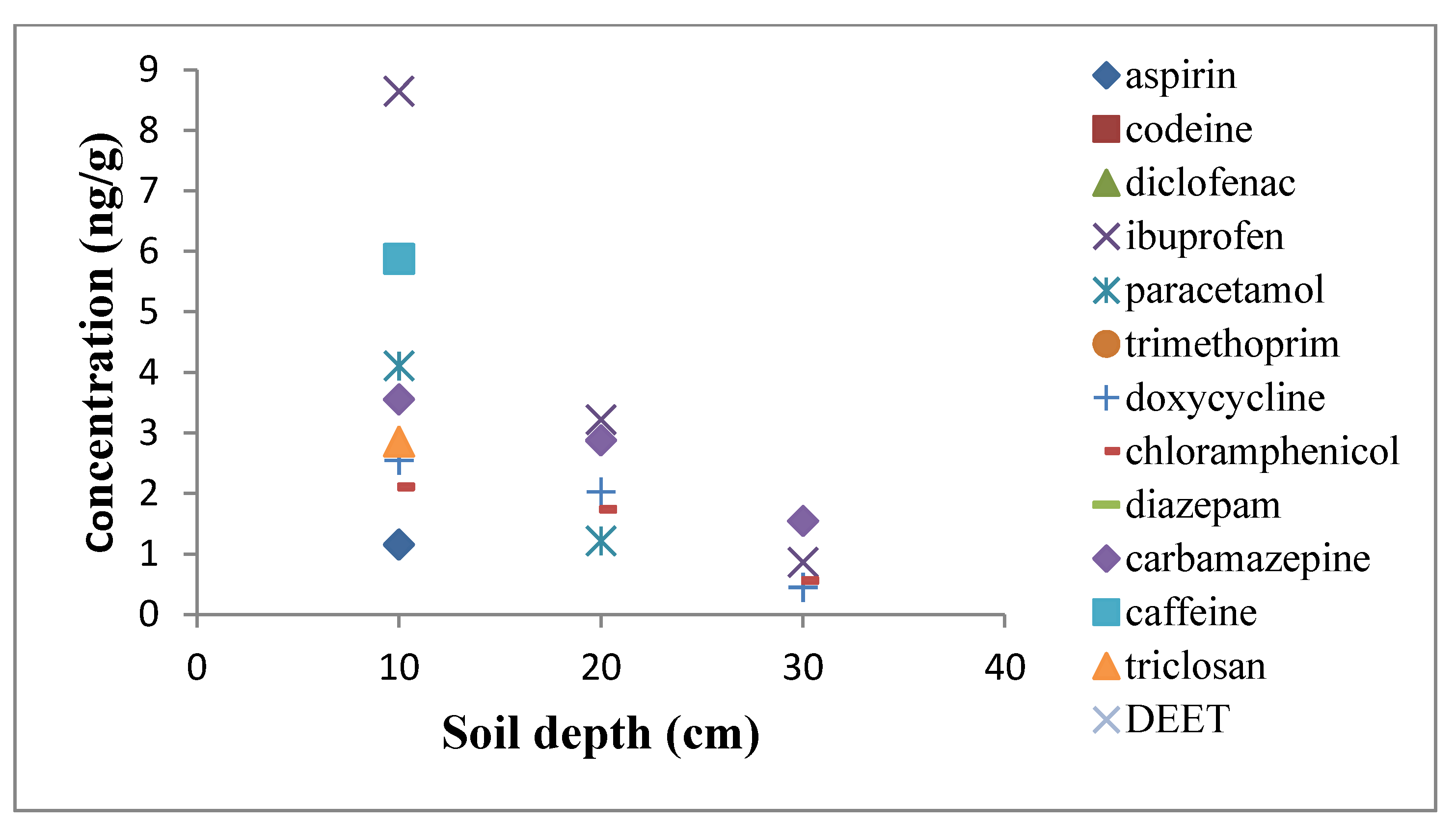
2. Results and Discussion
2.1. Analytical Performance for Optimization
2.2. Quality Assurance and Quality Control
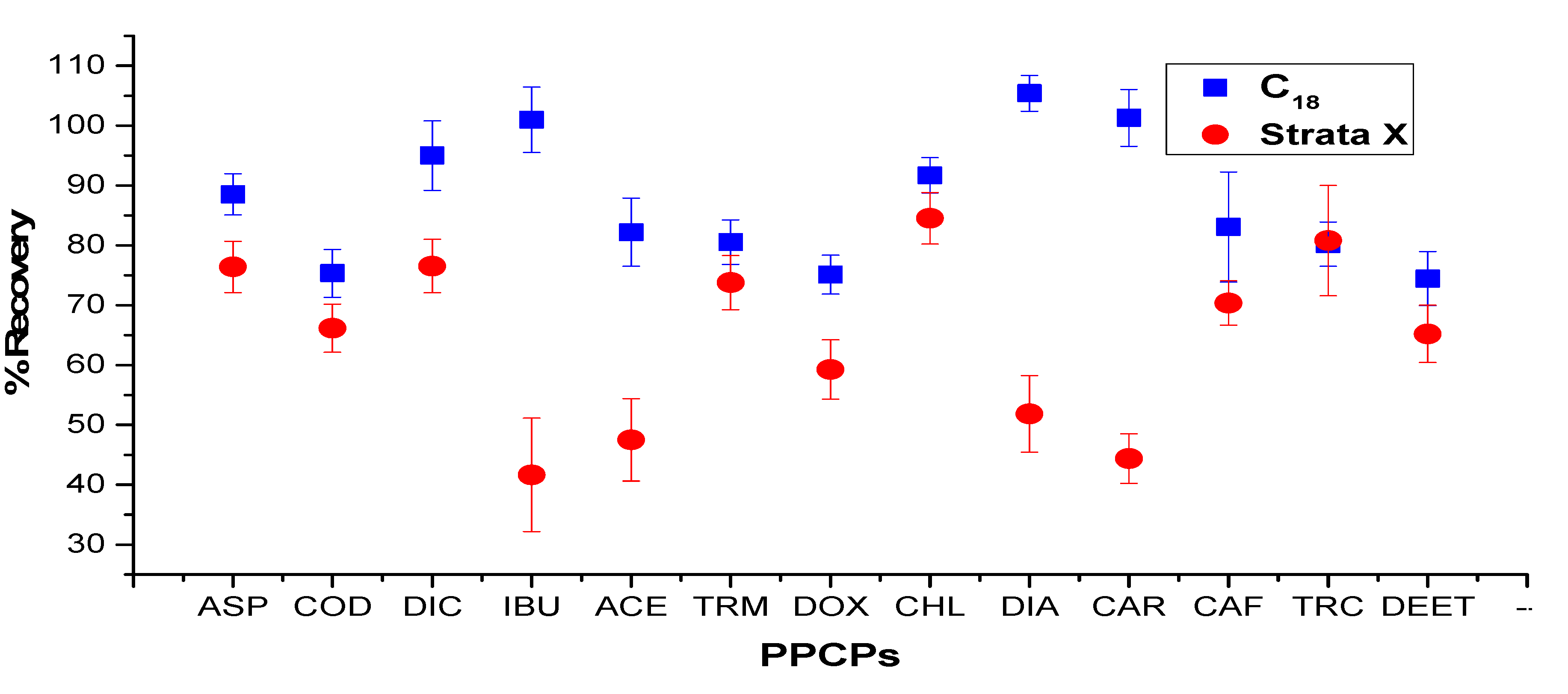
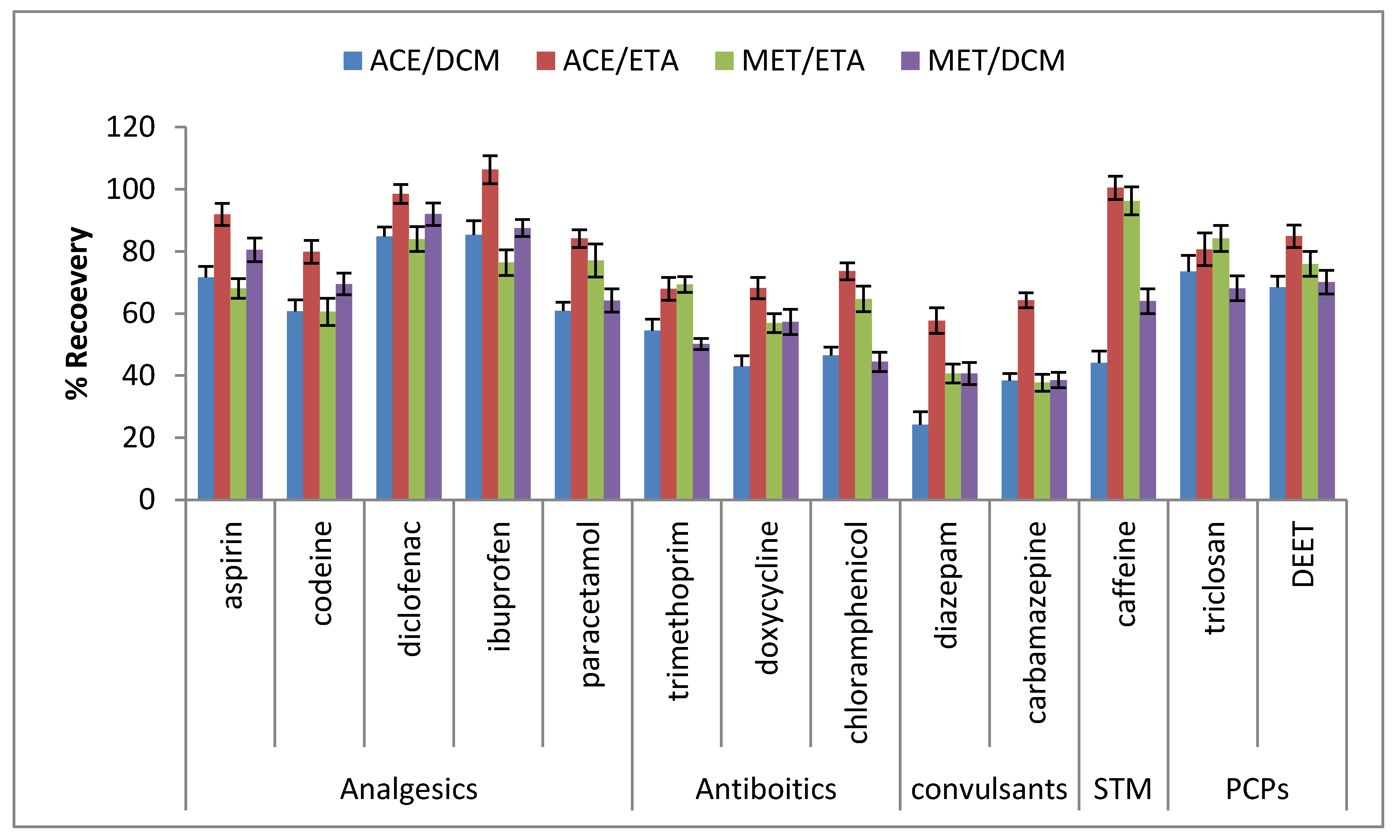
2.3. Optimization of Sonication-Assisted Extraction Procedure
2.4. Validation of Method
3. Experimental Procedures
3.1. Chemicals and Standards
3.2. Sampling
3.3. Extraction of Sludge and Soil Samples and Removal of Interference with Solid-Phase Extraction (SPE)
3.4. Derivatization Procedure
3.5. Gas Chromatography-Mass Spectrophotometry Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daughton, C.G.; Ternes, T.A. Pharmaceuticals and personal care products in the enviroment: Agents of subtle change? Environ. Health Perspect. 1999, 107, 907–938. [Google Scholar] [CrossRef] [PubMed]
- Kolpin, D.W.; Meyer, M.T. Pharmaceuticals, Hormones, and Other Organic Wastewater Contaminants in U.S. Streams, 1999–2000: A National Reconnaissance. Environ. Sci. Technol. 2002, 36, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Fent, K.; Weston, A.A.; Caminada, D. Ecotoxicology of human pharmaceuticals. Aquat. Toxicol. 2006, 76, 122–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Carlson, K. Temporal and spatial trends in the occurrence of human and veterinary antibiotics in aqueous and river sediment matrices. Environ. Sci. Technol. 2007, 41, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Focazio, M.J.; Kolpin, D.W.; Barnes, K.K.; Furlong, E.T.; Meyer, M.T.; Zaugg, S.D.; Barber, L.B.; Thurman, E.M. A national reconnaissance for pharmaceuticals and other organic wastewater contaminants in the United States—II) Untreated drinking water sources. Sci. Total Environ. 2008, 402, 201–216. [Google Scholar] [CrossRef]
- Li, W.; Shi, Y.; Gao, L.; Liu, J.; Cai, Y. Occurrence, distribution and potential affecting factors of antibiotics in sewage sludge of wastewater treatment plants in China. Sci. Total Environ. 2013, 445, 306–313. [Google Scholar] [CrossRef]
- Kosma, C.I.; Lambropoulou, D.A.; Albanis, T.A. Investigation of PPCPs in wastewater treat ment plants in Greece: Occurrence, removal and environmental risk assessment. Sci. Total Environ. 2014, 466–467, 421–438. [Google Scholar] [CrossRef]
- Díaz-Cruz, M.S.; García-Galán, M.J.; Guerra, P.; Jelic, A.; Postigo, C.; Eljarrat, E.; Farré, M.; López de Alda, M.J.; Petrovic, M.; Barceló, D.; et al. Analysis of selected emerging contaminants in sewage sludge. TrAC-Trends Anal. Chem. 2009, 28, 1263–1275. [Google Scholar] [CrossRef]
- Jelic, A.; Gros, M.; Ginebreda, A.; Cespedes-Sánchez, R.; Ventura, F.; Petrovic, M.; Barcelo, D. Occurrence, partition and removal of pharmaceuticals in sewage water and sludge during wastewater treatment. Water Res. 2011, 45, 1165–1176. [Google Scholar] [CrossRef]
- Azzouz, A.; Ballesteros, E. Influence of seasonal climate differences on the pharmaceutical, hormone and personal care product removal efficiency of a drinking water treatment plant. Chemosphere 2013, 93, 2046–2054. [Google Scholar] [CrossRef]
- Sun, Q.; Lv, M.; Hu, A.; Yang, X.; Yu, C.P. Seasonal variation in the occurrence and removal of pharmaceuticals and personal care products in a wastewater treatment plant in Xiamen, China. J. Hazard. Mater. 2014, 277. [Google Scholar] [CrossRef]
- Peng, X.; Yu, Y.; Tang, C.; Tan, J.; Huang, Q.; Wang, Z. Occurrence of steroid estrogens, endocrine-disrupting phenols, and acid pharmaceutical residues in urban riverine water of the Pearl River Delta, South China. Sci. Total Environ. 2008, 397, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Gao, X.; Chen, Y.P.; Peng, X.Y.; Zhang, Y.X.; Gan, X.M.; Zi, C.F.; Guo, J.S. Occurrence, fate and ecotoxicological assessment of pharmaceutically active compounds in wastewater and sludge from wastewater treatment plants in Chongqing, the Three Gorges Reservoir Area. Sci. Total Environ. 2014, 470, 618–630. [Google Scholar] [CrossRef]
- Hurtado, C.; Domínguez, C.; Pérez-Babace, L.; Cañameras, N.; Comas, J.; Bayona, J.M. Estimate of uptake and translocation of emerging organic contaminants from irrigation water concentration in lettuce grown under controlled conditions. J. Hazard. Mater. 2016, 305, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Santana, C.M.; Ferrera, Z.S.; Padrón, M.E.T.; Rodríguez, J.J.S. Methodologies for the extraction of phenolic compounds from environmental samples: New approaches. Molecules 2009, 14, 298–320. [Google Scholar] [CrossRef]
- Azzouz, A.; Ballesteros, E. Combined microwave-assisted extraction and continuous solid-phase extraction prior to gas chromatography-mass spectrometry determination of pharmaceuticals, personal care products and hormones in soils, sediments and sludge. Sci. Total Environ. 2012, 419, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Wang, B.; Huang, J.; Deng, S.; Yu, G. Pharmaceuticals and personal care products in the aquatic environment in China: A review. J. Hazard. Mater. 2013, 262, 189–211. [Google Scholar] [CrossRef]
- Huerta, B.; Rodriguez-Mozaz, S.; Nannou, C.; Nakis, L.; Ruhí, A.; Acuňa, V.; Sabater, S.; Barcelo, D. Determination of a broad spectrum of pharmaceuticals and endocrine disruptors in biofilm from a waste water treatment plant-impacted river. Sci. Total Environ. 2016, 540, 241–249. [Google Scholar] [CrossRef]
- Hiller, E.; Tatarková, V.; Šimonovičová, A.; Bartal’, M. Sorption, desorption, and degradation of (4-chloro-2-methylphenoxy) acetic acid in representative soils of the Danubian Lowland, Slovakia. Chemosphere 2012, 87, 437–444. [Google Scholar] [CrossRef]
- Ramón, A.R. Determination of Emerging Contaminants in Environmental Matrices. Ph.D. Thesis, Technical University of Madrid, Madrid, Spain, 2016; pp. 1–286. [Google Scholar]
- Vazquez-roig, P.; Segarra, R.; Blasco, C.; Andreu, V.; Picó, Y. Determination of pharmaceuticals in soils and sediments by pressurized liquid extraction and liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 2471–2483. [Google Scholar] [CrossRef]
- Chen, F.; Ying, G.G.; Kong, L.X.; Wang, L.; Zhao, J.L.; Zhou, L.J.; Zhang, L.J. Distribution and accumulation of endocrine-disrupting chemicals and pharmaceuticals in wastewater irrigated soils in Hebei, China. Environ. Pollut. 2011, 159, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, L.; Chen, W.; Chang, A.C. Simultaneous determination of pharmaceuticals, endocrine disrupting compounds and hormone in soils by gas chromatography-mass spectrometry. J. Chromatogr. A 2008, 1202, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Gatidou, G.; Thomaidis, N.S.; Stasinakis, A.S.; Lekkas, T.D. Simultaneous determination of the endocrine disrupting compounds nonylphenol, nonylphenol ethoxylates, triclosan and bisphenol A in wastewater and sewage sludge by gas chromatography-mass spectrometry. J. Chromatogr. A 2007, 1138, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Agunbiade:, F.O.; Moodley, B. Occurrence and distribution pattern of acidic pharmaceuticals in surface water, wastewater, and sediment of the Msunduzi River, Kwazulu-Natal, South Africa. Environ. Toxicol. Chem. 2016, 35, 36–46. [Google Scholar] [CrossRef]
- Azzouz, A.; Ballesteros, E. Determination of 13 endocrine disrupting chemicals in environmental solid samples using microwave-assisted solvent extraction and continuous solid-phase extraction followed by gas chromatography-mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 231–241. [Google Scholar] [CrossRef]
- Alvarino, T.; Suarez, S.; Katsou, E.; Vazquez-Padin, J.; Lema, J.M.; Omil, F. Removal of PPCPs from the sludge supernatant in a one stage nitritation/anam mox process. Water Res. 2015, 68, 701–709. [Google Scholar] [CrossRef]
- Peng, X.; Wang, Z.; Yang, C.; Chen, F.; Mai, B. Simultaneous determination of endocrine-disrupting phenols and steroid estrogens in sediment by gas chromatography-mass spectrometry. J. Chromatogr. A 2006, 1116, 51–56. [Google Scholar] [CrossRef]
- Vazquez-roig, P.; Andreu, V.; Blasco, C.; Picó, Y. Science of the Total Environment Risk assessment on the presence of pharmaceuticals in sediments, soils and waters of the Pego–Oliva Marshlands (Valencia, eastern Spain). Sci. Total Environ. 2012, 440, 24–32. [Google Scholar] [CrossRef]
- Dobor, J.; Varga, M.; Yao, J.; Chen, H.; Palkó, G.; Záray, G. A new sample preparation method for determination of acidic drugs in sewage sludge applying microwave assisted solvent extraction followed by gas chromatography–mass spectrometry. Microchem. J. 2010, 94, 36–41. [Google Scholar] [CrossRef]
- Sun, Q.; Li, M.; Ma, C.; Chen, X.; Xie, X.; Yu, C.P. Seasonal and spatial variations of PPCP occurrence, removal and mass loading in three wastewater treatment plants located in different urbanization areas in Xiamen, China. Environ. Pollut. 2016, 208, 371–381. [Google Scholar] [CrossRef]
- Loli, A.; Paíga, P.; Santos, L.H.M.L.M.; Ramos, S.; Correia, M.; Delerue-matos, C. Assessment of non-steroidal anti-in fl ammatory and analgesic pharmaceuticals in seawaters of North of Portugal: Occurrence and environmental risk. Sci. Total Environ. 2015, 508, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Golovko, O.; Kumar, V.; Fedorova, G.; Randak, T.; Grabic, R. Seasonal changes in antibiotics, antidepressants/psychiatric drugs, antihistamines and lipid regulators in a wastewater treatment plant. Chemosphere 2014, 111, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.R.; Shanmugam, G.; Velu, G.; Rengarajan, B.; Larsson, D.G.J. GC-MS analysis and ecotoxicological risk assessment of triclosan, carbamazepine and parabens in Indian rivers. J. Hazard. Mater. 2011, 186, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yamashita, N.; Tanaka, H.; Matsukawa, H.; Tanabe, K. Development of extraction method of pharmaceuticals and their occurrences found in Japanese wastewater treatment plants. Environ. Int. 2009, 35, 815–820. [Google Scholar] [CrossRef]
- Hibberd, A.; Maskaoui, K.; Zhang, Z.; Zhou, J.L. An improved method for the simultaneous analysis of phenolic and steroidal estrogens in water and sediment. Talanta 2009, 77, 1315–1321. [Google Scholar] [CrossRef]
- Azzouz, A.; Souhail, B.; Ballesteros, E. Continuous solid-phase extraction and gas chromatography-mass spectrometry determination of pharmaceuticals and hormones in water samples. J. Chromatogr. A 2010, 1217, 2956–2963. [Google Scholar] [CrossRef]
- Moldovan, Z. Occurrences of pharmaceutical and personal care products as micropollutants in rivers from Romania. Chemosphere 2006, 64, 1808–1817. [Google Scholar] [CrossRef]
- Bisceglia, K.J.; Yu, J.T.; Coelhan, M.; Bouwer, E.J.; Roberts, A.L. Trace determination of pharmaceuticals and other wastewater-derived micropollutants by solid phase extraction and gas chromatography/mass spectrometry. J. Chromatogr. A 2010, 1217, 558–564. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, L. Analysis of endocrine disrupting compounds, pharmaceuticals and personal care products in sewage sludge by gas chromatography-mass spectrometry. Talanta 2012, 89, 58–263. [Google Scholar] [CrossRef]
- Azzouz, A.; Ballesteros, E. Trace analysis of endocrine disrupting compounds in environmental water samples by use of solid-phase extraction and gas chromatography with mass spectrometry detection. J. Chromatogr. A 2014, 1360, 248–257. [Google Scholar] [CrossRef]
- Li, X.; Zheng, W.; Kelly, W.R. Occurrence and removal of pharmaceutical and hormone contaminants in rural wastewater treatment lagoons. Sci. Total Environ. 2013, 445, 22–28. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Range (ng·g−1) | R2 | Soils (ng·g−1) | Sludge (ng·g−1) | Others Findings in Soils (ng·g−1) | Others Finding in Sludge (ng·g−1) | Recovery (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LOD | LOQ | LOD | LOQ | LOD | LOQ | LOD | LOQ | sludge | soil | |||
| Aspirin | 10–400 | 0.9949 | 0.1 | 0.3 | 0.2 | 0.5 | 0.14 * | 0.38 * | 1.1 * | 3.6 * | 89 ± 5 * | 103 ± 4.2 * |
| Codeine | 10–400 | 0.9994 | 0.3 | 0.8 | 0.1 | 0.3 | na | na | na | na | 98 ± 4 ** | 91 ± 13 ** |
| Diclofenac | 10–400 | 0.9919 | 0.5 | 1.6 | 0.8 | 2.6 | 0.16 * | 0.48 * | 0.7 * | 2.3 * | 98 ± 7 * | 104.4 ± 3.3 * |
| Ibuprofen | 10–400 | 0.9994 | 0.2 | 0.6 | 0.2 | 0.5 | 0.07 * | 0.21 * | 1.0 * | 3.3 * | 95 ± 4 * | 104.4 ± 3.4 * |
| Paracetamol | 10–400 | 0.9922 | 0.6 | 1.9 | 0.9 | 2.8 | 0.07 * | 0.24 * | 2.5 * | 8.3 * | 92 ± 13 * | 86.2 ± 4.7 * |
| Chloramphenicol | 10–400 | 0.9945 | 0.4 | 1.2 | 0.7 | 1.7 | 0.8 **** | 2.7 **** | na | na | 93 ± 5 **** | 96 ± 5 **** |
| Doxycycline | 10–400 | 0.9991 | 1.4 | 4.6 | 1.7 | 5.1 | 0.80 *** | 2.67 *** | na | na | 68 ± 8 ** | 62 ± 10 ** |
| Trimethoprim | 10–400 | 0.9922 | 0.2 | 0.6 | 0.1 | 0.4 | 0.64 *** | 2.15 *** | na | na | 97 ± 7 ** | 105 ± 5 ** |
| Caffeine | 10–400 | 0.9959 | 0.3 | 1.1 | 0.2 | 0.6 | 0.09 * | 2.1 * | 1.7 * | 5.5 | 99 ± 11 * | 98 ± 6 * |
| Carbamazepine | 10–400 | 0.9985 | 0.3 | 1.0 | 0.6 | 1.7 | 0.16 * | 0.44 * | 1.5 | 5.0 | 98 ± 7 * | 82 ± 10 * |
| Diazepam | 10–400 | 0.9984 | 0.4 | 1.3 | 0.7 | 2.2 | na | na | na | na | 85 ± 3 ** | 79 ± 8 ** |
| DEET | 10–400 | 0.9975 | 0.5 | 1.5 | 1.2 | 3.3 | 0.89 * | 1.48 * | 0.58 * | 1.31 * | 86 ± 5 * | 89 ± 6 * |
| Triclosan | 10–400 | 0.9916 | 0.3 | 0.9 | 0.4 | 1.1 | 0.1 * | 0.3 * | 2.1 * | 7.2 * | 91 ± 4 * | 91 ± 6.6 * |
| Compounds | Leveling Spiking (Soils) Recovery (%) | Leveling Spiking (Sludge) Recovery (%) | ||||
|---|---|---|---|---|---|---|
| 50 ng | 100 ng | 200 ng | 50 ng | 100 ng | 200 ng | |
| Aspirin | 88.5 ± 5.1 | 95.4 ± 8.1 | 81.6 ± 7.2 | 78.3 ± 4.5 | 74.4 ± 6.4 | 82.3 ± 5.8 |
| Codeine | 71.2 ± 4.3 | 68.4 ± 3.8 | 75.5 ± 5.3 | 64.7 ± 3.6 | 67.8 ± 3.9 | 71.3 ± 3.9 |
| Diclofenac | 101.5 ± 6.3 | 106 ± 2.8 | 106 ± 6.4 | 88.5 ± 5.7 | 84.1 ± 4.3 | 89.4 ± 5.6 |
| Ibuprofen | 101 ± 7.3 | 96.8 ± 7.6 | 89.2 ± 5.4 | 87.2 ± 2.0 | 89.4 ± 4.7 | 78.5 ± 6.4 |
| Paracetamol | 88.4 ± 4.5 | 82.3 ± 3.2 | 76.9 ± 2.2 | 67.5 ± 3.2 | 62 ± 2.1 | 69.5 ± 3.4 |
| Chloramphenicol | 54.5 ± 4.8 | 62.7 ± 5.1 | 67.4 ± 2.7 | 55.4 ± 2.5 | 63.5 ± 5.5 | 66.4 ± 8.6 |
| Doxycycline | 71.1 ± 3.1 | 77.3 ± 3.4 | 79.5 ± 7.8 | 67.5 ± 5.2 | 72.6 ± 7.8 | 65.4 ± 4.5 |
| Trimethoprim | 54.5 ± 4.3 | 63.6 ± 8.1 | 49.8 ± 7.2 | 51.5 ± 3.3 | 55 ± 3.2 | 49.6 ± 2.1 |
| Caffeine | 89.6 ± 3.6 | 94.6 ± 1.9 | 102.7 ± 2.9 | 76.5 ± 2.5 | 77.9 ± 4.1 | 69.2 ± 1.8 |
| Carbamazepine | 56.6 ± 5.0 | 53.1 ± 3.4 | 60.7 ± 4.9 | 61.5 ± 4.9 | 63.2 ± 2.1 | 59.5 ± 1.1 |
| Diazepam | 45.6 ± 4.2 | 48.5 ± 3.8 | 53 ± 5.0 | 57.5 ± 4.4 | 60.1 ± 5.1 | 61.4 ± 4.2 |
| DEET | 60.4 ± 4.1 | 65.3 ± 3.3 | 70.2 ± 2.3 | 56.8 ± 4.1 | 53.1 ± 2.9 | 64.3 ± 7.6 |
| Triclosan | 76.2 ± 3.1 | 83.5 ± 6.9 | 72.6 ± 3.0 | 70.5 ± 4.5 | 74.5 ± 5.5 | 67.8 ± 3.2 |
| Compounds | Soil | Sludge | ||
|---|---|---|---|---|
| Repeatability RSD (%) n = 6 | Intermediate Precision RSD (%) n = 3, k = 3 | Repeatability RSD (%) n = 6 | Intermediate Precision RSD (%) n = 3, k = 3 | |
| Aspirin | 3.0 | 4.5 | 3.5 | 4.5 |
| Codeine | 8.5 | 9.0 | 11 | 13 |
| Diclofenac | 4.0 | 5.0 | 4.0 | 5.5 |
| Ibuprofen | 3.5 | 5.5 | 3.3 | 5.8 |
| Paracetamol | 2.0 | 3.0 | 2.0 | 3.5 |
| Chloramphenicol | 10 | 11.5 | 12 | 13 |
| D oxycycline | 8.5 | 9.5 | 9.2 | 9.5 |
| Trimethoprim | 7.0 | 8.5 | 8.5 | 9.0 |
| Caffeine | 5.0 | 5.5 | 5.5 | 8.0 |
| Carbamazepine | 5.0 | 6.0 | 4.5 | 6.0 |
| Diazepam | 4.5 | 5.0 | 5.0 | 7.5 |
| DEET | 3.5 | 4.5 | 4.4 | 4.5 |
| Triclosan | 3.0 | 4.5 | 4.0 | 5.7 |
| Therapeutic Groups/Abbreviation | Compounds/IUPAC Names | Structures | |
|---|---|---|---|
| 1 | Analgesic/anti-inflammatory/ASP | Aspirin/2-Acetoxybenzoic acid |  |
| COD | Codeine/ (5a, 6a) -3-Methoxy-17-methyl-7 |  | |
| DIC | Diclofenac/2-[2-(2,6-dichloroaniline) phenyl] acetic acid |  | |
| IBU | Ibuprofen/2, (4-isobutylphenyl) propanoic acid |  | |
| ACE | Paracetamol/N-(4-hydroxphenyl) ethanamide |  | |
| 2 | Antibiotics/CHL | Chloramphenicol/2,2-dichloro-N-(1,3-dihydroxy-1-) 4-nitrophe |  |
| DOX | Doxycycline/Pentahyroxy-6-methyl-1,11-dioxo |  | |
| TRM | Trimethoprim/5- (3,4,5-trimethoxybenzyl)-2, 4-pyrimidinediamine |  | |
| 3 | Anticonvulsants/CAR | Carbamazepine/5H-Dibenzo [b,f] azepine-5-carboxamide |  |
| DIA | Diazepam/7-chloro-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzediazepin 2-one |  | |
| 4 | Stimulant/CAF | Caffeine/1,3,7-trimethylpurine-2,6-dione |  |
| 5 | PCPs/TRC | Triclosan/5-chloro-2- (2,4-dichlorophenoxyl) phenol |  |
| DEET | DEET/N,N-Diethyl-meta-toluamide |  |
| Acetylation | Silylation | |||||||
|---|---|---|---|---|---|---|---|---|
| Compound Names | RT (RSD%) | CAS Number | MW: m/z Ions | Pka: logkd | Compound Names | RT (RSD%) | CAS Number | MW: m/z Ions |
| Analgesic/Anti-Inflammatories | ||||||||
| Aspirin | 10.470 (0.9) | 00050-78-2 | 180:120, 180, 43 | 3.5:1.19 | Aspirin | 10.412 (0.2) | 00050-78-2 | 252:120, 115, 210 |
| 2H-indol-2-one | 19.382 (0.77) | 015362-40-0 | 277:214, 242, 277 | 4.14:4.51 | Diclofenac | 21.256 (0.3) | 959106-20-8 | 367:214, 242 |
| Ibuprofen | 11.77 (0.8) | 061566-34 | 206:109, 161, 206 | 4.91:3.97 | Ibuprofen | 11.292 (0.3) | 015687-27-1 | 278:109, 161, 234 |
| Morpian-6-ol | 24.663 (0.6) | 006703-27-1 | 341:341, 282, 229 | 5.0:0.48 | Codeine | 23.840 (0.5) | 074367-14-9 | 299:299, 162, 229 |
| Acetaminophen | 13.737 (0.9) | 000103-90-2 | 194:109, 151, 194 | 9.38:0.46 | Paracetamol | 13.423 (0.4) | 041571-82-8 | 295:116, 206, 280 |
| Antibiotics | ||||||||
| Chloramphenicol | n.d. | 9.61:n/a | Chloramphenicol di (trimethylsily) | 24.988 (0.4) | 1000386-63-9 | 466, 225, 208, 242 | ||
| Trimethoprim | n.d. | 1.5:0.59 | Trimethoprim | 25.560 (0.3) | 000738 | 290:290, 259, 275 | ||
| Doxycycline | n.d. | Doxycycline | 15.992 (0.4) | 270:167, 255, 58.1 | ||||
| Anticonvulsants | ||||||||
| Diazepam | 23.734 (0.9) | 00439-14-5 | 284:256, 283, 221 | 0.10:2.8 | Diazepam | 23.733 (0.4) | 00439-14-5 | 284:256, 283, 221 |
| 5-Acety-5H-dibenz (b,f) azepine | 18.99 (0.9) | 015362-40-0 | 235:193, 235, 165 | 7:2.47 | Carbamazepine | 20.993 (0.3) | 000298-46-4 | 308:193, 235, 293 |
| Stimulant | ||||||||
| Caffeine | 14.612 (0.9) | 00058-08-2 | 194:194, 109, 67 | 2.0:−0.63 | Caffeine | 14.809 (0.3) | 00058-08-2 | 194:194, 109, 67 |
| PCPs | ||||||||
| Phenol, 5-chloro-2- | 19.522 (0.98) | 004623 | 330:288, 218, 146 | −0.95:4.8 | Triclosan | 19.499 (0.3) | 003380-34-5 | 362:347, 218, 310 |
| DEET | 10.663 (0.9) | 000134-62-3 | 199:190, 119, 91 | DEET | 10.659 (0.4) | 000134-62-3 | 199, 190, 119, 91 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ademoyegun, O.T.; Okoh, O.O.; Okoh, A.I. Method Validation and Investigation of the Levels of Pharmaceuticals and Personal Care Products in Sludge of Wastewater Treatment Plants and Soils of Irrigated Golf Course. Molecules 2020, 25, 3114. https://doi.org/10.3390/molecules25143114
Ademoyegun OT, Okoh OO, Okoh AI. Method Validation and Investigation of the Levels of Pharmaceuticals and Personal Care Products in Sludge of Wastewater Treatment Plants and Soils of Irrigated Golf Course. Molecules. 2020; 25(14):3114. https://doi.org/10.3390/molecules25143114
Chicago/Turabian StyleAdemoyegun, Olufemi Temitope, Omobola Oluranti Okoh, and Anthony Ifeanyi Okoh. 2020. "Method Validation and Investigation of the Levels of Pharmaceuticals and Personal Care Products in Sludge of Wastewater Treatment Plants and Soils of Irrigated Golf Course" Molecules 25, no. 14: 3114. https://doi.org/10.3390/molecules25143114
APA StyleAdemoyegun, O. T., Okoh, O. O., & Okoh, A. I. (2020). Method Validation and Investigation of the Levels of Pharmaceuticals and Personal Care Products in Sludge of Wastewater Treatment Plants and Soils of Irrigated Golf Course. Molecules, 25(14), 3114. https://doi.org/10.3390/molecules25143114