
Antimicrobial Activity of Different Artemisia Essential Oil Formulations

 ,
,  ,
, 
 , and
, and
Abstract
1. Introduction
2. Results
2.1. Artemisia Essential Oil and Its Components
2.2. Preparation and Stability Studies of O/W Type Pickering Nanoemulsions
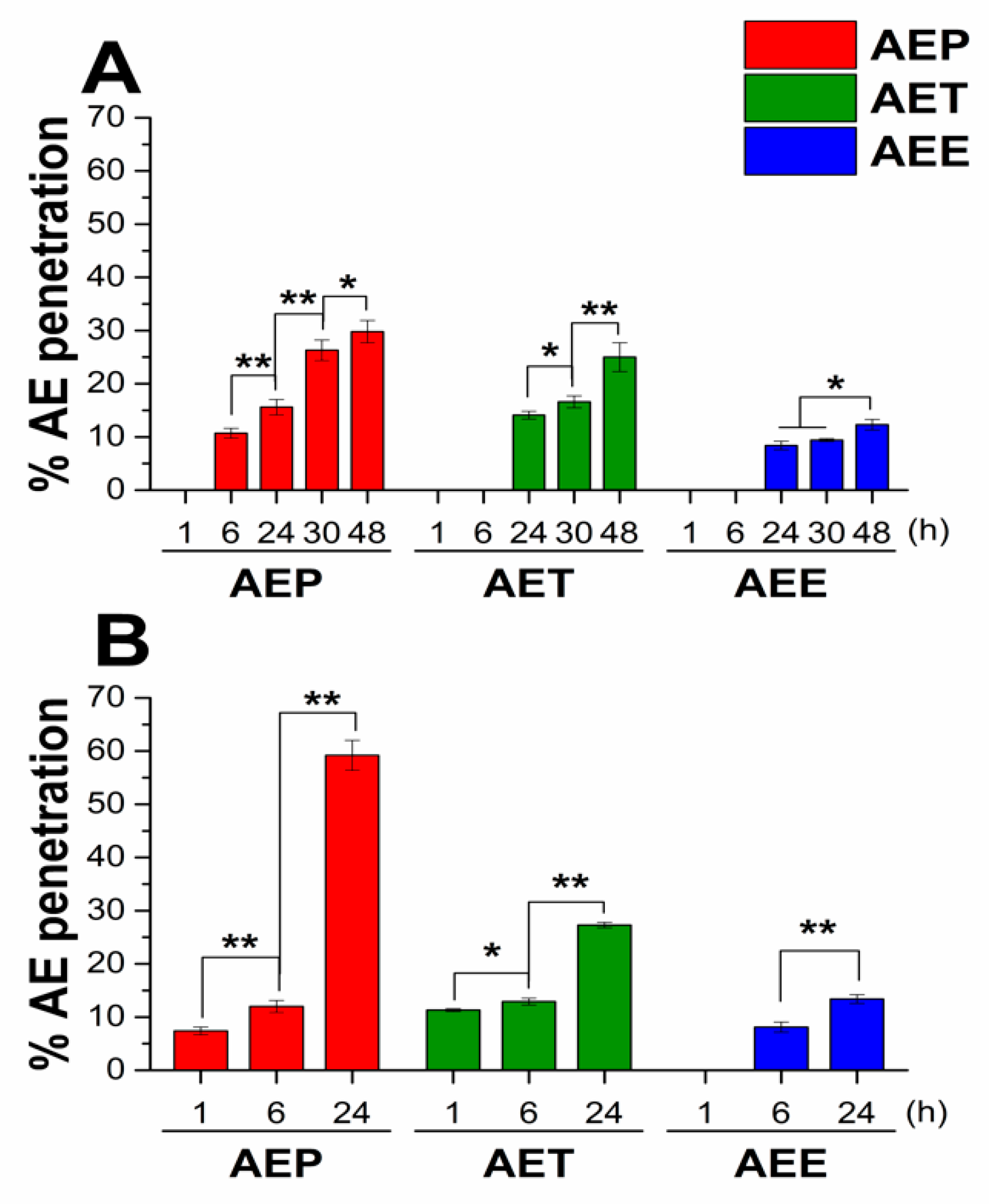
2.3. In Vitro Diffusion Study of Artemisia EO (AE) Formulations
2.4. Interaction Studies with Unilamellar Liposomes as Cellular Models
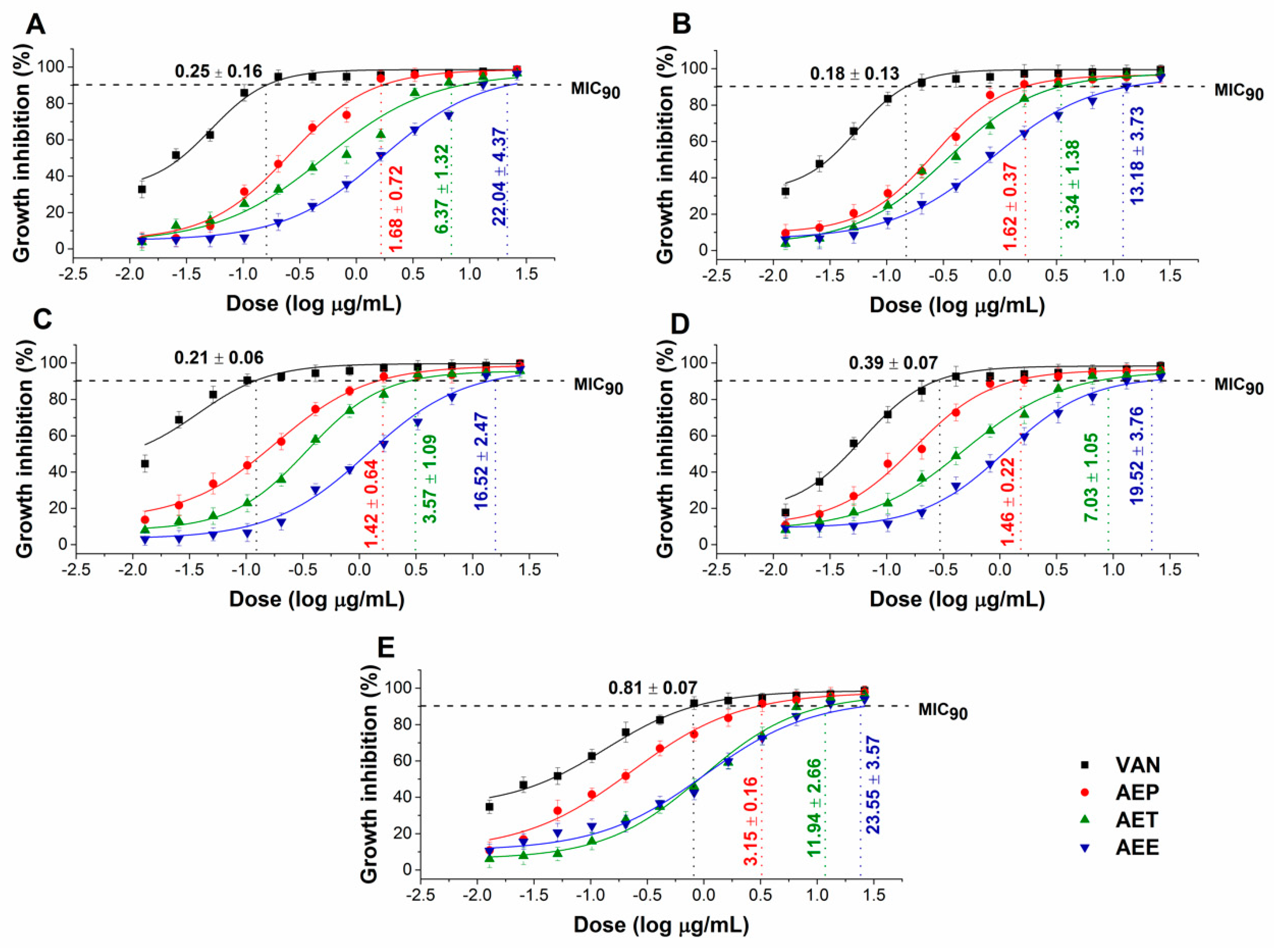
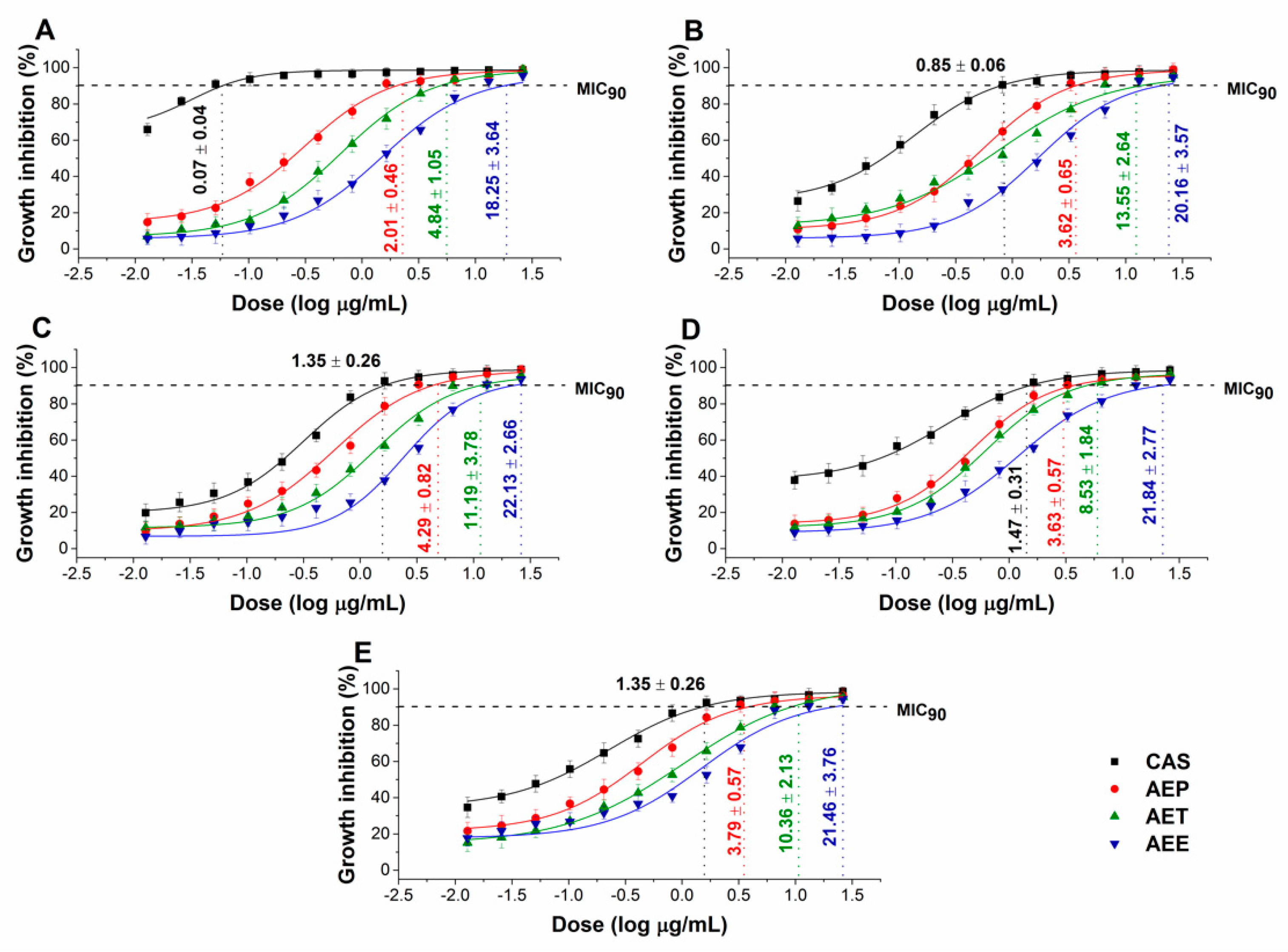
2.5. Antibacterial and Antifungal Activities of the Prepared Emulsions
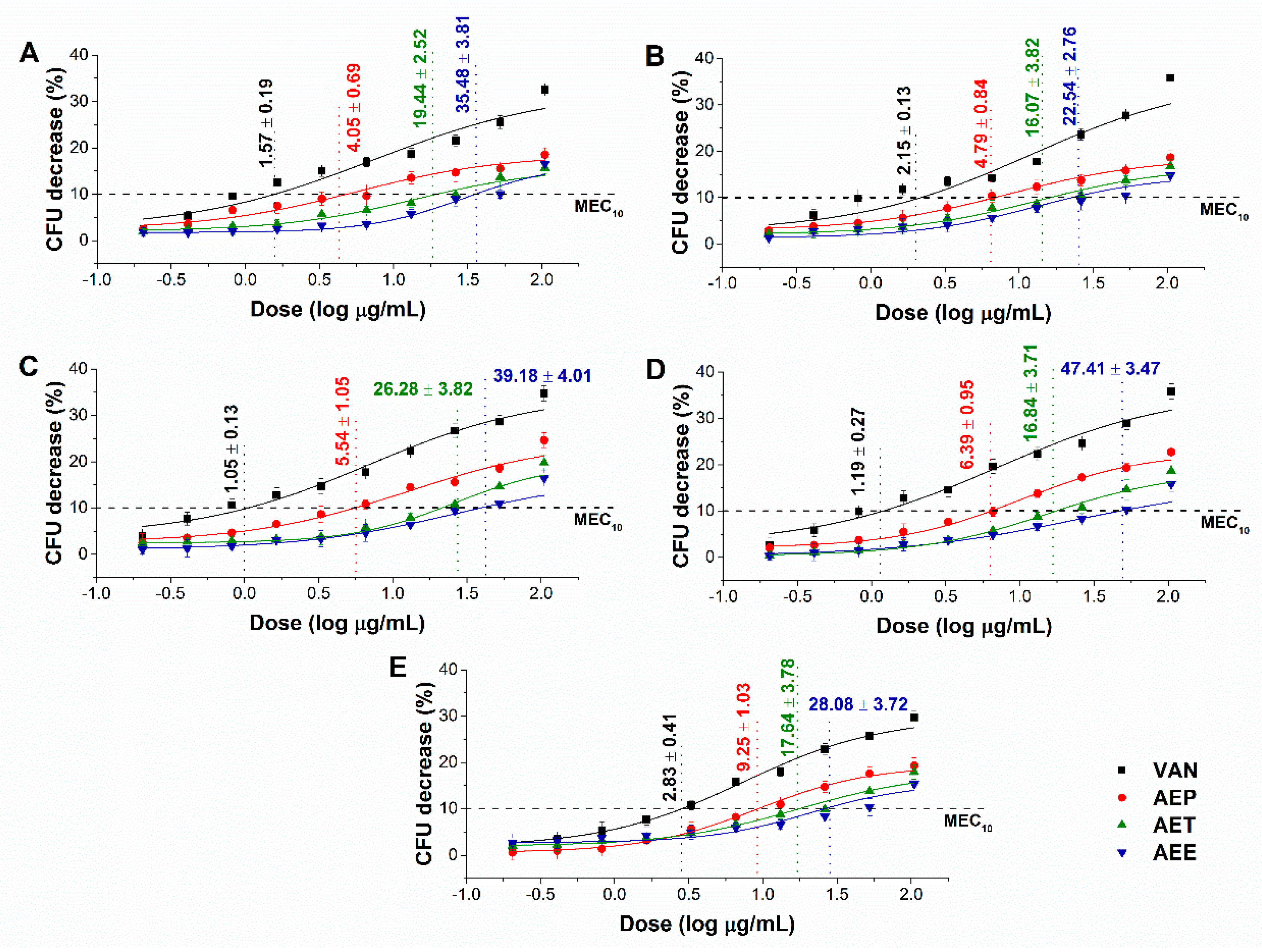
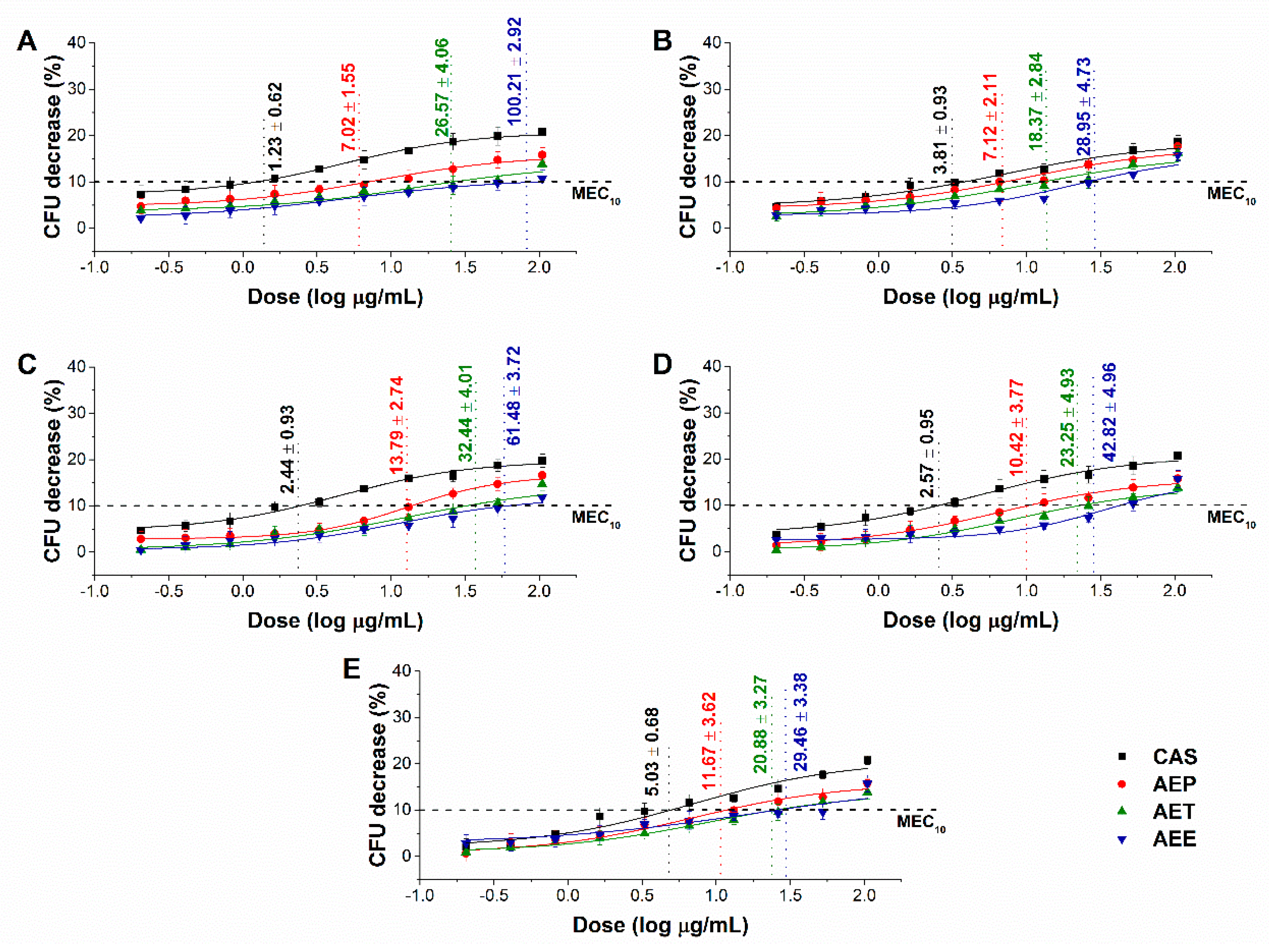
2.6. Effects of the Minimum Effective Concentration (MEC10) on Planktonic Microbial Cells
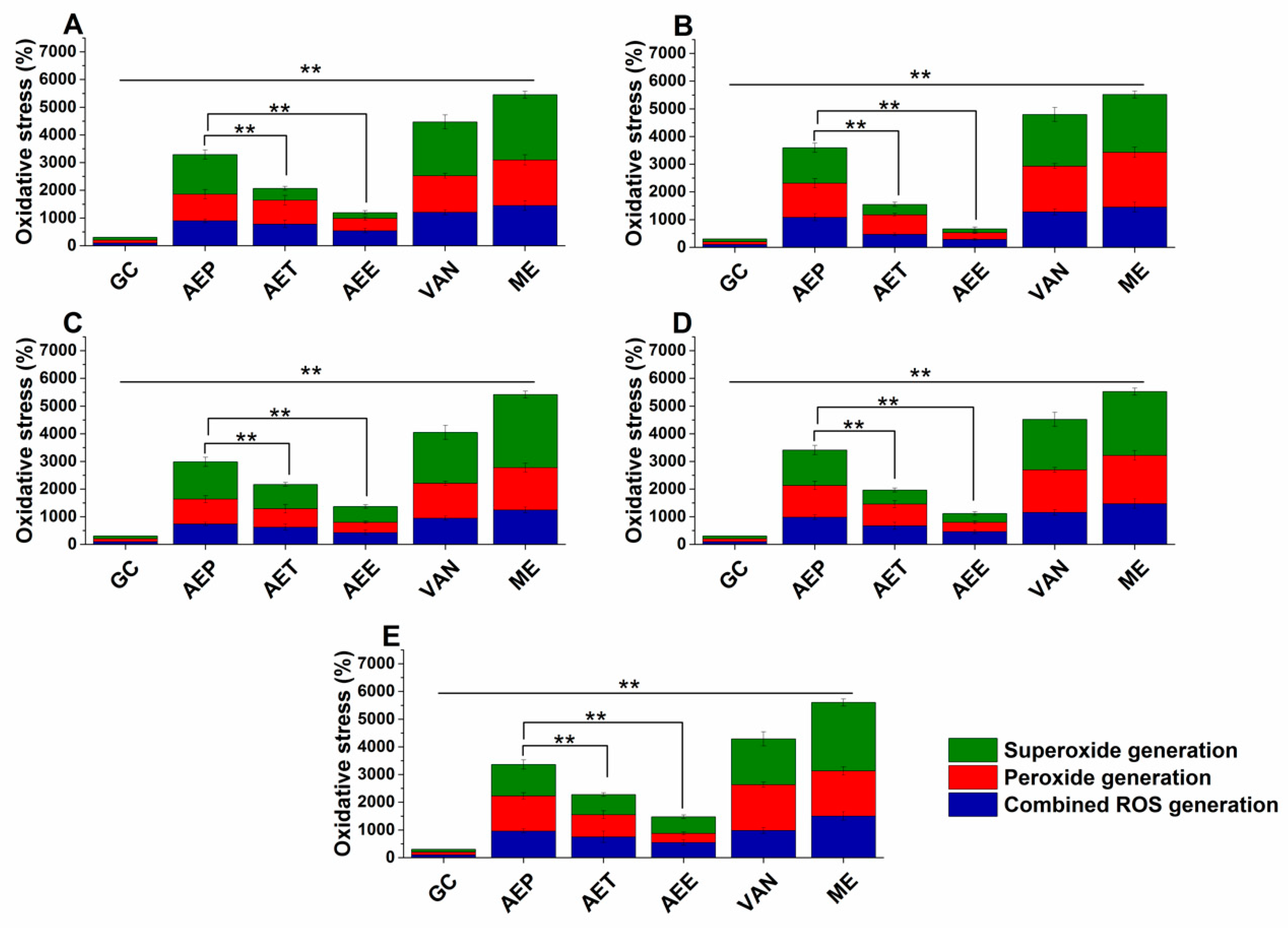
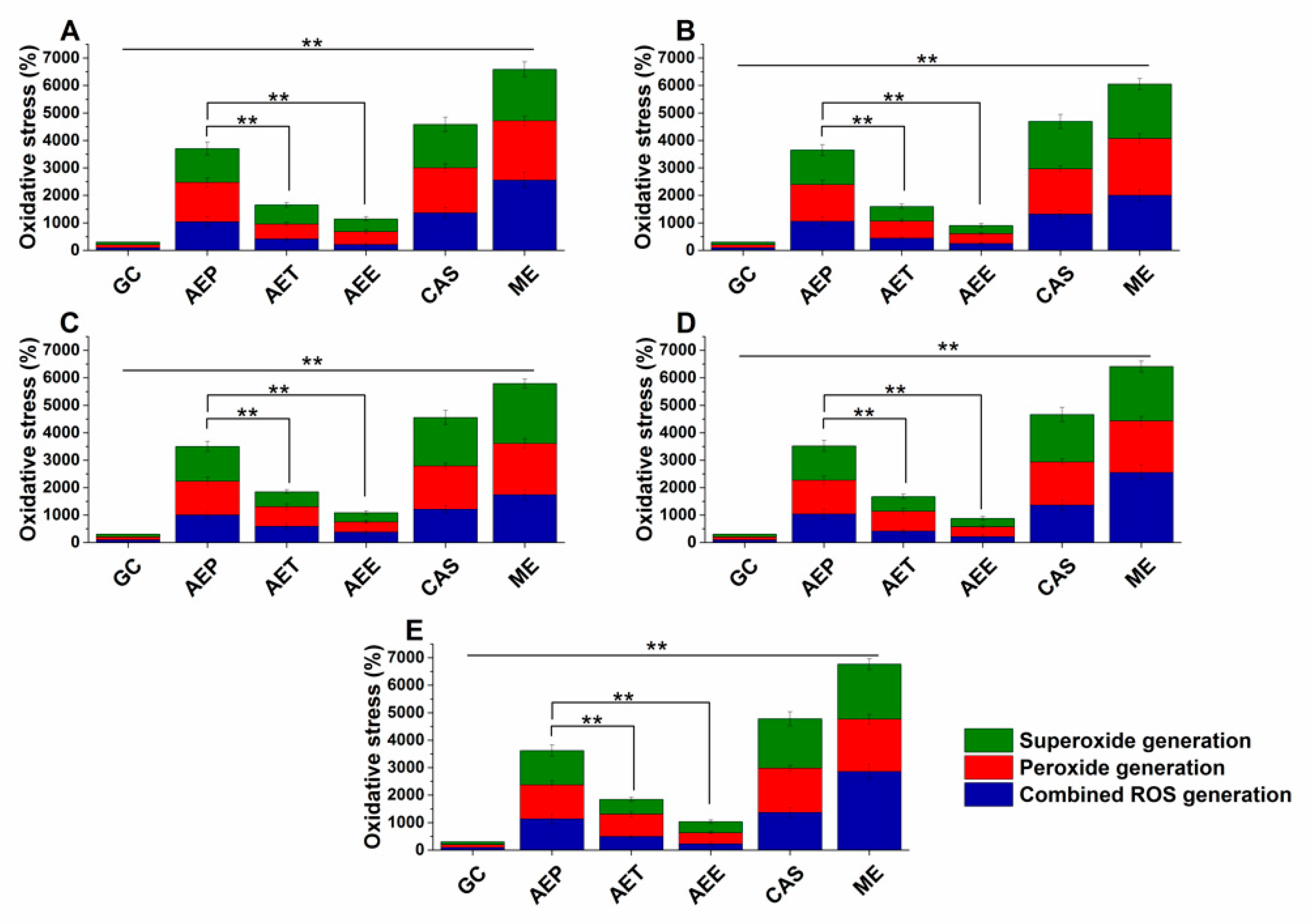
2.7. Effects on the Microbial Oxidative Balance
2.8. Effects on the Microbial Planktonics’ Behavior
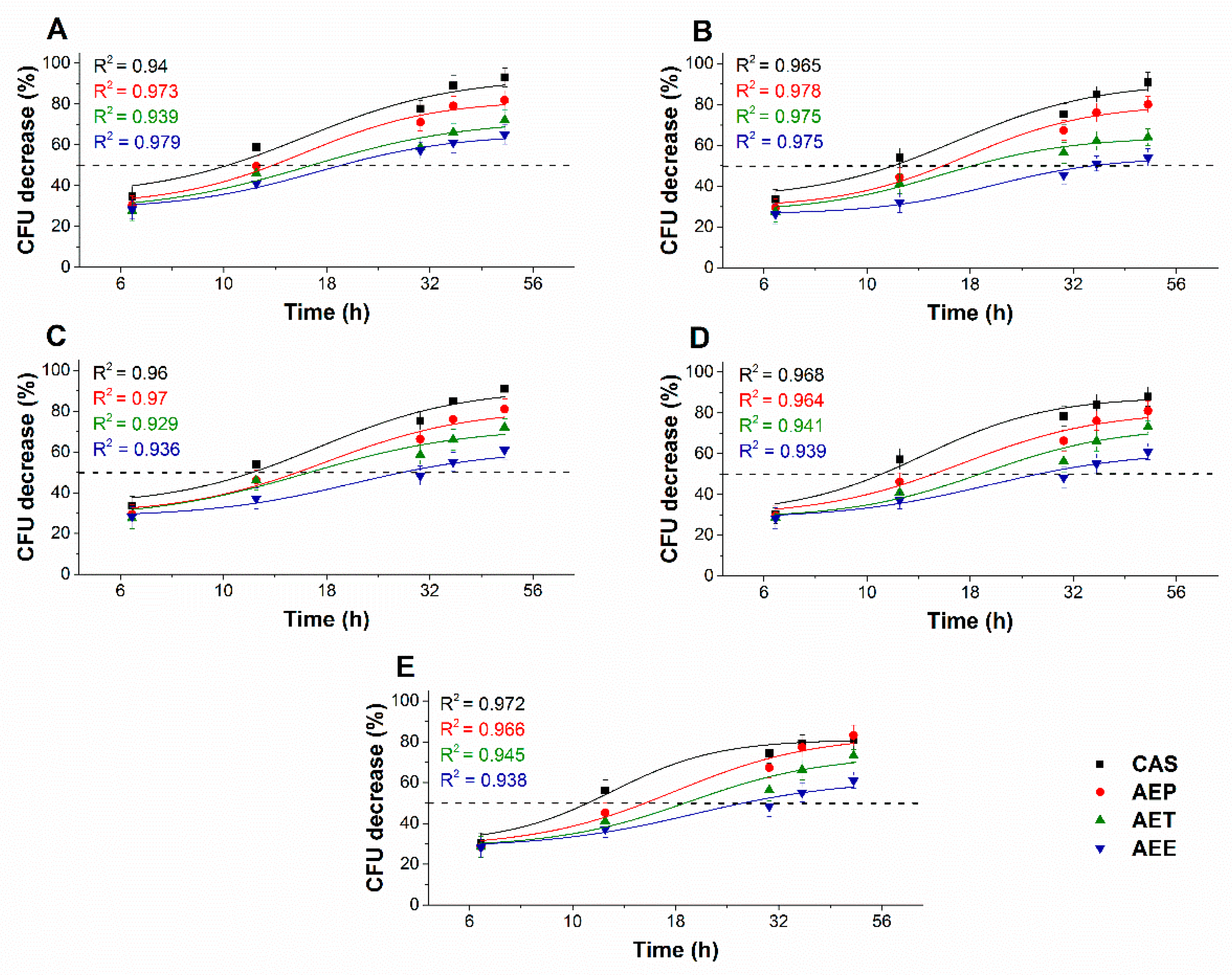
2.8.1. Colony Formation Changes
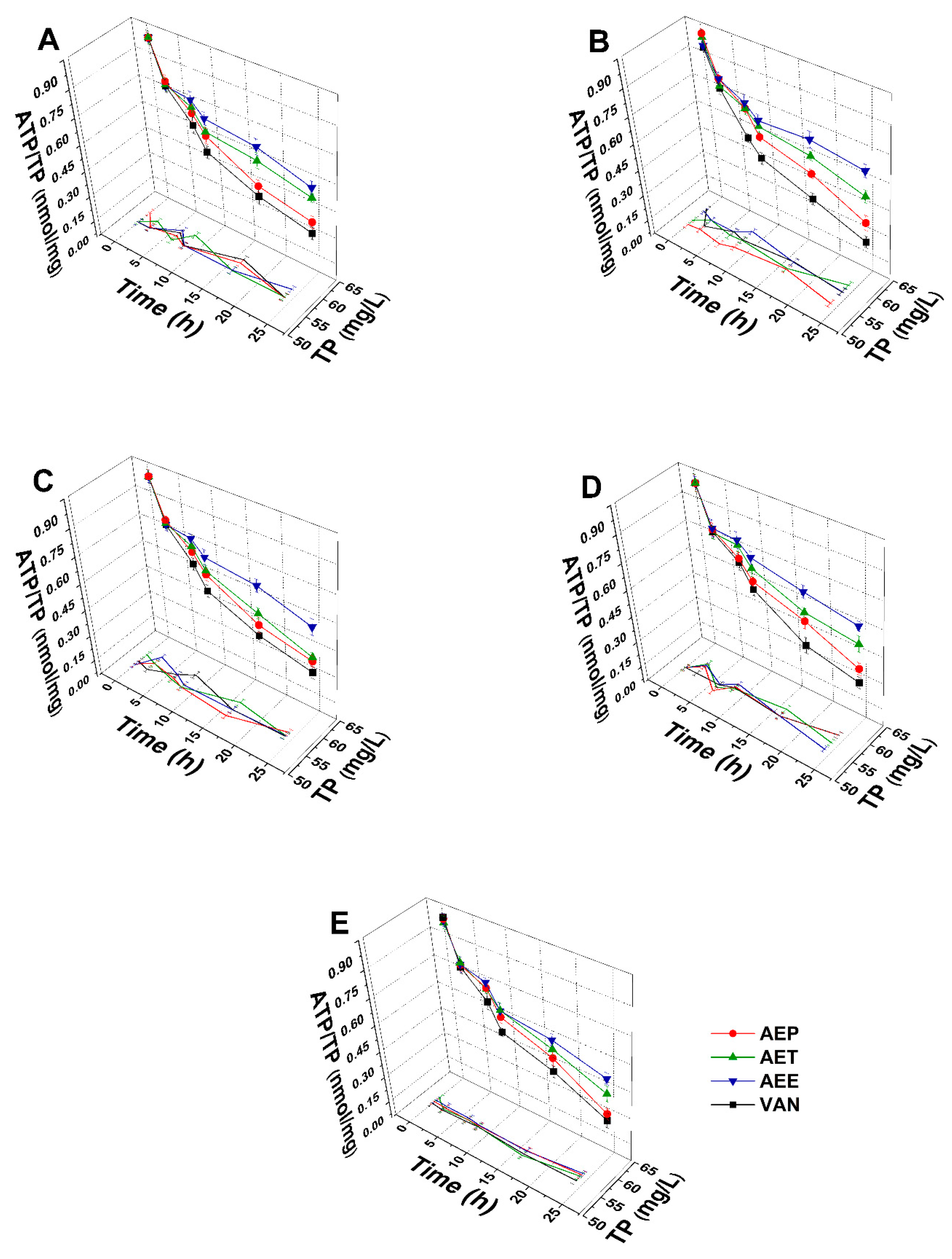
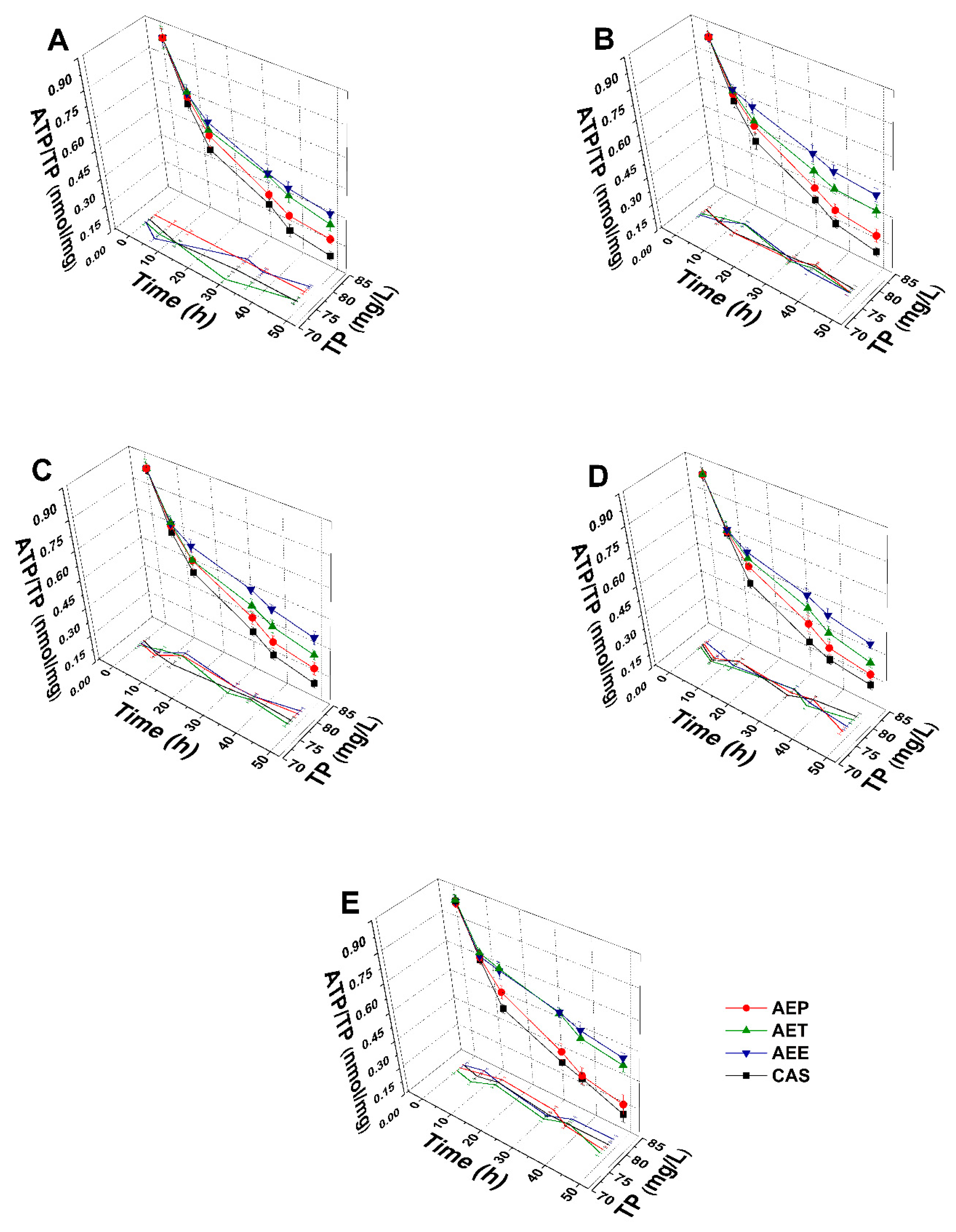
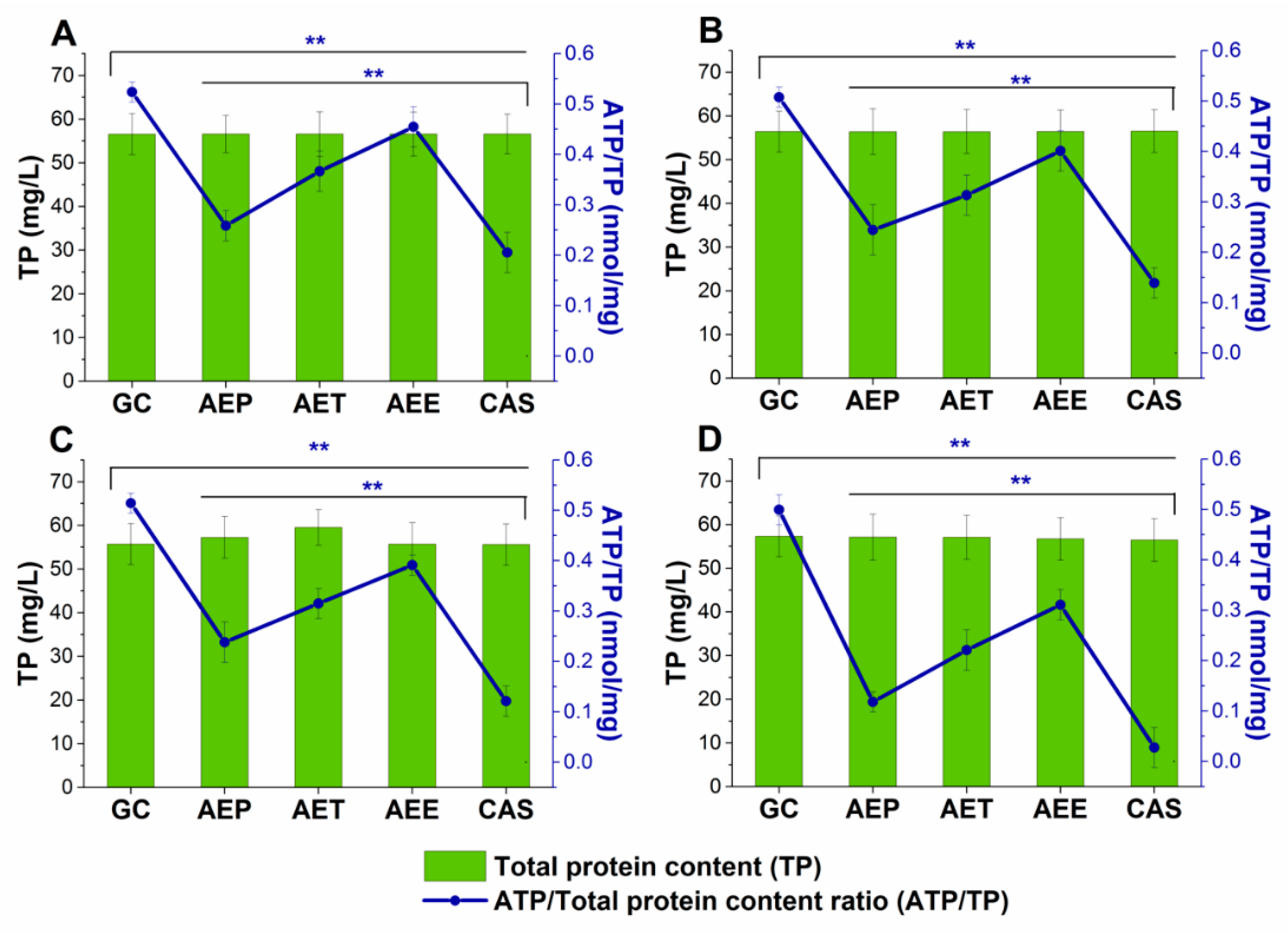
2.8.2. Variations in the Intracellular ATP to Total Protein Content Ratios
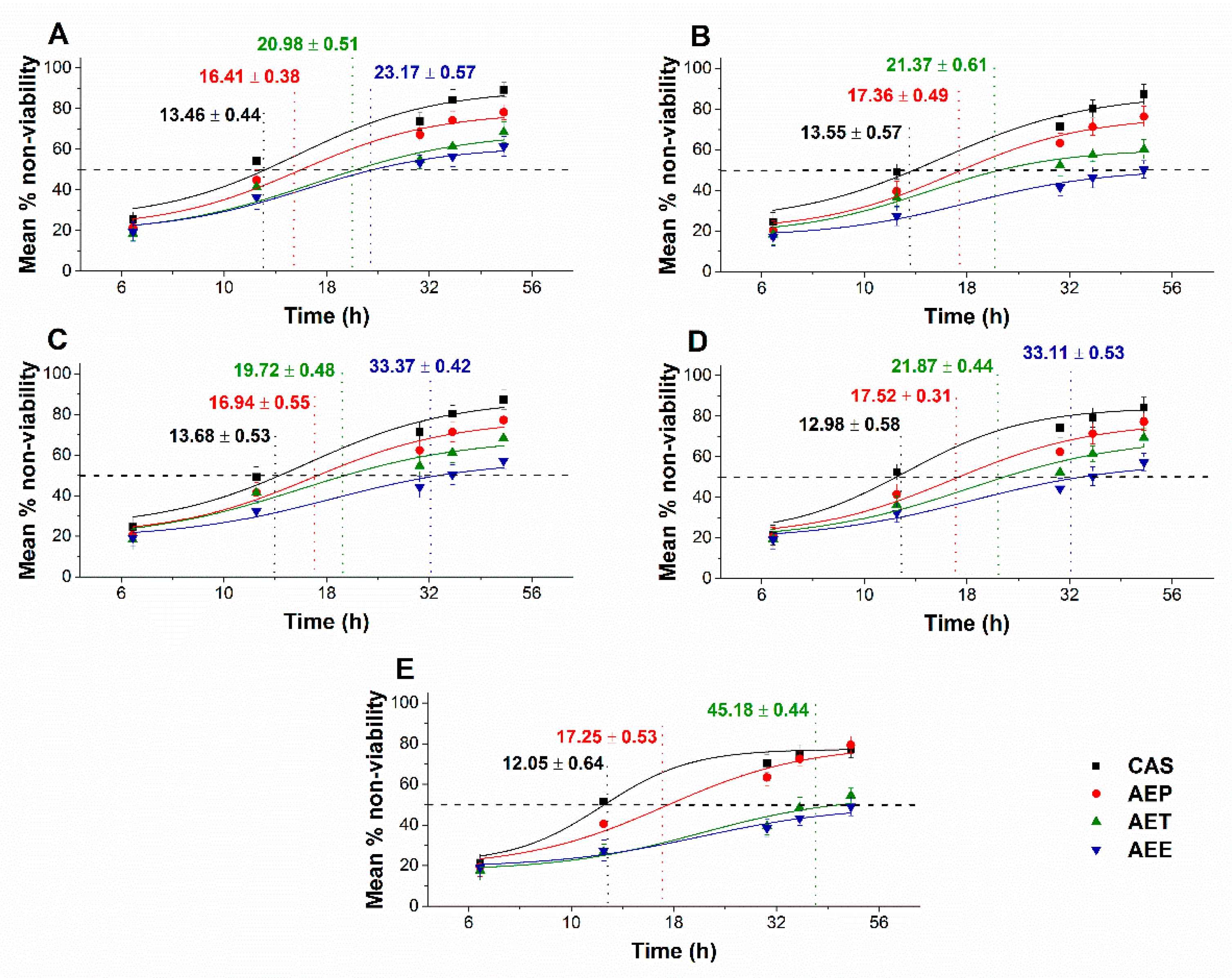
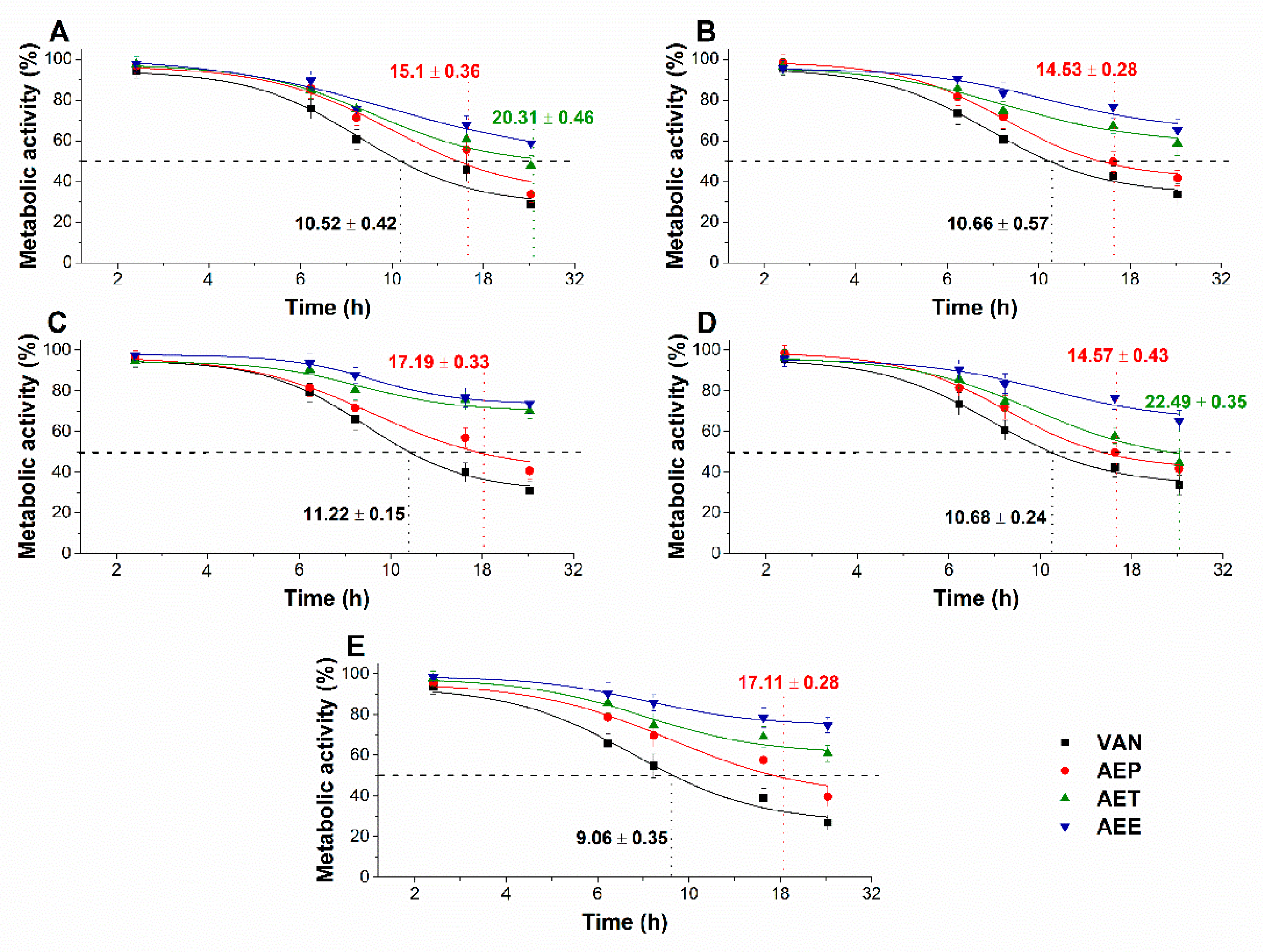
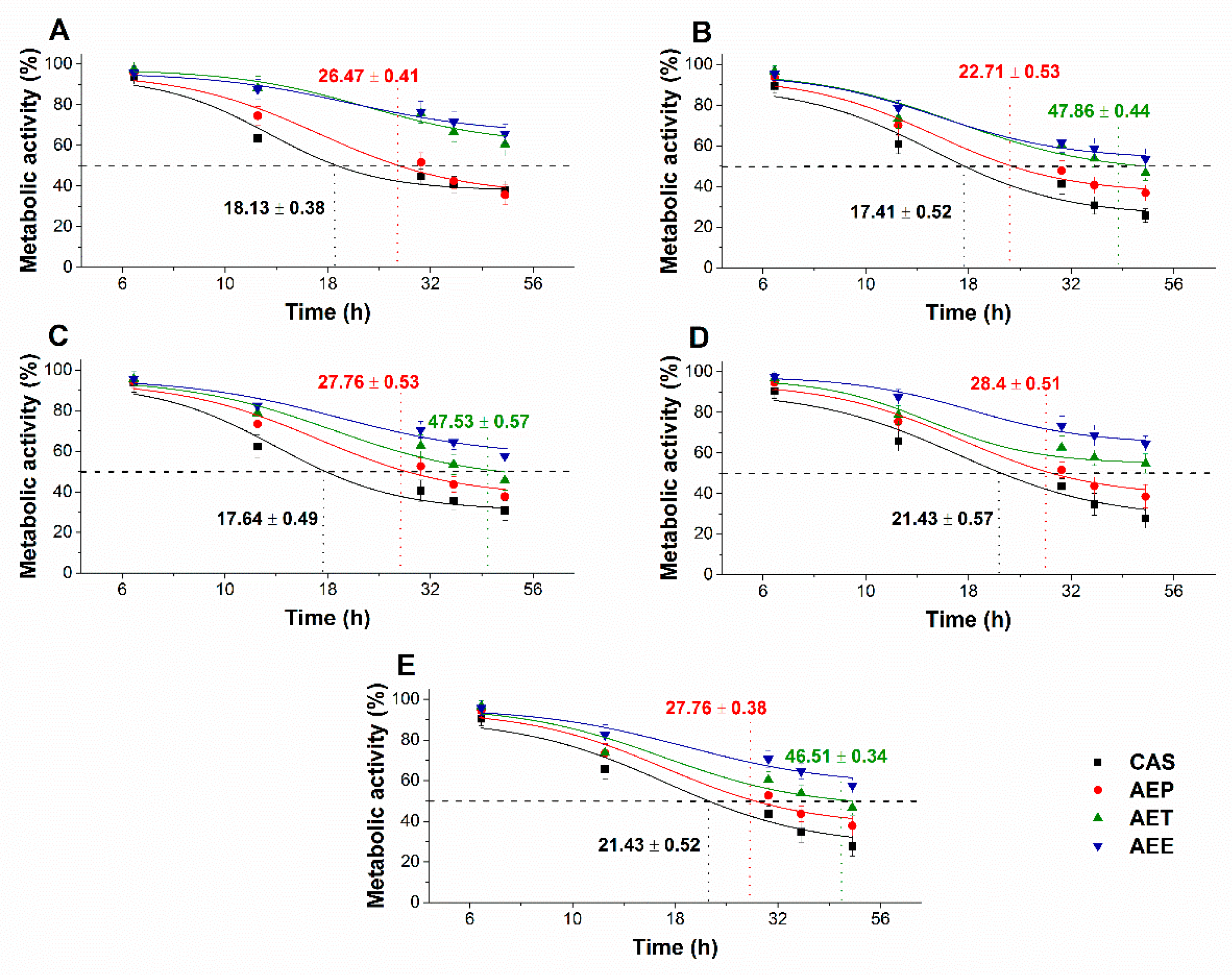
2.8.3. Effects on Microbial Cell Viability and Metabolic Activity
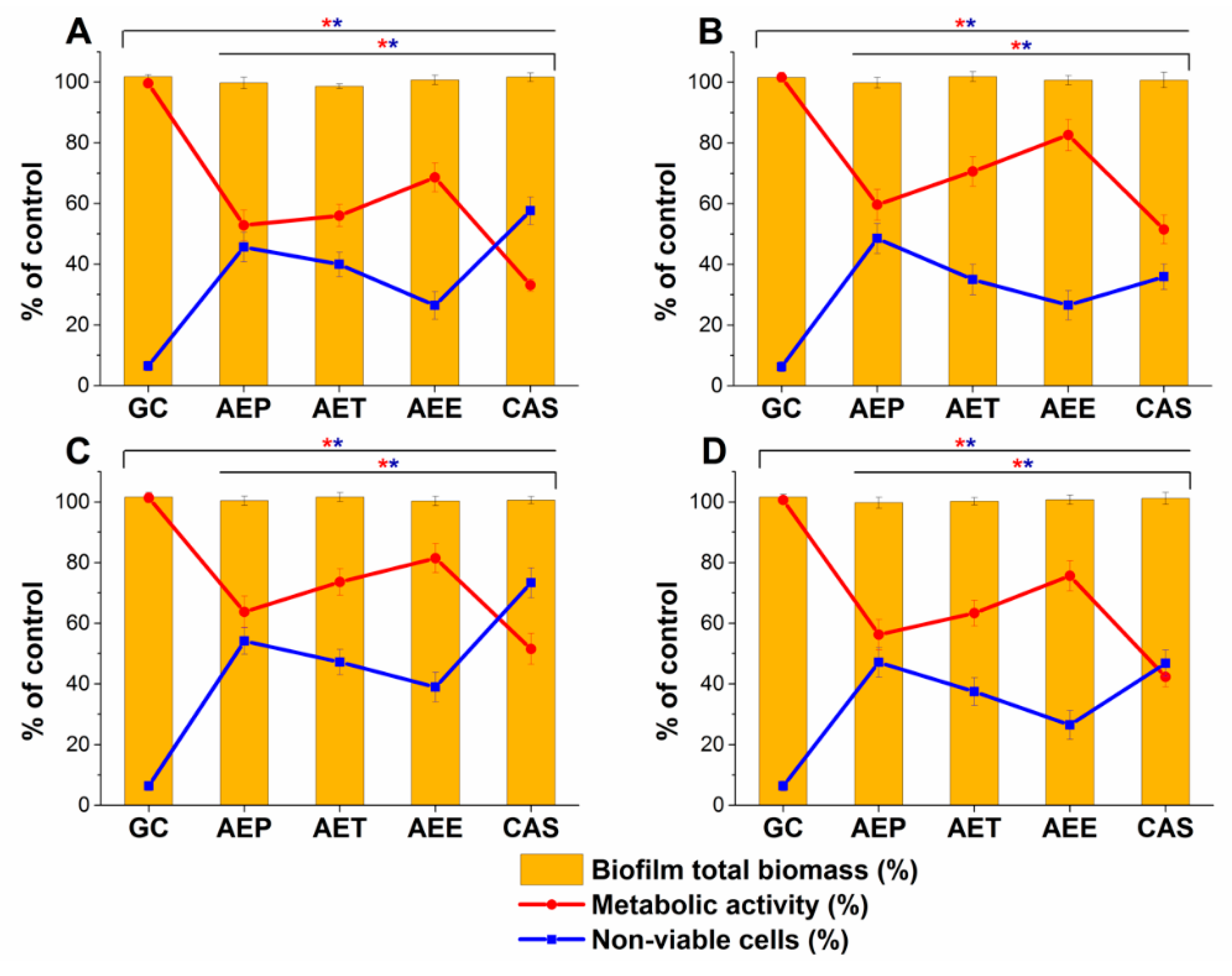
2.9. Effects on Mature Candida Spp. Biofilms
3. Discussion
4. Materials and Methods
4.1. Plant Collection and Extraction of Artemisia Essential Oil
4.2. GC-MS and GC-FID Studies
4.3. Preparation and Characterization of Pickering Nanoemulsion
4.4. In Vitro Diffusion Studies—Static Franz Diffusion Cell Method
4.5. Interaction Study between the Cellular Model (Unilamellar Liposomes) and Different Formulations of AE
4.6. Materials and Microorganisms Used for the Biological Experiments
4.7. Determination of Minimum Inhibitory Concentration (MIC90)
4.8. Determination of Minimum Effective Concentration (MEC10)
4.9. Quantification of Microbial Oxidative Stress Production in the Planktonic Cells
4.9.1. Combined ROS Generation Measurement
4.9.2. Detection of Peroxide (O22-) and Superoxide Anion (O2•-) Generation
4.10. Microbial Cytotoxicity and Viability Kinetic Assays of Planktonics
4.10.1. Determination of the Colony-Forming Unit (CFU/mL)
4.10.2. Quantification of the Planktonics’ Intracellular ATP and Total Protein Contents
4.10.3. Quantification of the Planktonics’ Metabolic Activity
4.10.4. Live/Dead Discrimination of the Planktonic Microbial Cells
4.11. Determination of the Effects on Preformed Mature Candida Biofilms
4.11.1. Evaluation of the Total Fungal Biomass
4.11.2. Metabolic Activity in the Biofilms
4.11.3. Viability Assay of the Biofilms
4.11.4. Determination of the Candida Biofilms’ Intracellular ATP and Total Protein Contents
4.12. Stastical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bilia, A.R.; Santomauro, F.; Sacco, C.; Bergonzi, M.C.; Donato, R. Essential Oil of Artemisia annua L.: An Extraordinary Component with Numerous Antimicrobial Properties. Evid. Based Complement. Alternat. Med. 2014, 2014, 1–7. [Google Scholar]
- Ćavar, S.; Maksimović, M.; Vidic, D.; Parić, A. Chemical composition and antioxidant and antimicrobial activity of essential oil of Artemisia annua L. from Bosnia. Ind. Crops Prod. 2012, 37, 479–485. [Google Scholar] [CrossRef]
- Dib, I.; Angenot, L.; Mihamou, A.; Ziyyat, A.; Tits, M. Artemisia campestris L.: Ethnomedicinal, phytochemical and pharmacological review. J. Herb. Med. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Das, S.; Czuni, L.; Báló, V.; Papp, G.; Gazdag, Z.; Papp, N.; Kőszegi, T. Cytotoxic Action of Artemisinin and Scopoletin on Planktonic Forms and on Biofilms of Candida Species. Molecules 2020, 25, 476. [Google Scholar] [CrossRef]
- Galal, A.M.; Ross, S.A.; Jacob, M.; ElSohly, M.A. Antifungal Activity of Artemisinin Derivatives. J. Nat. Prod. 2005, 68, 1274–1276. [Google Scholar] [CrossRef]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Gnonlonfin, G.J.B.; Sanni, A.; Brimer, L. Review Scopoletin—A Coumarin Phytoalexin with Medicinal Properties. Crit. Rev. Plant Sci. 2012, 31, 47–56. [Google Scholar] [CrossRef]
- De Cremer, K.; Lanckacker, E.; Cools, T.L.; Bax, M.; De Brucker, K.; Cos, P.; Cammue, B.P.A.; Thevissen, K. Artemisinins, New Miconazole Potentiators Resulting in Increased Activity against Candida albicans Biofilms. Antimicrob. Agents Chemother. 2015, 59, 421–426. [Google Scholar] [CrossRef]
- Ham, J.R.; Lee, H.-I.; Choi, R.-Y.; Sim, M.-O.; Choi, M.-S.; Kwon, E.-Y.; Yun, K.W.; Kim, M.-J.; Lee, M.-K. Anti-obesity and anti-hepatosteatosis effects of dietary scopoletin in high-fat diet fed mice. J. Funct. Foods 2016, 25, 433–446. [Google Scholar] [CrossRef]
- Loewenberg, J.R. Observations on scopoletin and scopolin metabolism. Phytochemistry 1970, 9, 361–366. [Google Scholar] [CrossRef]
- Héthelyi, E.B.; Cseko, I.B.; Grósz, M.; Márk, G.; Palinkás, J.J. Chemical Composition of the Artemisia annua Essential Oils from Hungary. J. Essent. Oil Res. 1995, 7, 45–48. [Google Scholar] [CrossRef]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S.F. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Radulović, N.S.; Randjelović, P.J.; Stojanović, N.M.; Blagojević, P.D.; Stojanović-Radić, Z.Z.; Ilić, I.R.; Djordjević, V.B. Toxic essential oils. Part II: Chemical, toxicological, pharmacological and microbiological profiles of Artemisia annua L. volatiles. Food Chem. Toxicol. 2013, 58, 37–49. [Google Scholar] [CrossRef]
- Wan, X.L.; Niu, Y.; Zheng, X.C.; Huang, Q.; Su, W.P.; Zhang, J.F.; Zhang, L.L.; Wang, T. Antioxidant capacities of Artemisia annua L. leaves and enzymatically treated Artemisia annua L. in vitro and in broilers. Anim. Feed Sci. Technol. 2016, 221, 27–34. [Google Scholar] [CrossRef]
- Bouyahya, A.; Abrini, J.; Dakka, N.; Bakri, Y. Essential oils of Origanum compactum increase membrane permeability, disturb cell membrane integrity, and suppress quorum-sensing phenotype in bacteria. J. Pharm. Anal. 2019, 9, 301–311. [Google Scholar] [CrossRef]
- Donsì, F.; Ferrari, G. Essential oil nanoemulsions as antimicrobial agents in food. J. Biotechnol. 2016, 233, 106–120. [Google Scholar] [CrossRef]
- Das, S.; Gazdag, Z.; Szente, L.; Meggyes, M.; Horváth, G.; Lemli, B.; Kunsági-Máté, S.; Kuzma, M.; Kőszegi, T. Antioxidant and antimicrobial properties of randomly methylated β cyclodextrin—captured essential oils. Food Chem. 2019, 278, 305–313. [Google Scholar] [CrossRef]
- Dhifi, W.; Bellili, S.; Jazi, S.; Bahloul, N.; Mnif, W. Essential Oils’ Chemical Characterization and Investigation of Some Biological Activities: A Critical Review. Medicines 2016, 3, 25. [Google Scholar] [CrossRef]
- Donato, R.; Santomauro, F.; Bilia, A.R.; Flamini, G.; Sacco, C. Antibacterial activity of Tuscan Artemisia annua essential oil and its major components against some foodborne pathogens. LWT Food Sci. Technol. 2015, 64, 1251–1254. [Google Scholar] [CrossRef]
- Das, S.; Horváth, B.; Šafranko, S.; Jokić, S.; Széchenyi, A.; Kőszegi, T. Antimicrobial Activity of Chamomile Essential Oil: Effect of Different Formulations. Molecules 2019, 24, 4321. [Google Scholar] [CrossRef]
- Pina-Barrera, A.M.; Alvarez-Roman, R.; Baez-Gonzalez, J.G.; Amaya-Guerra, C.A.; Rivas-Morales, C.; Gallardo-Rivera, C.T.; Galindo-Rodriguez, S.A. Application of a multisystem coating based on polymeric nanocapsules containing essential oil of Thymus vulgaris L. to increase the shelf life of table grapes (Vitis vinifera L.). IEEE Trans. NanoBioscience 2019, 18, 549–557. [Google Scholar] [CrossRef]
- Manion, C.R.; Widder, R.M. Essentials of essential oils. Am. J. Health. Syst. Pharm. 2017, 74, e153–e162. [Google Scholar] [CrossRef]
- Omonijo, F.A.; Ni, L.; Gong, J.; Wang, Q.; Lahaye, L.; Yang, C. Essential oils as alternatives to antibiotics in swine production. Anim. Nutr. 2018, 4, 126–136. [Google Scholar] [CrossRef]
- Abarca, R.L.; Rodríguez, F.J.; Guarda, A.; Galotto, M.J.; Bruna, J.E. Characterization of beta-cyclodextrin inclusion complexes containing an essential oil component. Food Chem. 2016, 196, 968–975. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.; Si, H.; Lin, L.; Chen, L. Release characteristics and antibacterial activity of solid state eugenol/β-cyclodextrin inclusion complex. J. Incl. Phenom. Macrocycl. Chem. 2011, 71, 207–213. [Google Scholar] [CrossRef]
- Alizadeh Behbahani, B.; Tabatabaei Yazdi, F.; Vasiee, A.; Mortazavi, S.A. Oliveria decumbens essential oil: Chemical compositions and antimicrobial activity against the growth of some clinical and standard strains causing infection. Microb. Pathog. 2018, 114, 449–452. [Google Scholar] [CrossRef]
- Tang, X.; Shao, Y.-L.; Tang, Y.-J.; Zhou, W.-W. Antifungal Activity of Essential Oil Compounds (Geraniol and Citral) and Inhibitory Mechanisms on Grain Pathogens (Aspergillus flavus and Aspergillus ochraceus). Mol. J. Synth. Chem. Nat. Prod. Chem. 2018, 23, 2108. [Google Scholar] [CrossRef]
- Alastruey-Izquierdo, A.; Gomez-Lopez, A.; Arendrup, M.C.; Lass-Florl, C.; Hope, W.W.; Perlin, D.S.; Rodriguez-Tudela, J.L.; Cuenca-Estrella, M. Comparison of Dimethyl Sulfoxide and Water as Solvents for Echinocandin Susceptibility Testing by the EUCAST Methodology. J. Clin. Microbiol. 2012, 50, 2509–2512. [Google Scholar] [CrossRef]
- Inouye, S.; Tsuruoka, T.; Uchida, K.; Yamaguchi, H. Effect of Sealing and Tween 80 on the Antifungal Susceptibility Testing of Essential Oils. Microbiol. Immunol. 2001, 45, 201–208. [Google Scholar] [CrossRef]
- Memar, M.Y.; Ghotaslou, R.; Samiei, M.; Adibkia, K. Antimicrobial use of reactive oxygen therapy: Current insights. Infect. Drug Resist. 2018, 11, 567–576. [Google Scholar] [CrossRef]
- Dohare, S.; Dubey, S.D.; Kalia, M.; Verma, P.; Pandey, H.; Singh, K.; Agarwal, V. Anti-biofilm activity of Eucalyptus globulus oil encapsulated silica nanoparticles against E. coli biofilm. Int. J. Pharm. Sci. Res. 2014, 5, 5013–5018. [Google Scholar]
- Astray, G.; Gonzalez-Barreiro, C.; Mejuto, J.C.; Rial-Otero, R.; Simal-Gándara, J. A review on the use of cyclodextrins in foods. Food Hydrocoll. 2009, 23, 1631–1640. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, S.; Bei, W.; Zahi, M.R.; Yuan, Q.; Liang, H. Preparation and antimicrobial activity of oregano essential oil Pickering emulsion stabilized by cellulose nanocrystals. Int. J. Biol. Macromol. 2018, 112, 7–13. [Google Scholar] [CrossRef]
- Horváth, B.; Balázs, V.L.; Horváth, G.; Széchenyi, A. Preparation and in vitro diffusion study of essential oil Pickering emulsions stabilized by silica nanoparticles for Streptococcus mutans biofilm inhibition. Flavour Frag. J. 2018, 33, 385–396. [Google Scholar] [CrossRef]
- Bankier, C.; Cheong, Y.; Mahalingam, S.; Edirisinghe, M.; Ren, G.; Cloutman-Green, E.; Ciric, L. A comparison of methods to assess the antimicrobial activity of nanoparticle combinations on bacterial cells. PLoS ONE 2018, 13, e0192093. [Google Scholar] [CrossRef]
- Pape, W.J.; Pfannenbecker, U.; Hoppe, U. Validation of the red blood cell test system as in vitro assay for the rapid screening of irritation potential of surfactants. Mol. Toxicol. 1987, 1, 525–536. [Google Scholar]
- Aveyard, R.; Binks, B.P.; Clint, J.H. Emulsions stabilised solely by colloidal particles. Adv. Colloid Interface Sci. 2003, 100–102, 503–546. [Google Scholar] [CrossRef]
- Drobna, E.; Gazdag, Z.; Culakova, H.; Dzugasova, V.; Gbelska, Y.; Pesti, M.; Subik, J. Overexpression of the YAP1, PDE2, and STB3 genes enhances the tolerance of yeast to oxidative stress induced by 7-chlorotetrazolo[5,1-c]benzo[1,2,4]triazine. FEMS Yeast Res. 2012, 12, 958–968. [Google Scholar] [CrossRef]
- Fujs, Š.; Gazdag, Z.; Poljšak, B.; Stibilj, V.; Milacic, R.; Pesti, M.; Raspor, P.; Batic, M. The oxidative stress response of the yeast Candida intermedia to copper, zinc, and selenium exposure. J. Basic Microbiol. 2005, 45, 125–135. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS Using Oxidized DCFDA and Flow-Cytometry. In Advanced Protocols in Oxidative Stress II; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; Volume 594, pp. 57–72. ISBN 978-1-60761-410-4. [Google Scholar]
- Orosz, E.; Antal, K.; Gazdag, Z.; Szabó, Z.; Han, K.-H.; Yu, J.-H.; Pócsi, I.; Emri, T. Transcriptome-Based Modeling Reveals that Oxidative Stress Induces Modulation of the AtfA-Dependent Signaling Networks in Aspergillus nidulans. Int. J. Genomics 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Ács, K.; Bencsik, T.; Böszörményi, A.; Kocsis, B.; Horváth, G. Essential Oils and Their Vapors as Potential Antibacterial Agents against Respiratory Tract Pathogens. Nat. Prod. Commun. 2016, 11, 1702–1712. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Sanders, E.R. Aseptic Laboratory Techniques: Plating Methods. J. Vis. Exp. JoVE 2012, 63, 3064. [Google Scholar] [CrossRef] [PubMed]
- Pernodet, N.; Maaloum, M.; Tinland, B. Pore size of agarose gels by atomic force microscopy. Electrophoresis 1997, 18, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Moscho, A.; Orwar, O.; Chiu, D.T.; Modi, B.P.; Zare, R.N. Rapid preparation of giant unilamellar vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 11443–11447. [Google Scholar] [CrossRef]
- Stromájer-Rácz, T.; Gazdag, Z.; Belágyi, J.; Vágvölgyi, C.; Zhao, R.Y.; Pesti, M. Oxidative stress induced by HIV-1 F34IVpr in Schizosaccharomyces pombe is one of its multiple functions. Exp. Mol. Pathol. 2010, 88, 38–44. [Google Scholar] [CrossRef]
- Sali, N.; Nagy, S.; Poór, M.; Kőszegi, T. Multiparametric luminescent cell viability assay in toxicology models: A critical evaluation. J. Pharmacol. Toxicol. Methods 2016, 79, 45–54. [Google Scholar] [CrossRef]
- Kőszegi, T.; Sali, N.; Raknić, M.; Horváth-Szalai, Z.; Csepregi, R.; Končić, M.Z.; Papp, N.; Poór, M. A novel luminol-based enhanced chemiluminescence antioxidant capacity microplate assay for use in different biological matrices. J. Pharmacol. Toxicol. Methods 2017, 88, 153–159. [Google Scholar] [CrossRef]
- Dobson, A.J.; Boulton-McDonald, R.; Houchou, L.; Svermova, T.; Ren, Z.; Subrini, J.; Vazquez-Prada, M.; Hoti, M.; Rodriguez-Lopez, M.; Ibrahim, R.; et al. Longevity is determined by ETS transcription factors in multiple tissues and diverse species. PLoS Genet. 2019, 15, e1008212. [Google Scholar] [CrossRef]
- Yu, T.; Zhou, Y.J.; Huang, M.; Liu, Q.; Pereira, R.; David, F.; Nielsen, J. Reprogramming Yeast Metabolism from Alcoholic Fermentation to Lipogenesis. Cell 2018, 174, 1549–1558.e14. [Google Scholar] [CrossRef]
Sample Availability: Samples of AEP, AET and AEE are available from the authors. |



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilizing Agent | coil (mg/mL) | Ddroplet (nm) ± SD | Stability |
|---|---|---|---|
| SNP | 0.2–3.5 | 160 ± 2.2–670 ± 37.2 | 2–3 months |
| Tw80 | 0.2–3.5 | 130 ± 0.9–590 ± 19.6 | 2–3 months |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, S.; Vörös-Horváth, B.; Bencsik, T.; Micalizzi, G.; Mondello, L.; Horváth, G.; Kőszegi, T.; Széchenyi, A. Antimicrobial Activity of Different Artemisia Essential Oil Formulations. Molecules 2020, 25, 2390. https://doi.org/10.3390/molecules25102390
Das S, Vörös-Horváth B, Bencsik T, Micalizzi G, Mondello L, Horváth G, Kőszegi T, Széchenyi A. Antimicrobial Activity of Different Artemisia Essential Oil Formulations. Molecules. 2020; 25(10):2390. https://doi.org/10.3390/molecules25102390
Chicago/Turabian StyleDas, Sourav, Barbara Vörös-Horváth, Tímea Bencsik, Giuseppe Micalizzi, Luigi Mondello, Györgyi Horváth, Tamás Kőszegi, and Aleksandar Széchenyi. 2020. "Antimicrobial Activity of Different Artemisia Essential Oil Formulations" Molecules 25, no. 10: 2390. https://doi.org/10.3390/molecules25102390
APA StyleDas, S., Vörös-Horváth, B., Bencsik, T., Micalizzi, G., Mondello, L., Horváth, G., Kőszegi, T., & Széchenyi, A. (2020). Antimicrobial Activity of Different Artemisia Essential Oil Formulations. Molecules, 25(10), 2390. https://doi.org/10.3390/molecules25102390