
Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2)

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
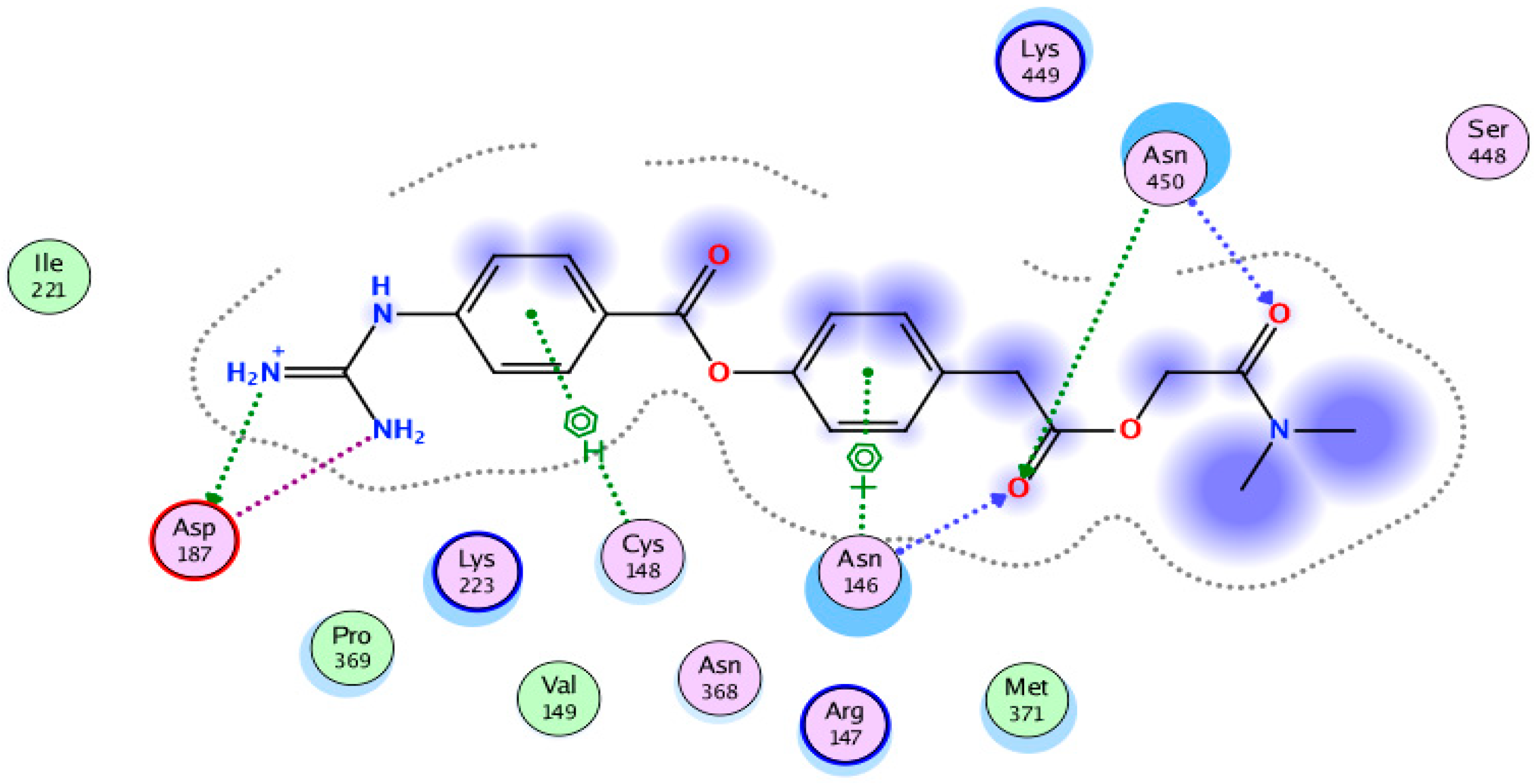
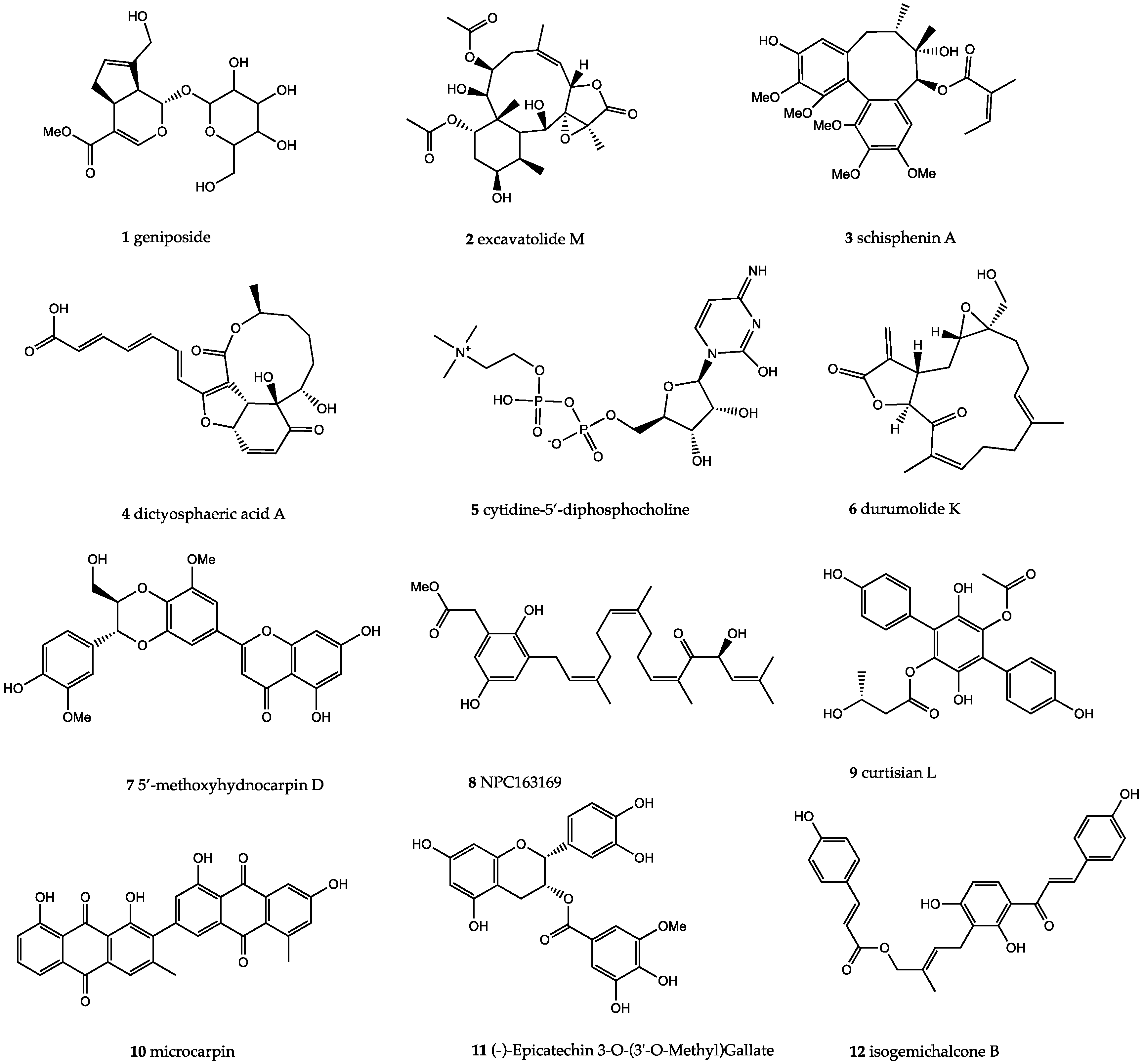
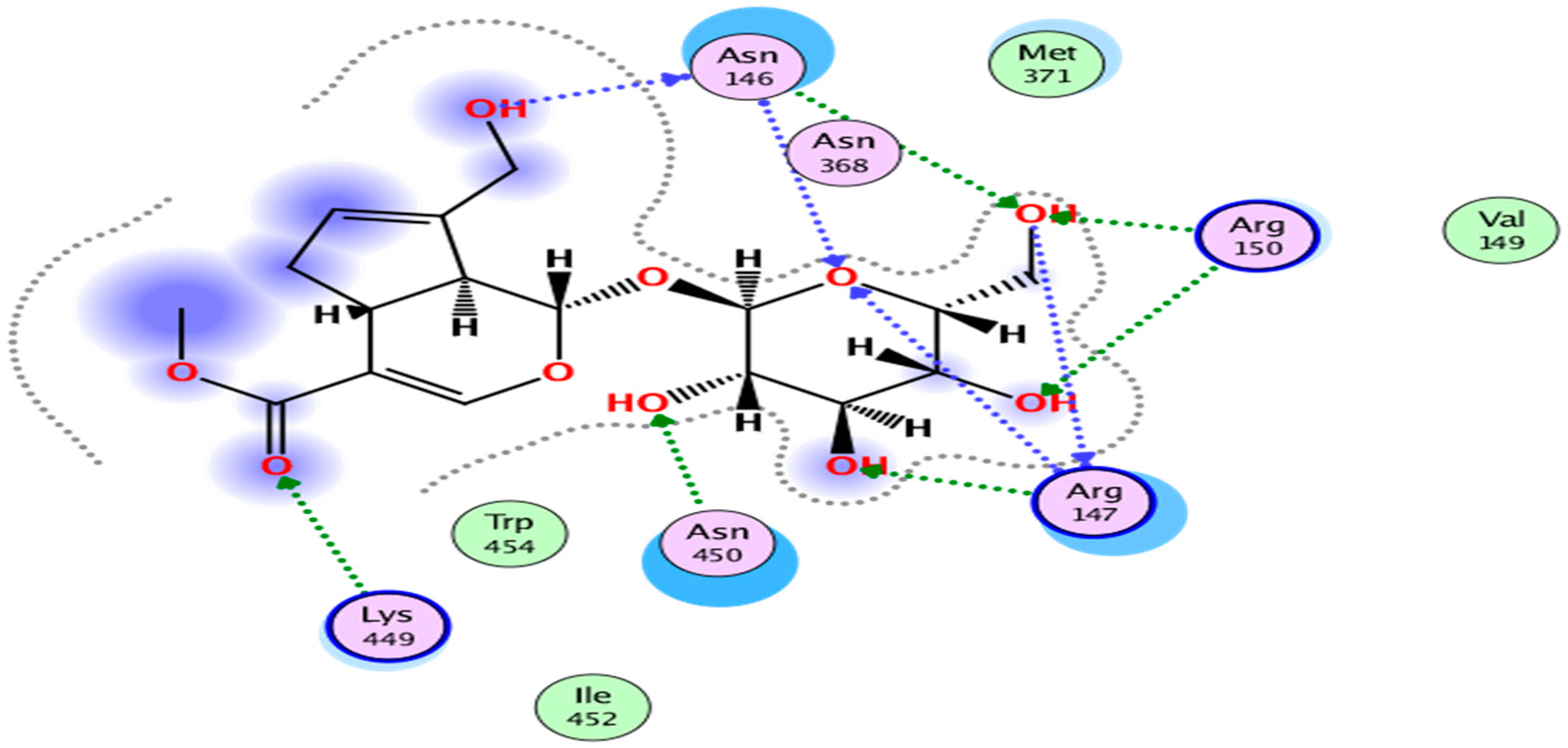
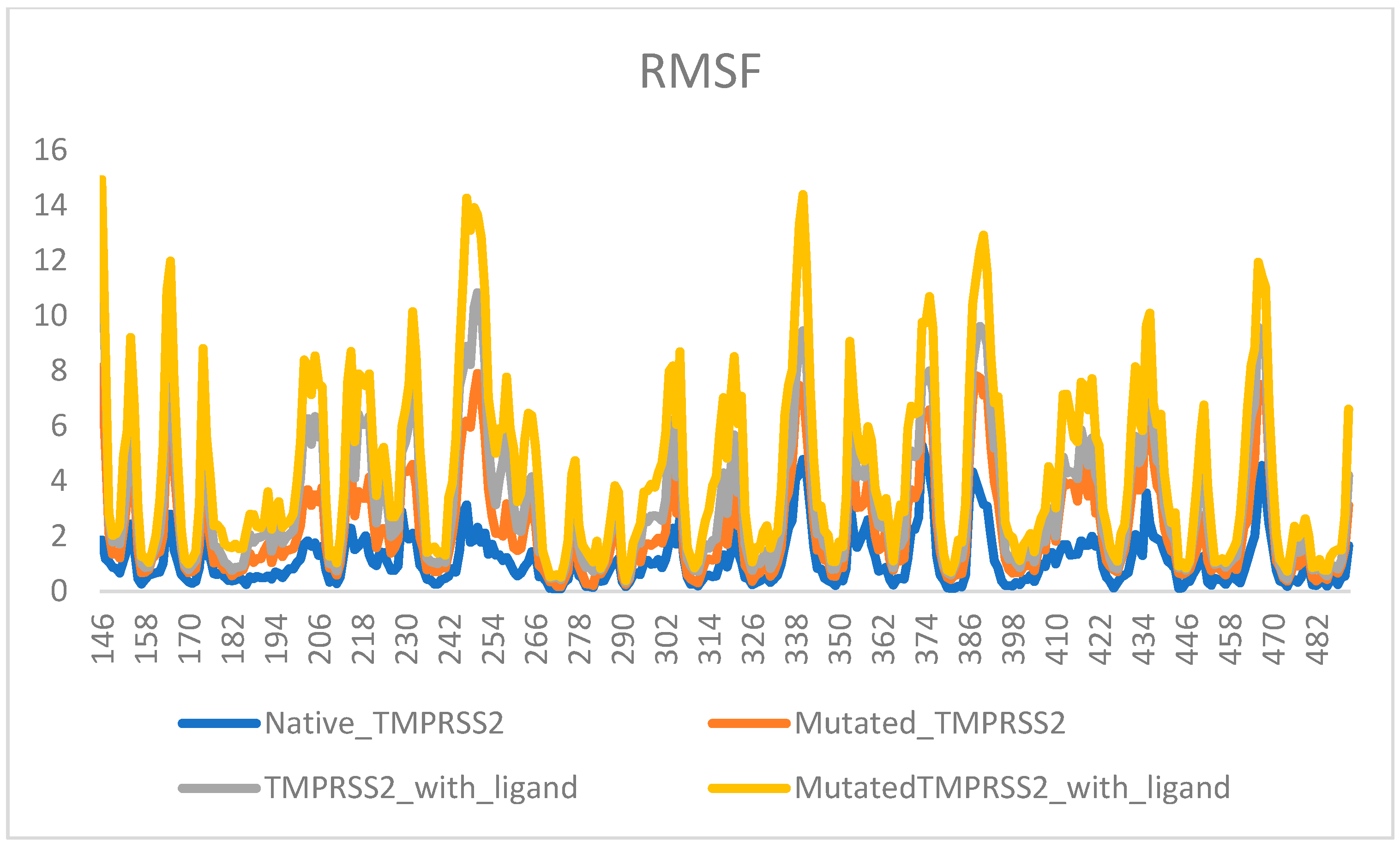
2. Results
3. Discussion
4. Methods
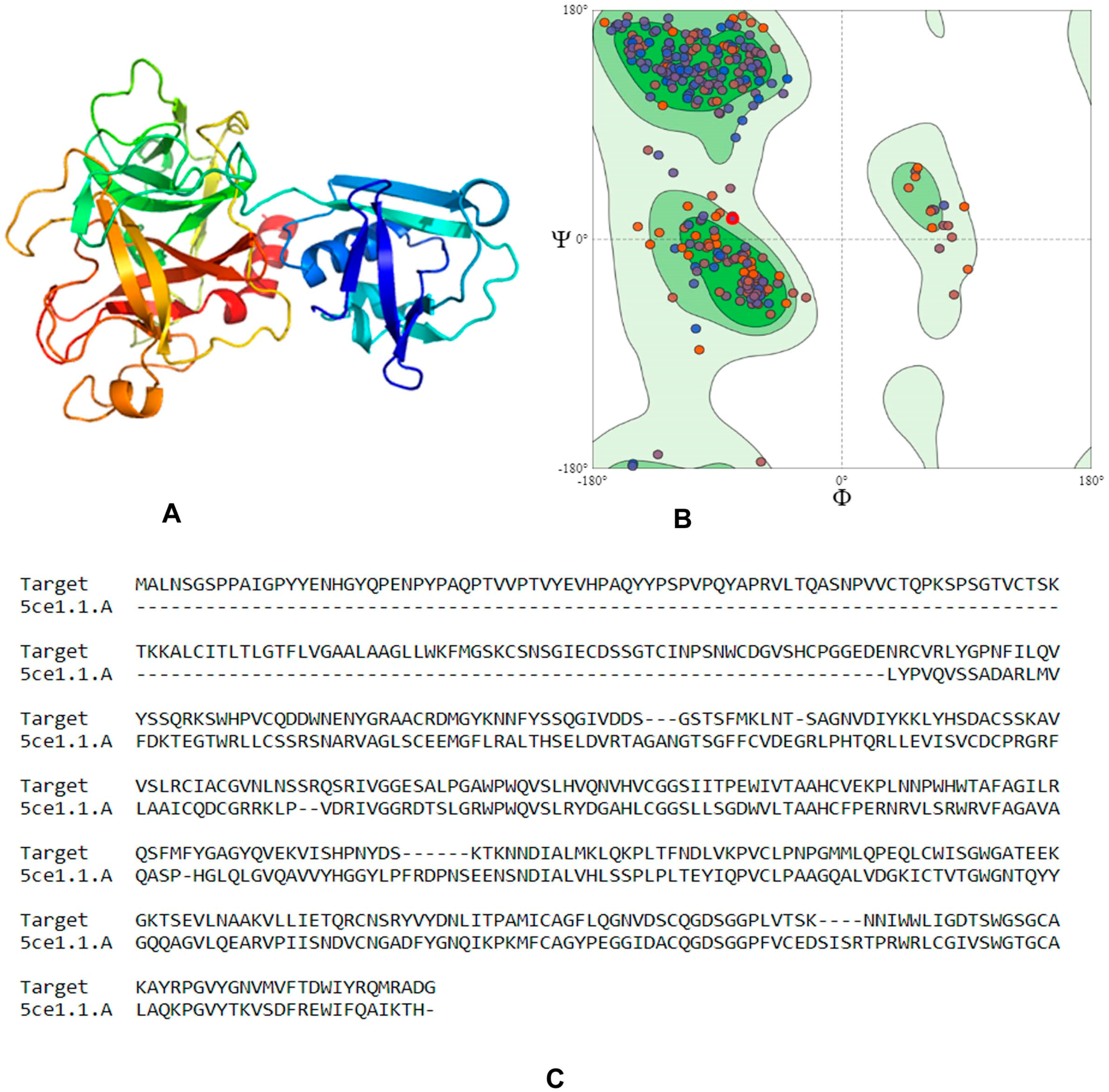
4.1. Homology Modeling
4.2. Compounds Library Screening
4.3. Molecular Docking and Downstream Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [PubMed]
- Reusken, C.B.; Broberg, E.K.; Haagmans, B.; Meijer, A.; Corman, V.M.; Papa, A.; Leitmeyer, K.; Charrel, R.; Drosten, C.; Koopmans, M. Laboratory readiness and response for novel coronavirus (2019-nCoV) in expert laboratories in 30 EU/EEA countries, January 2020. Eurosurveillance 2020, 25, 2000082. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed]
- Rest, J.S.; Mindell, D.P. SARS associated coronavirus has a recombinant polymerase and coronaviruses have a history of host-shifting. Infect. Genet. Evol. 2003, 3, 219–225. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003. 2003. Available online: http://www.who.int/csr/sars/country/table2004_04_21/en/index.html (accessed on 31 December 2003).
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Venkatagopalan, P.; Daskalova, S.M.; Lopez, L.A.; Dolezal, K.A.; Hogue, B.G. Coronavirus envelope (E) protein remains at the site of assembly. Virology 2015, 478, 75–85. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef]
- De Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [PubMed]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [PubMed]
- Song, H.C.; Seo, M.Y.; Stadler, K.; Yoo, B.J.; Choo, Q.L.; Coates, S.R.; Uematsu, Y.; Harada, T.; Greer, C.E.; Polo, J.M.; et al. Synthesis and characterization of a native, oligomeric form of recombinant severe acute respiratory syndrome coronavirus spike glycoprotein. J. Virol. 2004, 78, 10328–10335. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Arpin, N.; Talbot, P.J. Molecular characterization of the 229E strain of human coronavirus. In Coronaviruses and Their Diseases; Springer: Boston, MA, USA, 1990; pp. 73–80. [Google Scholar]
- Moore, M.J.; Dorfman, T.; Li, W.; Wong, S.K.; Li, Y.; Kuhn, J.H.; Coderre, J.; Vasilieva, N.; Han, Z.; Greenough, T.C.; et al. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J. Virol. 2004, 78, 10628–10635. [Google Scholar] [CrossRef]
- Sui, J.; Li, W.; Murakami, A.; Tamin, A.; Matthews, L.J.; Wong, S.K.; Moore, M.J.; Tallarico, A.S.; Olurinde, M.; Choe, H.; et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA 2004, 101, 2536–2541. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543–12547. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Wata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J. Virol. 2019, 93, e01815–e01818. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. 2020. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, P.; He, W.; Qin, C.; Chen, S.; Tao, L.; Wang, Y.; Tan, Y.; Gao, D.; Wang, B.; et al. NPASS: Natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018, 46, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Izuhara, R.; Kaneko, K.; Koshihara, Y.; Fujimoto, Y. 5-Lipoxygenase inhibitors isolated from Gardeniae fructus. Chem. Pharm. Bull. 1988, 36, 87–95. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, X.; Huang, Z.; Zhang, Y.; Zhou, Z. Geniposide reverses multidrug resistance in vitro and in vivo by inhibiting the efflux function and expression of P-glycoprotein. Exp. Ther. Med. 2017, 13, 437–442. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, N.; Tan, H.Y.; Guo, W.; Chen, F.; Zhong, Z.; Zhong, Z.; Man, K.; Tsao, S.W.; Lao, L.; et al. Direct inhibition of TLR4/MyD88 pathway by geniposde suppresses HIF1α-independent VEGF expression and hepatocellular carcinoma angiogenesis. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Huang, B.; Chen, P.; Huang, L.; Li, S.; Zhu, R.; Sheng, T.; Yu, W.; Chen, Z.; Wang, T. Geniposide attenuates post-ischaemic neurovascular damage via GluN2A/AKT/ERK-dependent mechanism. Cell. Physiol. Biochem. 2017, 43, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Yu, S.; Yan, H.; Guo, S.; Xiao, W.; Wang, Z.; Zhang, L.; Ding, A.; Wu, Q.; Li, S.F.Y. A review on the phytochemistry, pharmacology, pharmacokinetics and toxicology of geniposide, a natural product. Molecules 2017, 22, 1689. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.R.; McAllister, G.D.; Shanahan, S.E.; Taylor, R.J. Total Synthesis and Structural Reassignment of (+)-Dictyosphaeric Acid A: A Tandem Intramolecular Michael Addition/Alkene Migration Approach. Angew. Chem. Int. Ed. 2010, 49, 5574–5577. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective effects of a new C-Jun N-terminal kinase inhibitor in the model of global cerebral ischemia in rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef]
- Stermitz, F.R.; Lorenz, P.; Tawara, J.N.; Zenewicz, L.A.; Lewis, K. Synergy in a medicinal plant: Antimicrobial action of berberine potentiated by 5′-methoxyhydnocarpin, a multidrug pump inhibitor. Proc. Natl. Acad. Sci. USA 2000, 97, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.L.; Rana, A.C. Withania somnifera (Ashwagandha): A review. Pharmacogn. Rev. 2007, 1, 129–136. [Google Scholar]
- Celano, R.; Piccinelli, A.L.; Pagano, I.; Roscigno, G.; Campone, L.; De Falco, E.; Russo, M.; Rastrelli, L. Oil distillation wastewaters from aromatic herbs as new natural source of antioxidant compounds. Food Res. Int. 2017, 99, 298–307. [Google Scholar] [CrossRef]
- Menachery, V.D.; Dinnon, K.H.; Yount, B.L.; McAnarney, E.T.; Gralinski, L.E.; Hale, A.; Graham, R.L.; Scobey, T.; Anthony, S.J.; Wang, L.; et al. Trypsin treatment unlocks barrier for zoonotic bat coronavirus infection. J. Virol. 2020, 94, e01774-19. [Google Scholar] [CrossRef] [PubMed]
- Bugge, T.H.; Antalis, T.M.; Wu, Q. Type II transmembrane serine proteases. J. Biol. Chem. 2009, 284, 23177–23181. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kawase, M.; Matsuyama, S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013, 87, 12552–12561. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.no. | Compound ID | Docking Score | Toxicity | M.Wt (g/mol) | H-Bond Donor | H-bond Acceptor | LogP | LogS |
|---|---|---|---|---|---|---|---|---|
| 1 | NPC306344 | −14.69 | No | 388.37 | 5 | 9 | −1.45 | −0.72 |
| 2 | NPC473877 | −14.38 | Yes epoxide | 482.53 | 3 | 7 | 0.38 | −3.04 |
| 3 | NPC470916 | −14.27 | No | 516.59 | 2 | 8 | 4.16 | −5.62 |
| 4 | NPC66108 | −14.02 | No | 416.63 | 3 | 7 | 1.8 | −3.78 |
| 5 | NPC328914 | −13.96 | No | 488.33 | 5 | 12 | −3.94 | −0.53 |
| 6 | NPC476270 | −13.92 | Yes epoxide | 346.42 | 1 | 4 | 2.67 | −3.75 |
| 7 | NPC84324 | −13.59 | No | 494.45 | 4 | 9 | 3.41 | −5.43 |
| 8 | NPC163169 | −13.55 | No | 484.63 | 3 | 5 | 7.03 | −7.00 |
| 9 | NPC155015 | −13.38 | No | 454.43 | 5 | 7 | 3.84 | −5.22 |
| 10 | NPC19631 | −13.31 | No | 506.47 | 4 | 8 | 6.47 | −8.21 |
| 11 | NPC53889 | −13.10 | No | 456.40 | 6 | 9 | 2.03 | −4.05 |
| 12 | NPC19622 | −13.07 | No | 486.52 | 4 | 6 | 5.47 | −6.55 |
| 13 | Camostat mesylate(standard) | −11.06 | No | 399.43 | 3 | 3 | 0.23 | −2.83 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. https://doi.org/10.3390/molecules25102271
Rahman N, Basharat Z, Yousuf M, Castaldo G, Rastrelli L, Khan H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules. 2020; 25(10):2271. https://doi.org/10.3390/molecules25102271
Chicago/Turabian StyleRahman, Noor, Zarrin Basharat, Muhammad Yousuf, Giuseppe Castaldo, Luca Rastrelli, and Haroon Khan. 2020. "Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2)" Molecules 25, no. 10: 2271. https://doi.org/10.3390/molecules25102271
APA StyleRahman, N., Basharat, Z., Yousuf, M., Castaldo, G., Rastrelli, L., & Khan, H. (2020). Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules, 25(10), 2271. https://doi.org/10.3390/molecules25102271